

Abstract
Hepatitis C virus (HCV) infection is a global health problem characterized by a high rate of chronic infection, which may in part be due to a defect in myeloid dendritic cells (mDCs). This defect appears to be remedied by treatment with interferon-α (IFN-α) -based antiviral therapies; however, the molecular mechanisms underlying mDC dysfunction in HCV infection and restoration by IFN-α treatment are unclear. The ubiquitin-editing protein A20 plays a crucial role in controlling the maturation, cytokine production and immunostimulatory function of mDCs. We propose that the expression of A20 correlates with the function of mDCs during HCV infection and IFN-α therapy. In this study, we observed that A20 expression in mDCs isolated from chronically HCV-infected subjects was significantly higher than healthy subjects or subjects achieving sustained virological responses (SVR) following antiviral treatment. Notably, A20 expression in mDCs from HCV patients during IFN-α treatment was significantly lower than for untreated patients, SVR patients, or healthy subjects. Besides, A20 expression in mDCs stimulated by polyI:C differed between HCV patients and healthy subjects, and this difference could be abrogated by the treatment with IFN-α in vitro. Additionally, A20 expression by polyI:C-activated mDCs, with or without IFN-α treatment, negatively correlated with the expression of HLA-DR, CD86 and CCR7, and the secretion of interleukin-12 (IL-12), but positively associated with the production of IL-10. Importantly, silencing A20 expression using small interfering RNAs increased the production of IL-12 in mDCs of chronically HCV-infected individuals. These findings suggest that A20 plays a crucial role in negative regulation of innate immune responses during chronic viral infection.
Keywords: A20, chronic infection, hepatitis C virus, interferon-α, myeloid dendritic cells
Introduction
Hepatitis C virus (HCV) is a major health problem, with an estimated 170 million individuals infected worldwide.1 Over 80% of HCV-infected individuals develop chronic infection, which is associated with liver cirrhosis and hepatocellular carcinoma.2 The effective clearance of HCV depends on a robust and prolonged antiviral T-cell response.3,4 However, chronically HCV-infected individuals are often characterized by attenuated HCV-specific CD4+ and CD8+ T-cell responses, which allows HCV to evade immune surveillance and persist in infected individuals.5–7 Dendritic cells (DCs) are the most potent professional antigen-presenting cells, and play a pivotal role in initiating and regulating innate and adaptive immune responses to viral infection.8 There are two DC subsets: myeloid DCs (mDCs) and lymphoid DCs. The mDCs are derived from a myeloid bone marrow precursor and their primary function is uptake, processing and presentation of endogenous and exogenous antigens.9 Dysfunction of mDCs may be the primary cause of impaired T-cell responses in HCV-infected individuals.10 Indeed, mDCs isolated from chronically HCV-infected patients show a reduced capacity to induce a mixed lymphocyte response, impaired maturation and decreased interleukin-12 (IL-12) production.11–16 Intriguingly, recent studies indicate that treatment with interferon-α (IFN-α)/ribavirin (RBV) restores the function of mDCs in HCV patients, a finding that correlates with higher antiviral T-cell responses.17–21 However, the mechanisms underlying mDC dysfunction in chronic HCV infection and functional restoration by IFN-α-based therapy remain largely unknown.
A20 is a potent anti-inflammatory protein that negatively regulates nuclear factor-κB signalling via its de-ubiquitinating, E3 ligase and ubiquitin-binding functions.22,23 Several genetic and biochemical studies show that A20 inhibits tumour necrosis factor (TNF), Toll-like receptor (TLR), nucleotide-binding oligomerization domain protein and CD40-mediated activation of nuclear factor-κB signalling by editing the ubiquitin chains on key signalling proteins that transduce these signals.24–27 A20-deficient mice spontaneously develop cachexia and severe inflammatory disease.28 Single nucleotide polymorphisms in the human A20 locus (also known as TNFAIP3) correlate with several human autoimmune diseases, including psoriasis, systemic lupus erythematosus and rheumatoid arthritis.29–31 Additionally, a recent study shows that A20-silenced DCs induce robust anti-tumour immune responses, resulting in the rejection of tumours engrafted into immunized mice.32 Similarly, A20-silenced human DCs show an increased capacity to polarize IFN-γ-producing CD4+ T-cell responses, and to prime tumour-specific CD8+ T cells.33 These data indicate that A20 plays a critical role in controlling DC function, and subsequently T-cell responses.
Here we hypothesize that A20 expression might be up-regulated and associated with mDC dysfunction in patients with chronic hepatitis C and could be corrected by the treatment with IFN-α. To test this hypothesis, we examined A20 expression in mDCs isolated from treatment-naive patients, patients during IFN-α therapy, patients achieving sustained virological responses (SVR) after therapy, and healthy subjects. We also examined A20 expression and its function in human mDCs stimulated with TLR3 ligand-PolyI:C, with or without IFN-α, which are known to synergistically induce IL-12 production. Our study demonstrated that A20 expression in mDCs from chronically HCV-infected patients was significantly higher than that in healthy subjects or patients achieving SVR following antiviral treatment. However, A20 expression in mDCs from patients during IFN-α treatment was significantly lower when compared with treatment-naive patients, healthy subjects, or patients who achieved SVR. We also found that A20 was over-expressed in both unstimulated and polyI:C-stimulated mDCs from HCV patients when compared with healthy subjects, and its expression level was negatively associated with IL-12 production. Notably, IFN-α significantly decreased A20 expression in mDCs, and restored the HCV-mediated suppression of IL-12, a key cytokine linking innate and adaptive immunity. Importantly, silencing A20 expression using small interfering RNAs (siRNAs) increased the production of IL-12 in mDCs of chronically HCV-infected individuals.
Materials and methods
Subjects
Fifty-seven HCV-infected patients and 15 healthy subjects were enrolled in this study. The characteristics of all subjects are listed in Table 1. Group one comprised 31 HCV-infected patients that had never been treated (treatment-naive patients). The second group comprised 16 patients that had been undergoing IFN-α/RBV antiviral therapy for 1–6 months (patients receiving antiviral treatment); the viral load was significantly lower in these patients. The third group contained 10 patients who had achieved SVR after antiviral treatment (patients achieved SVR). All HCV patients were negative for HIV and hepatitis B virus (HBV). Testing of the HCV viral load was performed by the Department of Infectious Diseases at Tangdu Hospital. The fourth group comprised 15 healthy subjects who were negative for HCV, HBV and HIV. Written informed consent was obtained from all subjects. This study was approved by the institutional review boards of the Forth Military Medical University and Tangdu Hospital.
Table 1.
Characteristics of the study subjects
| Study subjects | Healthy subjects (n = 15) | Treatment-naive patients (n = 31) | Patients receiving antiviral treatment(n = 16) | Patients achieving SVR (n = 10) |
|---|---|---|---|---|
| Age (years), mean (range) | 32·1 (22–43) | 34·3 (17–46) | 37·8 (21–50) | 39·6 (27–52) |
| Gender (male/female) | 10/5 | 22/9 | 10/6 | 7/3 |
| Genotype (G1/G2) | N/A | 15/16 | 9/7 | 4/6 |
| HCV viral load (105 IU/μl), mean (range) | N/A | 66·86 (1·30–371) | N/A | N/A |
HCV, hepatitis C virus; N/A, not applicable; SVR, sustained virological response.
Isolation and culture of mDCs
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by density gradient centrifugation using Ficoll–Hypaque (Sigma-Aldrich, St Louis, MO). PBMCs were then viably frozen in a solution comprising 90% fetal bovine serum (Gibco, Grand Island, NY) and 10% dimethyl sulphoxide (Invitrogen, Carlsbad, CA) and stored in liquid nitrogen until use. Myeloid DCs were isolated from PBMCs using the BDCA-1+ Dendritic Cell Isolation Kit (Miltenyi Biotech, Auburn, CA) according to the manufacturer’s instructions. The mDCs isolated from 10 treatment-naive patients and 10 healthy subjects were cultured separately at 2 × 105 cells/ml in 0·2 ml of culture medium in 96-well round bottom plates with RPMI-1640 (Gibco) containing 5% fetal bovine serum, 100 μg/ml penicillin-streptomycin and 2 mm l-glutamine (Gibco). Myeloid DCs were cultured in medium alone, or supplemented with 50 μg/ml polyI:C (Invivogen, San Diego, CA) for 12 hr, or supplemented with IFN-α (1000 U/ml; Roche, Shanghai, China) for 48 hr, or supplemented with IFN-α (1000 U/ml) for 36 hr, followed by the addition of polyI:C (50 μg/ml) for a further 12 hr (48 hr in total).
Flow cytometry
To examine DC maturation, cells were cultured in vitro and stained with allophycocyanin-conjugated anti-HLA-DR, R-phycoerythrin-Cy5-conjugated anti-CD86, and phycoerythrin-conjugated anti-CCR7 antibodies (BD Biosciences, San Jose, CA). Staining of intracellular A20 was performed using an Inside Stain kit (eBioscience, San Diego, CA) and the DyLight488-conjugated anti-A20 antibody (Abcam, Cambridge, MA). Isotype-matched control antibodies (Abcam) were used to determine the levels of background staining. The mDCs were analysed on a FACS Aria II flow cytometer (BD, Piscataway, NJ) and the data were analysed using cell quest software (BD, San Jose, CA).
Quantitative real-time PCR
A20 mRNA expression was measured by a relative real-time quantitative PCR assay. Briefly, total RNA was isolated from 2 × 105 mDCs using the RNeasy Minikit (Qiagen, Hilden, Germany) and reverse transcribed into cDNA using oligo(dT) primers and the Revert Aid First Strand cDNA Synthesis Kit (Fermentas, Burlington, Canada). Quantitative real-time PCR was performed using the ABI 7500 system (Applied Biosystems, Carlsbad, CA) and the SYBR Premix Ex TaqII kit (Takara, Otsu, Shiga, Japan) according to the manufacturer’s instructions. The expression of β-actin was measured to normalize the A20 mRNA levels. The following primers were used: A20 sense, 5′-GGAAGCTTGTGGCGCTGAA-3′; and A20 antisense, 5′-TCCAGTTGCCAGCGGAATTTA-3′. The primers were designed and synthesized by Takara (Dalian, China).
Cytokine ELISA
The levels of IL-12p70 and IL-10 secreted into the culture supernatant by mDCs were measured by using Quantikine M ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
A20 siRNA silencing
A total of 3 × 105 mDCs isolated from chronically HCV-infected patients were incubated with 10 μmol A20 siRNA duplex or control siRNA in 200 ml siRNA Transfection Medium in a six-well plate according to the manufacturer’s instructions (Santa Cruz Biotechnology, Dallas, TX). After 6 hr of incubation at 37°, normal growth medium containing 2× concentrations of normal serum and antibiotics was added. The transfected cells were then incubated for 18–72 hr, followed by detection of A20, IL-12p70 and IL-10 as described previously.
Statistical analysis
Student’s t-test was used to analyse the significance of differences. Spearman’s correlation analysis was also performed. All tests were two-tailed and P values < 0·05 were considered significant.
Results
A20 expression is up-regulated in mDCs from HCV-infected patients compared with healthy subjects and down-regulated by IFN-α therapy
As an initial approach to characterize the role of A20 in mDCs from HCV-infected individuals and its response to IFN treatment, mDCs were isolated from PBMCs of four groups of subjects by sorting with anti-BDCA-1 magnetic beads, to a purity of > 90%. A20 mRNA and protein expression were measured using quantitative real-time PCR and flow cytometry. As shown in Fig. 1, A20 was found to be constitutively expressed in mDCs of all study subjects. Chronically HCV-infected individuals (treatment-naive patients) exhibited significantly elevated A20 mRNA and protein expression in mDCs when compared with healthy subjects. Conversely, A20 expression in patients undergoing IFN-α-based antiviral therapy was significantly lower than that in treatment-naive patients and healthy subjects, suggesting that A20 expression is down-regulated by IFN-α-based antiviral therapy. Moreover, A20 expression in patients achieving SVR was lower than HCV treatment-naive patients, but higher than patients undergoing IFN-α therapy, and had no significant difference with healthy subjects (Fig. 1a–c). These results suggested that A20 may be up-regulated by HCV infection but down-regulated by IFN-α therapy.
Figure 1.
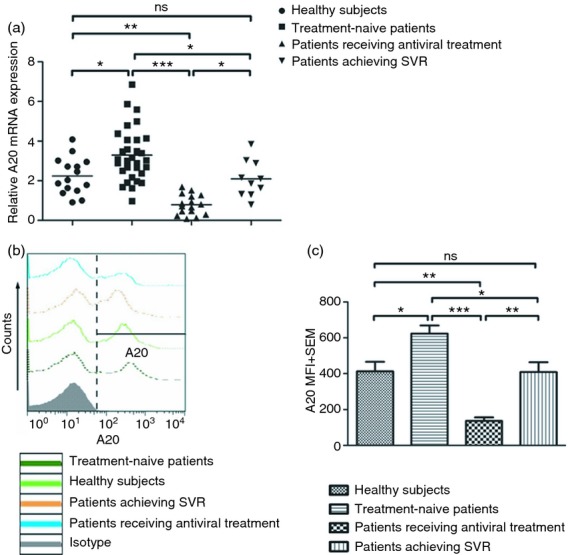
A20 expression increased in myeloid dendritic cells (mDCs) isolated from patients chronically infected with hepatitis C virus (HCV) compared with healthy subjects and is decreased in mDCs isolated from HCV patients receiving antiviral therapy. (a) Quantitative real-time PCR was used to detect A20 mRNA levels in mDCs isolated from treatment-naive patients (n = 31), patients receiving antiviral treatment (n = 16), patients achieving sustained virological responses (n = 10) and healthy subjects (n = 15). Each symbol represents an individual subject, and the horizontal bars represent the median values. (b, c) A20 expression in mDCs was assessed by flow cytometry. Corresponding changes in % gated and mean fluorescence intensity (MFI) are shown with isotype control in representative histograms (b). Summary data of corresponding changes in MFI from subjects from the four groups are shown by bar figure (c). *P < 0·05, **P < 0·05, ***P < 0·001, ns: not significant.
A20 mRNA expression in mDCs is down-regulated by IFN-α treatment in vitro
Myeloid DC dysfunction has been reported in chronic HCV patients, but the underlying mechanisms remain unclear.11–16 Since A20 regulates the immunological function of mDCs,32,33 we hypothesized that the expression of A20 might be dysregulated in mDCs by HCV infection. To test this hypothesis, we isolated mDCs from HCV-infected individuals and healthy subjects, stimulated with polyI:C (a ligand for TLR3), followed by measuring A20 mRNA levels by quantitative PCR. As shown in Fig. 2(a), the level of A20 mRNA in mDCs from healthy subjects was significantly reduced upon stimulation with 50 μg/ml polyI:C for 12 hr. In contrast, there was no significant decrease of A20 mRNA expression by mDCs isolated from HCV patients following polyI:C stimulation. This result suggested that A20 expression in mDCs may be dysregulated by chronic HCV infection. To determine the effect of IFN-α on A20 expression, mDCs were cultured with 1000 U/ml IFN-α or with IFN-α/polyI:C. A20 mRNA expression by mDCs from both treatment-naive patients and healthy subjects was markedly down-regulated upon stimulation with either IFN-α (Fig. 2b) or IFN-α/polyI:C (Fig. 2c). Moreover, A20 mRNA expression of mDCs stimulated with IFN-α/polyI:C was down-regulated more than when stimulated with IFN-α alone. These data suggest that in addition to directly affecting A20 expression, IFN-α synergistically sensitizes mDCs to TLR stimulation.
Figure 2.
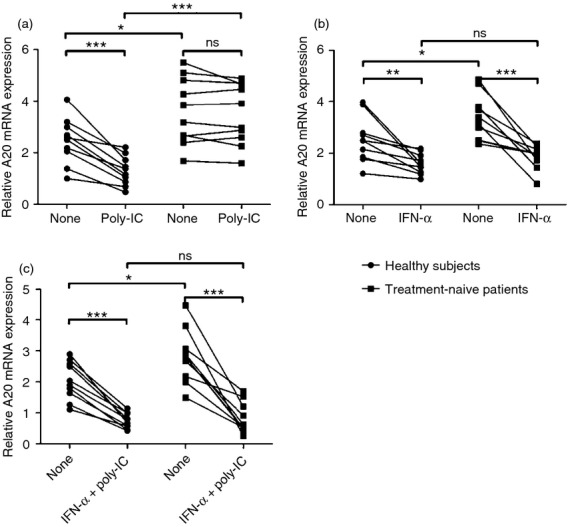
Interferon-α (IFN-α) down-regulates the expression of A20 mRNA in myeloid dendritic cells (mDCs) cultured in vitro. The mDCs purified from treatment-naive patients (n = 10) and healthy subjects (n = 10) were cultured in the presence of poly-IC, IFN-α, or IFN-α/poly-IC. A20 mRNA levels were detected by quantitative real-time PCR. (a) A20 mRNA expression in mDCs isolated from treatment-naive patients and healthy subjects and then treated for 12 hr with polyI:C (50 μg/ml). (b) A20 mRNA expression in mDCs isolated from healthy subjects and treatment-naive patients and then cultured with IFN-α (1000 U/ml) for 48 hr. (c) Myeloid DCs isolated from healthy subjects and treatment-naive patients and then cultured with IFN-α (1000 U/ml) for 48 hr. PolyI:C (50 μg/ml) was added to the culture for the final 12 hr. Each symbol represents an individual subject. *P < 0·05, **P < 0·01, ***P < 0·001, ns: not significant.
A20 protein expression is down-regulated, whereas HLA-DR, CD86 and CCR7 expressions are up-regulated, in mDCs by IFN-α treatment in vitro
To further characterize the relationship between A20 expression and mDC function in patients with HCV infection and undergoing IFN-α treatment, we examined the expression of A20 protein and several phenotypic markers in mDCs by flow cytometry. Myeloid DCs isolated from 10 treatment-naive patients and 10 healthy subjects were stimulated with polyI:C, IFN-α, or IFN-α/polyI:C as described above. As shown in Fig. 3(a), the expression of A20 protein mirrored the A20 mRNA level. More than 90% of mDCs expressed the MHC Class II molecule, HLA-DR; however, polyI:C-activated mDCs from healthy subjects showed higher HLA-DR expression than that of HCV patients (Fig. 3b). Similarly, the expression of the co-stimulatory molecule CD86 and chemokine receptor CCR7 was found to be consistently higher in mDCs isolated from healthy subjects than those from HCV patients after stimulation with polyI:C (Fig. 3c,d). Conversely, treatment with either IFN-α or IFN-α/polyI:C up-regulated HLA-DR, CD86 and CCR7 expression to a similar extent in mDCs from treatment-naive patients and healthy subjects (Fig. 3b–d). These data suggest that mDCs from treatment-naive patients are in an immature state; and that IFN-α-induced phenotypic changes of mDCs from HCV patients may be associated with A20 expression.
Figure 3.
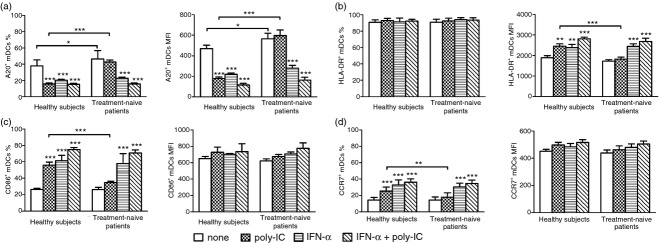
Interferon-α (IFN-α) down-regulates the expression of A20 and up-regulates the expression of HLA-DR, CD86 and CCR7 in myeloid dendritic cells (mDCs). The mDCs isolated from healthy subjects (n = 10) and treatment-naive patients (n = 10) were cultured in medium alone, or in medium containing polyI:C, IFN-α, or IFN-α/polyI:C. The expression of A20, HLA-DR, CD86 and CCR7 was then detected by flow cytometry. The percentage (mean ± SD) and the mean fluorescence intensity (MFI) of mDCs expressing A20 (a), HLA-DR (b), CD86 (c) or CCR7 (d) are shown. *P < 0·05; **P < 0·01; ***P < 0·001.
IL-12 production is improved, whereas IL-10 is inhibited, in mDCs by IFN-α treatment in vitro
We next examined the production of IL-12 and IL-10 by mDCs treated with polyI:C, IFN-α, or IFN-α/poly-I:C in vitro. As shown in Fig. 4(a), mDCs isolated from HCV patients secreted much less IL-12 than those from healthy subjects. Treatment with IFN-α or IFN-α/polyI:C increased IL-12 expression by mDCs from HCV patients to levels similar to those of healthy subjects. In contrast, mDCs from HCV patients exhibited overall low levels of IL-10 production, but their levels were still significantly higher than those from healthy subjects (Fig. 4b). Exposure of mDCs from HCV patients to IFN-α/polyI:C reduced IL-10 production to levels similar to those observed in healthy subjects. We also examined the association between A20 expression and mDC phenotype or cytokine production in HCV-infected individuals by Pearson correlation analysis. As shown in Fig. 5, A20 expression was found to negatively correlate with the expression of HLA-DR (Fig. 5a), CD86 (Fig. 5b), CCR7 (Fig. 5c) and IL-12 secretion (Fig. 5d), but positively correlate with IL-10 production (Fig. 5e).
Figure 4.
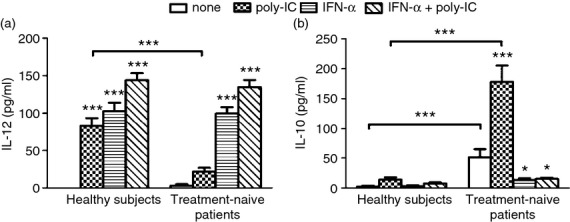
Myeloid dendritic cells (mDCs) show increased production of interleukin-12 (IL-12) and decreased production of IL-10 upon treatment with interferon-α (IFN-α). The mDCs isolated from healthy subjects (n = 10) and treatment-naive patients (n = 10) were cultured in medium alone or in medium containing polyI:C, IFN-α, or IFN-α/polyI:C. The levels of IL-12 (a) or IL-10 (b) secreted into the culture supernatants were measured by ELISA. Data represent the mean ± SD. *P < 0·05; ***P < 0·001.
Figure 5.
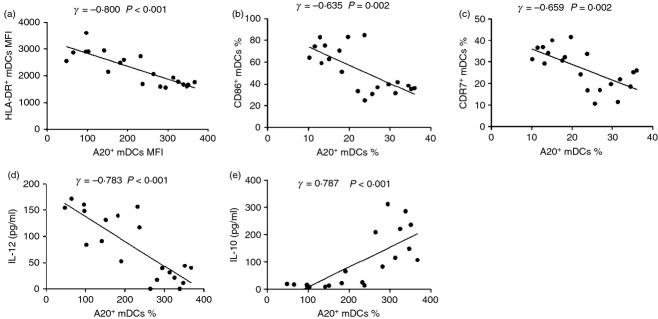
A20 expression was negatively correlated with the expression of HLA-DR, CD86, CCR7 and interleukin-12 (IL-12), but positively correlated with IL-10 production. Immunophenotype molecules HLA-DR, CD86, CCR7 and cytokines IL-12, IL-10 in myeloid dendritic cells isolated from treatment-naive patients and activated with poly-I:C in the presence or absence of interferon-α were detected and their correlations with A20 were analysed by Spearman’s correlation analysis with two-tailed significance denoted in the upper right corner of each analysis. **P < 0·01, ***P < 0·001.
Silencing A20 affects the production of IL-12 and IL-10 in mDCs from treatment-naive patients
To determine whether A20 was truly involved in HCV-induced IL-12 suppression and IL-10 production, we silenced A20 gene expression in mDCs isolated from treatment-naive patients by siRNA transfection. Compared with control siRNA, mDCs transfected with A20 siRNA displayed inhibition of A20 mRNA (Fig. 6a) and protein expression (Fig. 6b). The inhibition of A20 by siRNA was found to be more robust at 48 hr, and most significant at 72 hr after transfection. Correspondingly, HCV-mediated inhibition of IL-12 expression by mDCs was corrected (Fig. 6c) and the production of IL-10 was decreased (Fig. 6d), most notably at 72 hr, following transfection of A20 siRNA compared with control siRNA. Collectively, these data suggest that HCV up-regulates A20 expression, and A20 negatively regulates IL-12 expression in mDCs.
Figure 6.
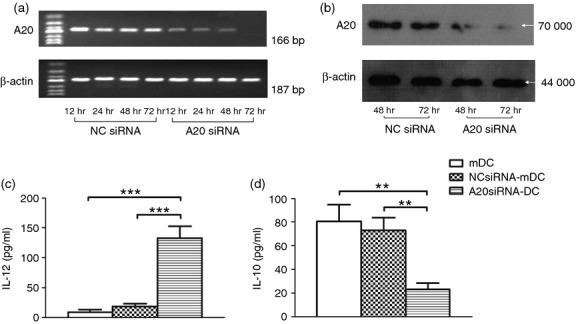
Silencing A20 rescues myeloid dendritic cell (mDC) interleukin-12 (IL-12) suppression and decreases IL-10 production in mDCs from treatment-naive patients. Myeloid DCs transfected with A20 small interfering RNA (siRNA) or control siRNA were subjected to RT-PCR and Western blot detection of A20 expression and ELISA of IL-12p70 and IL-10 secreted into the culture supernatant by mDCs. A20 siRNA inhibited hepatitis C virus (HCV) infection induced A20 expression by RT-PCR (a) and Western blot detection (b), a stronger silencing of A20 protein was achieved 48 hr and 72 hr after transfection. HCV-mediated IL-12 inhibition was partially restored (c) and the IL-10 production was decreased (d) in mDCs transfected with A20 siRNA. The data were reproducible in three repeated experiments.**P < 0·01, ***P < 0·001.
Discussion
A20, also known as TNFAIP3, is a newly identified negative immunomodulator and plays a crucial role in controlling the maturation, cytokine production, and immunostimulatory potency of DCs.34 So far, no studies have examined the expression and function of A20 in mDCs during chronic HCV infection or in response to IFN-α-based antiviral therapy. The present study found that the expression of A20 was significantly higher in mDCs isolated from chronically infected HCV patients when compared with healthy subjects and down-regulated by IFN-α therapy. A20 mRNA expression in mDCs isolated from treatment-naive patients and healthy subjects was markedly down-regulated upon stimulation by IFN-α, and more significantly by IFN-α/polyI:C. Moreover, A20 expression in polyI:C-activated mDCs, with or without IFN-α treatment, negatively correlated with the expression of HLA-DR, CD86 and CCR7, as well as the secretion of IL-12, but positively correlated with the production of IL-10. Importantly, silencing A20 rescued IL-12 inhibition and decreased IL-10 production in mDCs isolated from treatment-naive patients. Taken together, these data suggest that A20 is up-regulated and associated with mDC dysfunction in chronically HCV-infected patients, and can be corrected by the IFN-α treatment.
A20 was initially identified as a primary response gene in human umbilical vein endothelial cells stimulated with TNF-α; resting human umbilical vein endothelial cells do not express A20.35 The expression of A20 is up-regulated by stimulators, such as TNF-α or lipopolysaccharide, resulting in inhibition of nuclear factor-κB signalling, supporting the idea that A20 is an inducible molecule.25,27,36–38 However, most previous studies have examined the regulation of A20 expression in cell lines, or in primary cells that do not express A20 in the resting state.36–38 In contrast, our results showed that significant levels of A20 were expressed in the resting mDCs purified ex vivo from healthy subjects and A20 expression was remarkably down-regulated in polyI:C-activated mDCs characterized by up-regulation of HLA-DR, CD86 and CCR7 expression and IL-12 production. Interestingly, previous studies have shown that A20 expression is down-regulated in mature thymocytes and in activated peripheral T cells, thereby initiating antigen-induced immune responses; whereas the loss of A20 mRNA upon activation is not stimulus-specific, and can be induced by pharmacological stimuli, including phytohaemagglutinin, IL-2 or anti-CD3 monoclonal antibody.39 Immune homeostasis in unchallenged animals is thought to be the result of the absence of infectious agents or inflammatory stimuli; however, recent studies suggest that immunostimulatory ligands, such as microbial molecules, may be sensed in non-perturbed animals.40 Hence, we conclude that resting immune cells (such as DCs and T cells) that encountered commensal microbes and host inflammatory factors constitutively express certain levels of A20 to prevent inappropriate activation, thereby maintaining immune homeostasis. When the strength and duration of the stimulus attain a certain level, A20 expression is down-regulated in these cells, allowing them to actively participate in the immune response. This notion needs to be confirmed by more studies using primary immune cells.
Previous studies indicate that mDCs isolated from chronically HCV-infected patients show a decreased immune capacity11–16 and the treatment with IFN-α/RBV restores the immune function of mDCs.17–21 The present study suggests that mDCs from chronically HCV-infected patients express high levels of A20 compared with healthy subjects, and that A20 expression is markedly down-regulated by IFN-based antiviral therapy. Physiological doses of RBV do not alter the phenotype of mDCs,17 whereas IFN-α induces mDC maturation.41 Our study shows that mDCs from treatment-naive patients exhibit almost no response to polyI:C treatment and a restored immunophenotype upon stimulation with IFN-α. Interferon-α, a type-1 IFN, has potent antiviral and immunomodulatory properties and shares a common receptor on eukaryotic cells.42 Cellular responses to IFN-α are mediated by the coordinated activation of different signalling pathways, including the Janus-activated kinase-signal transducer and activator of transcription pathway;43 however, the mechanism underlying the effects of IFN-α on DC homeostasis in HCV patients has yet to be identified. Interestingly, a recent study shows that A20 is a candidate negative regulator of the signalling cascade upstream of IFN regulatory factor 3 during innate antiviral responses, suggesting that A20 physically inhibits the dimerization of IFN regulatory factor 3 after TLR3 is bound by dsRNA or Newcastle disease virus, thereby suppressing IFN production.44 Hence, it is possible that A20 interacts with IFN-α in some form of a negative feedback loop. On the one hand, IFN-α may activate mDCs (and immune responses) by inhibiting the expression of A20. On the other hand, it may inhibit the function of mDCs, thereby inhibiting the immune response by either directly or indirectly reducing the production of IFN-α. Our data indicate that the expression of A20 by polyI:C-activated mDCs, with or without IFN-α treatment, negatively correlated with the expression of HLA-DR, CD86 and CCR7, as well as the secretion of IL-12, and positively correlated with the production of IL-10. HLA-DR and CD86 play pivotal roles in antigen presentation and immune activation. CCR7 controls DC migration to inflammatory sites and homing to secondary lymphoid organs.45 Immature mDCs express CCR1, CCR2 and CCR5; the ligands for these receptors are produced at sites of inflammation and act as chemoattractants for DCs.46 After binding and taking up antigens in the periphery, mDCs mature and up-regulate the expression of MHC class I and II molecules, adhesion molecules and co-stimulatory molecules. As they mature, they down-regulate the expression of receptors for inflammatory chemokines, and increase their expression of CCR7. This induces the cells to migrate towards chemokines constitutively expressed in the secondary lymphoid organs: the site of T-cell stimulation.47 Given the data presented herein, A20 may alter the function of mDCs through regulation of these molecules during chronic HCV infection. Finally, we investigated the effect of A20 on mDC production of IL-12 and IL-10. It is well known that IL-12 promotes inflammation, whereas IL-10 has opposing effects. The mDCs isolated from HCV patients and cultured with polyI:C secrete significantly lower amounts of IL-12, and larger amounts of IL-10, than mDCs isolated from healthy subjects. These findings are supported by previous studies showing that IL-12 activates NK cells48 and induces the differentiation of Th1 cells, which are the main producers of endogenous IFN,49 whereas IL-10 inhibits cytokine secretion by activated monocytes50 and Th1 cells.48 Furthermore, blockade of the A20 pathway significantly enhances IL-12 production and decreases IL-10 production in mDCs of HCV-infected subjects, suggesting that the A20 pathway plays a crucial role in suppressing mDC functions. Our previous studies show that IL-12 expression is affected by several negative immunomodulators, such as T cell immunoglobulin and mucin domain-containing protein-3(Tim-3), programmed death-1(PD-1), suppressor of cytokine signaling-1(SOCS-1).51–53 These observations imply that negative immunomodulators, such as A20, along with Tim-3, PD-1 and SOCS-1, are associated with or linked in an inhibitory circuit or form a cluster to prevent ubiquitin degradation, and exert an integrated role in suppressing cell functioning.
In conclusion, this study demonstrates that the expression of A20 is up-regulated and closely correlated with mDC function in HCV-infected patients. This presents a novel and crucial role for A20 as a negative regulator of DC function in innate immune responses to HCV infection. These results may help to clarify the molecular mechanisms underlying the immune response to chronic HCV infection, and identify new targets for immune therapy of HCV infection.
Disclosures
This work was supported by National Natural Science Foundation of China to Zhansheng Jia (81170389, 81273218). The authors have no financial conflicts of interest.
References
- Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–67. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann Intern Med. 2000;132:296–305. doi: 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- Lechner F, Wong DK, Dunbar PR, et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. 2000;191:1499–512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194:1395–406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KM, Thimme R, Melpolder JJ, et al. Differential CD4+ and CD8+ T-cell responsiveness in hepatitis C virus infection. Hepatology. 2001;33:267–76. doi: 10.1053/jhep.2001.21162. [DOI] [PubMed] [Google Scholar]
- Lechner F, Gruener NH, Urbani S, et al. CD8+ T lymphocyte responses are induced during acute hepatitis C virus infection but are not sustained. Eur J Immunol. 2000;30:2479–87. doi: 10.1002/1521-4141(200009)30:9<2479::AID-IMMU2479>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Gerlach JT, Diepolder HM, Jung MC, et al. Recurrence of hepatitis C virus after loss of virus-specific CD4+ T-cell response in acute hepatitis C. Gastroenterology. 1999;117:933–41. doi: 10.1016/s0016-5085(99)70353-7. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Piccioli D, Tavarini S, Borgogni E, et al. Functional specialization of human circulating CD16 and CD1c myeloid dendritic-cell subsets. Blood. 2007;109:5371–9. doi: 10.1182/blood-2006-08-038422. [DOI] [PubMed] [Google Scholar]
- Dolganiuc A, Paek E, Kodys K, Thomas J, Szabo G. Myeloid dendritic cells of patients with chronic HCV infection induce proliferation of regulatory T lymphocytes. Gastroenterology. 2008;135:2119–27. doi: 10.1053/j.gastro.2008.07.082. [DOI] [PubMed] [Google Scholar]
- Bain C, Fatmi A, Zoulim F, Zarski JP, Trepo C, Inchauspe G. Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology. 2001;120:512–24. doi: 10.1053/gast.2001.21212. [DOI] [PubMed] [Google Scholar]
- Auffermann-Gretzinger S, Keeffe EB, Levy S. Impaired dendritic cell maturation in patients with chronic, but not resolved, hepatitis C virus infection. Blood. 2001;97:3171–6. doi: 10.1182/blood.v97.10.3171. [DOI] [PubMed] [Google Scholar]
- Ulsenheimer A, Gerlach JT, Gruener NH, et al. Detection of functionally altered hepatitis C virus-specific CD4 T cells in acute and chronic hepatitis C. Hepatology. 2003;37:1189–98. doi: 10.1053/jhep.2003.50194. [DOI] [PubMed] [Google Scholar]
- Tsubouchi E, Akbar SM, Horiike N, Onji M. Infection and dysfunction of circulating blood dendritic cells and their subsets in chronic hepatitis C virus infection. J Gastroenterol. 2004;39:754–62. doi: 10.1007/s00535-003-1385-3. [DOI] [PubMed] [Google Scholar]
- Anthony DD, Yonkers NL, Post AB, Asaad R, Heinzel FP, Lederman MM, Lehmann PV, Valdez H. Selective impairments in dendritic cell-associated function distinguish hepatitis C virus and HIV infection. J Immunol. 2004;172:4907–16. doi: 10.4049/jimmunol.172.8.4907. [DOI] [PubMed] [Google Scholar]
- Averill L, Lee WM, Karandikar NJ. Differential dysfunction in dendritic cell subsets during chronic HCV infection. Clin Immunol. 2007;123:40–9. doi: 10.1016/j.clim.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes E, Salio M, Cerundolo V, Medlin J, Murphy S, Dusheiko G, Klenerman P. Impact of α interferon and ribavirin on the function of maturing dendritic cells. Antimicrob Agents Chemother. 2004;48:3382–9. doi: 10.1128/AAC.48.9.3382-3389.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi E, Akbar SM, Murakami H, Horiike N, Onji M. Isolation and functional analysis of circulating dendritic cells from hepatitis C virus (HCV) RNA-positive and HCV RNA-negative patients with chronic hepatitis C: role of antiviral therapy. Clin Exp Immunol. 2004;137:417–23. doi: 10.1111/j.1365-2249.2004.02544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicinnati VR, Kang J, Sotiropoulos GC, Hilgard P, Frilling A, Broelsch CE, Gerken G, Beckebaum S. Altered chemotactic response of myeloid and plasmacytoid dendritic cells from patients with chronic hepatitis C: role of α interferon. J Gen Virol. 2008;89:1243–53. doi: 10.1099/vir.0.83517-0. [DOI] [PubMed] [Google Scholar]
- Pachiadakis I, Chokshi S, Cooksley H, Farmakiotis D, Sarrazin C, Zeuzem S, Michalak TI, Naoumov NV. Early viraemia clearance during antiviral therapy of chronic hepatitis C improves dendritic cell functions. Clin Immunol. 2009;131:415–25. doi: 10.1016/j.clim.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Sacchi A, Agrati C, D’Offizi G, Vlassi C, Rozera G, Abbate I, Capobianchi MR, Martini F. The basal activation state of DC subsets correlates with anti-HCV treatment outcome in HCV/HIV co-infected patients. Clin Immunol. 2011;138:178–86. doi: 10.1016/j.clim.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Verstrepen L, Verhelst K, van Loo G, Carpentier I, Ley SC, Beyaert R. Expression, biological activities and mechanisms of action of A20 (TNFAIP3) Biochem Pharmacol. 2010;80:2009–20. doi: 10.1016/j.bcp.2010.06.044. [DOI] [PubMed] [Google Scholar]
- Vereecke L, Beyaert R, van Loo G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009;30:383–91. doi: 10.1016/j.it.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science. 2000;289:2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–60. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- Hitotsumatsu O, Ahmad RC, Tavares R, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity. 2008;28:381–90. doi: 10.1016/j.immuni.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares RM, Turer EE, Liu CL, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 2010;33:181–91. doi: 10.1016/j.immuni.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turer EE, Tavares RM, Mortier E, et al. Homeostatic MyD88-dependent signals cause lethal inflammation in the absence of A20. J Exp Med. 2008;205:451–64. doi: 10.1084/jem.20071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair RP, Duffin KC, Helms C, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RR, Cotsapas C, Davies L, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:1059–61. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton PR, Clayton DG, Cardon LR, et al. Association scan of 14 500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39:1329–37. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song XT, Evel-Kabler K, Shen L, Rollins L, Huang XF, Chen SY. A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T-cell-mediated suppression. Nat Med. 2008;14:258–65. doi: 10.1038/nm1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckpot K, Aerts-Toegaert C, Heirman C, Peeters U, Beyaert R, Aerts JL, Thielemans K. Attenuated expression of A20 markedly increases the efficacy of double-stranded RNA-activated dendritic cells as an anti-cancer vaccine. J Immunol. 2009;182:860–70. doi: 10.4049/jimmunol.182.2.860. [DOI] [PubMed] [Google Scholar]
- Hong B, Song XT, Rollins L, Berry L, Huang XF, Chen SY. Mucosal and systemic anti-HIV immunity controlled by A20 in mouse dendritic cells. J Clin Invest. 2011;121:739–51. doi: 10.1172/JCI42656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit VM, Green S, Sarma V, et al. Tumor necrosis factor-α induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. J Biol Chem. 1990;265:2973–8. [PubMed] [Google Scholar]
- He KL, Ting AT. A20 inhibits tumor necrosis factor (TNF) α-induced apoptosis by disrupting recruitment of TRADD and RIP to the TNF receptor 1 complex in Jurkat T cells. Mol Cell Biol. 2002;22:6034–45. doi: 10.1128/MCB.22.17.6034-6045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YY, Li L, Han KJ, Zhai Z, Shu HB. A20 is a potent inhibitor of TLR3- and Sendai virus-induced activation of NF-κB and ISRE and IFN-β promoter. FEBS Lett. 2004;576:86–90. doi: 10.1016/j.febslet.2004.08.071. [DOI] [PubMed] [Google Scholar]
- Arvelo MB, Cooper JT, Longo C, et al. A20 protects mice from d-galactosamine/lipopolysaccharide acute toxic lethal hepatitis. Hepatology. 2002;35:535–43. doi: 10.1053/jhep.2002.31309. [DOI] [PubMed] [Google Scholar]
- Tewari M, Wolf FW, Seldin MF, O’Shea KS, Dixit VM, Turka LA. Lymphoid expression and regulation of A20, an inhibitor of programmed cell death. J Immunol. 1995;154:1699–706. [PubMed] [Google Scholar]
- Hammer GE, Turer EE, Taylor KE, et al. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol. 2011;12:1184–93. doi: 10.1038/ni.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luft T, Pang KC, Thomas E, Hertzog P, Hart DN, Trapani J, Cebon J. Type I IFNs enhance the terminal differentiation of dendritic cells. J Immunol. 1998;161:1947–53. [PubMed] [Google Scholar]
- Bekisz J, Schmeisser H, Hernandez J, Goldman ND, Zoon KC. Human interferons α β and ω. Growth Factors. 2004;22:243–51. doi: 10.1080/08977190400000833. [DOI] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon mediated signalling. Nat Rev Immunol. 2005;5:375–86. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- Saitoh T, Yamamoto M, Miyagishi M, et al. A20 is a negative regulator of IFN regulatory factor 3 signaling. J Immunol. 2005;174:1507–12. doi: 10.4049/jimmunol.174.3.1507. [DOI] [PubMed] [Google Scholar]
- Jang MH, Sougawa N, Tanaka T, et al. CCR7 is critically important for migration of dendritic cells in intestinal lamina propria to mesenteric lymph nodes. J Immunol. 2006;176:803–10. doi: 10.4049/jimmunol.176.2.803. [DOI] [PubMed] [Google Scholar]
- Dieu MC, Vanbervliet B, Vicari A, et al. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med. 1998;188:373–86. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- O’Garra A, Murphy KM. From IL-10 to IL-12: how pathogens and their products stimulate APCs to induce TH1 development. Nat Immunol. 2009;10:929–32. doi: 10.1038/ni0909-929. [DOI] [PubMed] [Google Scholar]
- Alli RS, Khar A. Interleukin-12 secreted by mature dendritic cells mediates activation of NK cell function. FEBS Lett. 2004;559:71–6. doi: 10.1016/S0014-5793(04)00026-2. [DOI] [PubMed] [Google Scholar]
- Liu BS, Groothuismink ZM, Janssen HL, Boonstra A. Role for IL-10 in inducing functional impairment of monocytes upon TLR4 ligation in patients with chronic HCV infections. J Leukoc Biol. 2011;89:981–8. doi: 10.1189/jlb.1210680. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ma CJ, Wang JM, Ji XJ, Wu XY, Moorman JP, Yao ZQ. Tim-3 regulates pro- and anti-inflammatory cytokine expression in human CD14+ monocytes. J Leukoc Biol. 2012;91:189–96. doi: 10.1189/jlb.1010591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ma CJ, Wang JM, Ji XJ, Wu XY, Jia ZS, Moorman JP, Yao ZQ. Tim-3 negatively regulates IL-12 expression by monocytes in HCV infection. PLoS ONE. 2011;6:e19664. doi: 10.1371/journal.pone.0019664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ma CJ, Ni L, et al. Cross-talk between programmed death-1 and suppressor of cytokine signaling-1 in inhibition of IL-12 production by monocytes/macrophages in hepatitis C virus infection. J Immunol. 2011;186:3093–103. doi: 10.4049/jimmunol.1002006. [DOI] [PubMed] [Google Scholar]