

Abstract
Mutations in leucine-rich repeat kinase 2 (LRRK2) represent the most frequent genetic lesions associated with Parkinson’s disease (PD). LRRK2 function and the pathogenic mechanisms of LRRK2 genetic mutations remain poorly understood. Cho et al report in this issue of The EMBO Journal a new interaction between LRRK2 and the ER exit site (ERES) protein Sec16A, important for anterograde transport of secretory proteins. Moreover, a disease-associated mutation disrupts LRRK2–Sec16A interaction and ERES function.
See also: HJ Cho et al (October 2014)
PD is characterized by the degeneration of dopaminergic neurons (DNs) in substantia nigra of the midbrain, although other brain regions are also affected. The cause of DN degeneration is largely unknown, although mitochondrial dysfunction, protein misfolding, oxidative stress, and ER stress have been frequently observed in patients or animal models (Dexter & Jenner, 2013). The identification of genetic mutations associated with familial forms of PD has offered the much needed entry point to pry into the mysterious disease process. Dominant mutations in LRRK2 are found in both familial and sporadic PD cases and represent the most frequent genetic lesions associated with PD (Dachsel & Farrer, 2010). The normal function of LRRK2 relevant to disease pathogenesis remains a contentious topic in the field.
One approach to elucidating the cellular function of LRRK2 is through identification of its interacting proteins or substrates. Cho et al (2014) used mass spectrometry-based proteomic screening to identify Sec16A as a new interacting partner of LRRK2 (REF). Sec16A is a conserved protein required for the formation of ERES, a specialized ER domain mediating the formation of coat protein complex II (COPII) vesicles in the early phase of ER-to-Golgi transport (Hughes et al, 2009). Using a variety of approaches, including co-IP and co-localization of endogenous LRRK2 and Sec16A proteins, and fractionation of LRRK2 and Sec16A containing protein complexes, the authors verified the LRRK2–Sec16A association. To probe the physiological significance of this interaction, the authors made use of LRRK2−/− mouse cells and tissues in a series of elegant cell biological studies encompassing immunohistochemistry, EM, and live imaging of COPII-mediated vesicle transport. They showed that in the absence of LRRK2, the localization of Sec16A to juxtanuclear ERES is compromised. As a result, the organization of ERES and the formation and transport of COPII vesicles are impaired. To assess the relevance of the LRRK2–Sec16A interaction to PD pathogenesis, the authors analyzed the interaction between Sec16A and a number of pathogenic forms of LRRK2. Strikingly, the R1441C mutation located in the ROC domain of LRRK2 significantly affected LRRK2–Sec16A interaction, whereas other mutations including those altering the kinase activity of LRRK2 had no effect. These results offer further support for the notion that different LRRK2 pathogenic mutations cause the disease through distinct mechanisms, and suggest that the LRRK2–Sec16A interaction is mediated or regulated by the GTPase activity of LRRK2. Using hippocampal neurons in culture to further address the potential relevance of the observed interaction for neurodegeneration, the authors demonstrated that LRRK2 and Sec16A interact and co-colocalize at dendritic ERES (dERES), that Sec16A delocalizes from dERES in the absence of LRRK2, and that the dERES-dependent transport of NMDA receptors is compromised in LRRK2−/− neurons. Overall, the study by Cho et al (2014) makes a convincing case that LRRK2 regulates an early step in the ER-to-Golgi secretory pathway (REF). Interestingly, overexpression of α-Syn, the main component of Lewy bodies and dominant mutations in which have also been linked to PD, affects a later step in the ER-to-Golgi transport (Cooper et al, 2006).
Interference with ER-to-Golgi transport by genetic lesions in LRRK2 and α-Syn would be consistent with the strong genetic interactions between these two genes in animal models (Lin et al, 2009) and highlight the prominent role of this basic cellular process in DN maintenance. It would be interesting to test how other identified cellular and molecular functions of LRRK2 might be integrated into this process (Fig 1). For example, LRRK2 regulates the dynamics of microtubules (Parisiadou & Cai, 2010), which are critical for ERES function (Watson et al, 2005). LRRK2 has also been implicated in interacting with AGO2 and to influence translational repression (Gehrke et al, 2010). In Drosophila, a translational repressor complex was shown to be required for ERES formation and secretion of developmental regulators (Wilhelm et al, 2005), and AGO2 has been implicated in ER-associated translational quality control (Karamyshev et al, 2014). Thus, LRRK2 could serve as a regulator of these processes in a coordinated manner.
Figure 1. A diagram depicting possible mechanisms of LRRK2-mediated regulation of ER-to-Golgi transport of secretory proteins.
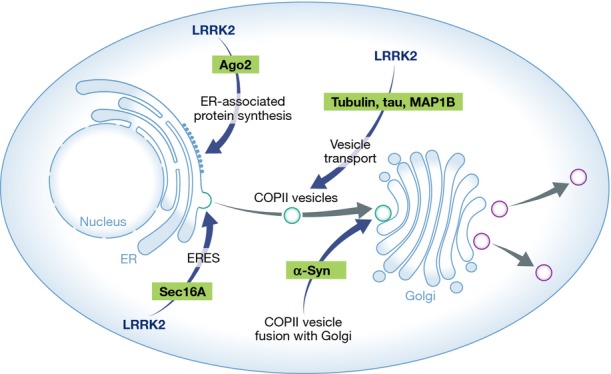
LRRK2 may regulate ER-associated protein synthesis through its interaction with Ago2, the organization of ERES and the formation of COPII vesicles through interaction with Sec16A, and microtubule-based transport of COPII vesicles through its regulation of tubulin or microtubule-associated proteins (e.g. tau and MAP1B). The regulation of COPII vesicle fusion with the trans-Golgi network by α-synuclein is also depicted.
Many interesting questions are raised by the study by Cho et al (2014): (i) How does LRRK2-R1441C affect ERES function? The R1441C mutation is shown to reduce LRRK2–Sec16A interaction in this study. The LRRK2–Sec16A interaction competes with Sec13A binding to Sec16A, indicating that an increased Sec13A–Sec16A interaction in LRRK2-R1441C condition somehow impairs ERES function (REF). The underlying mechanism awaits future investigation. (ii) Is LRRK2 regulating secretory proteins in general or only a subset of secretory proteins? Different cargos transported by the COPII vesicles are known to use different ancillary factors. In this study, the LRRK2–Sec16A interaction is shown to regulate the transport of Nicastrin and NMDARs. Future studies evaluating whether defective transport of these proteins contribute to LRRK2 pathogenesis will be interesting. (iii) What is the underlying molecular mechanism of LRRK2 GTPase activity regulation of ERES? Does it involve interaction with SAR1, another GTPase that also interacts with Sec16 and is required early in ERES formation? (iv) Is there ER stress when ERES function is compromised? (v) Are DNs selectively vulnerable to ERES impairment? The vesicular transport of dopamine could be involved, an intrinsically unstable neurotransmitter that can generate toxic by-products. Is mitochondrial biogenesis, known to depend on ER–mitochondrial interactions, affected? Attempts to answer these various questions will keep PD researchers busy for a while.
References
- Cho HJ, Yu J, Xie C, Rudrabhatla P, Chen X, Wu J, Parisiadou L, Liu G, Sun l, Ma B, Ding J, Liu Z, Cai H. Leucine-rich Repeat Kinase 2 regulates Sec16A at ER exit sites to allow ER-Golgi export. EMBO J. 2014;33:2314–2331. doi: 10.15252/embj.201487807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dachsel JC, Farrer MJ. LRRK2 and Parkinson disease. Arch Neurol. 2010;67:542–547. doi: 10.1001/archneurol.2010.79. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Jenner P. Parkinson disease: from pathology to molecular disease mechanisms. Free Radical Biol Med. 2013;62:132–144. doi: 10.1016/j.freeradbiomed.2013.01.018. [DOI] [PubMed] [Google Scholar]
- Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466:637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes H, Budnik A, Schmidt K, Palmer KJ, Mantell J, Noakes C, Johnson A, Carter DA, Verkade P, Watson P, Stephens DJ. Organisation of human ER-exit sites: requirements for the localisation of Sec16 to transitional ER. J Cell Sci. 2009;122:2924–2934. doi: 10.1242/jcs.044032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamyshev AL, Patrick AE, Karamysheva ZN, Griesemer DS, Hudson H, Tjon-Kon-Sang S, Nilsson I, Otto H, Liu Q, Rospert S, von HG, Johnson AE, Thomas PJ. Inefficient SRP interaction with a nascent chain triggers a mRNA quality control pathway. Cell. 2014;156:146–157. doi: 10.1016/j.cell.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Cai H. LRRK2 function on actin and microtubule dynamics in Parkinson disease. Commun Integr Biol. 2010;3:396–400. doi: 10.4161/cib.3.5.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson P, Forster R, Palmer KJ, Pepperkok R, Stephens DJ. Coupling of ER exit to microtubules through direct interaction of COPII with dynactin. Nat Cell Biol. 2005;7:48–55. doi: 10.1038/ncb1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm JE, Buszczak M, Sayles S. Efficient protein trafficking requires trailer hitch, a component of a ribonucleoprotein complex localized to the ER in Drosophila. Dev Cell. 2005;9:675–685. doi: 10.1016/j.devcel.2005.09.015. [DOI] [PubMed] [Google Scholar]