

Abstract
Neutral competition, an emerging feature of stem cell homeostasis, posits that individual stem cells can be lost and replaced by their neighbors stochastically, resulting in chance dominance of a clone at the niche. A single stem cell with an oncogenic mutation could bias this process and clonally spread the mutation throughout the stem cell pool. The Drosophila testis provides an ideal system for testing this model. The niche supports two stem cell populations that compete for niche occupancy. Here, we show that cyst stem cells (CySCs) conform to the paradigm of neutral competition and that clonal deregulation of either the Hedgehog (Hh) or Hippo (Hpo) pathway allows a single CySC to colonize the niche. We find that the driving force behind such behavior is accelerated proliferation. Our results demonstrate that a single stem cell colonizes its niche through oncogenic mutation by co-opting an underlying homeostatic process.
Keywords: competition, Hedgehog, Hippo, stem cell, testis
See also: ER Morrissey & L Vermeulen (October 2014)
Introduction
The ability of a stem cell to continually generate offspring for tissue maintenance depends on its ability to remain and renew at the niche. A critical consideration is whether stem cells are eternal and always divide invariantly or whether they function as members of an equipotent population, within which a single stem cell could be lost and replaced stochastically by a neighbor. Recent work has revealed that the latter, termed neutral competition, is an emerging feature of stem cell homeostasis. This model states that individual stem cells can be stochastically lost and replaced by their neighbors, resulting in chance dominance of a clone at the niche. Neutral competition has been established for both vertebrates and invertebrates and in several different tissues (Clayton et al, 2007; Klein et al, 2010; Lopez-Garcia et al, 2010; Snippert et al, 2010; Doupe et al, 2012; de Navascues et al, 2012). However, the fact that loss and gain of stem cells occurs opens the possibility of a transformed stem cell exploiting this process in its favor and achieving clonal dominance. Such behavior theoretically could underlie the observation of tumor-initiating cells in certain types of cancer (Reya et al, 2001) and has recently been reported for mouse intestinal stem cells (Vermeulen et al, 2013; Snippert et al, 2014).
The Drosophila testis provides an ideal system for analyzing single stem cell behavior. The niche (called the hub) supports two stem cell populations, germ line stem cells (GSCs) and somatic cyst stem cells (CySCs) (Fig 1A and de Cuevas & Matunis, 2011; Hardy et al, 1979). GSCs give rise to sperm, while CySCs produce somatic cyst cells, which ensheath developing germ cells and are required for germ cell differentiation. Each testis niche harbors approximately 9–14 GSCs, which divide with oriented mitosis perpendicular to the niche, such that one offspring, likely to remain in contact with the niche, self-renews while the other, physically displaced from niche signals, begins differentiation (Yamashita et al, 2003; Sheng & Matunis, 2011). Serially reconstructed electron micrographs of wild-type testes revealed ∼13 somatic cells, presumed to be the CySCs, in contact with the hub in young adults (Hardy et al, 1979). Most current studies rely on immunofluorescence of nuclear factors in presumptive CySCs and their daughters. The best molecular marker of CySCs is Zfh1, which labels the nucleus of ∼44 cells in wild-type testes (Leatherman & Dinardo, 2008; Inaba et al, 2011; Amoyel et al, 2013). This value substantially overestimates the true number of CySCs and includes post-mitotic daughter cells that no longer contact the niche. Finally, there is no evidence for oriented division among CySCs (Cheng et al, 2011), raising the possibility that this population may be subject to different regulation than GSCs. Stem cell loss and replacement has been observed in Drosophila gonads, in both somatic and germ lineages, but its significance remains under debate (Margolis & Spradling, 1995; Xie & Spradling, 1998, 2000; Zhang & Kalderon, 2001; Wallenfang et al, 2006; Nystul & Spradling, 2007). It remains to be resolved whether loss of stem cells reflects their loss of fitness or represents a normal homeostatic process of neutral competition.
Figure 1. Characterizing the CySC pool.
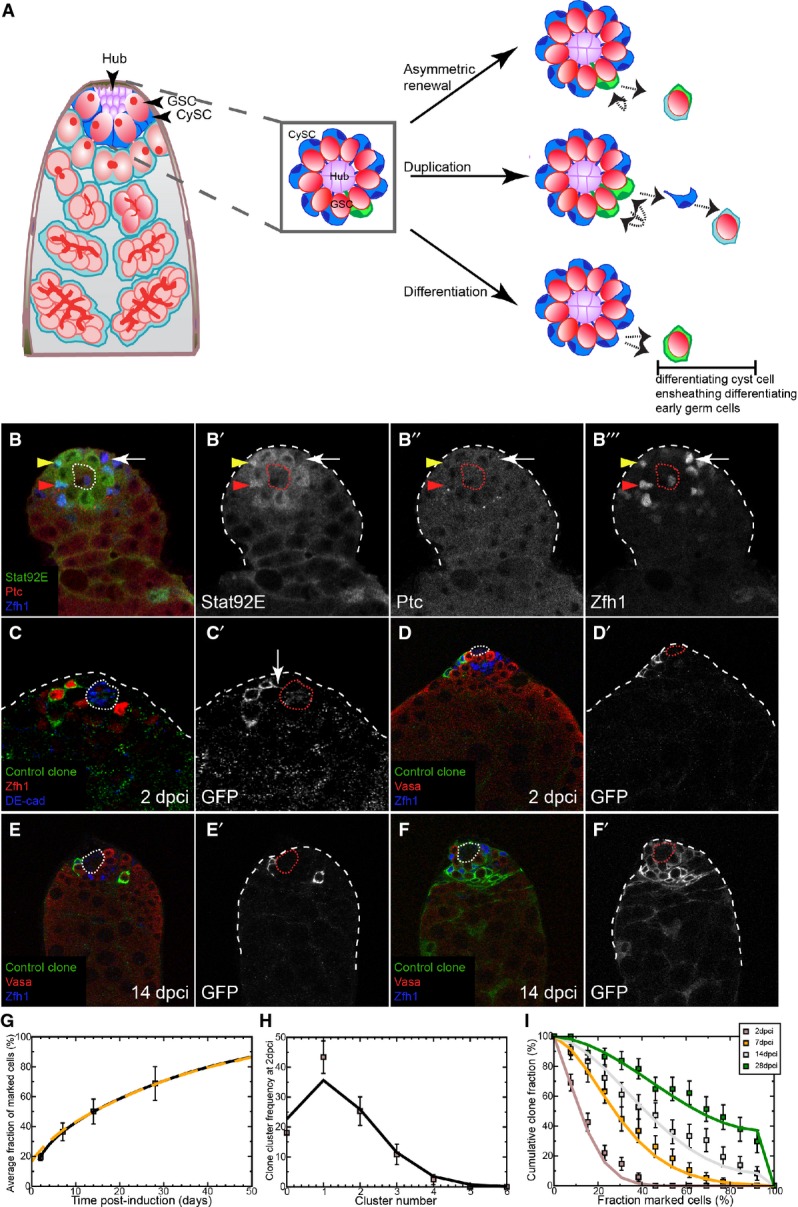
A Left: Schematic of the apical tip of the Drosophila testis. GSCs (red) and CySCs (dark blue) contact the hub (purple). Differentiating progeny move away from the hub to form germ cysts (red), which are ensheathed by two cyst cells (light blue). Center: Boxed enlargement showing that CySCs form a ring around the hub and contact the hub in between the GSCs. The CySC nucleus (dark blue) resides just ‘behind’ the row of GSCs. A marked CySC (green) will undergo division with possible outcomes depicted at right. Right: In asymmetric renewal (top), the two daughters of the clone give rise to one CySC and one differentiating cyst cell, which ensheaths a gonialblast along with an unmarked cyst cell (light blue). In duplication (middle), both marked daughters remain at the niche as CySCs, displacing an unmarked CySC (blue) in the process. This displaced unmarked cell differentiates into an ensheathing cyst cell. In differentiation (bottom), both daughters of the marked CySC differentiate into cyst cells, resulting in no marked CySCs at the hub.
B A control testis labeled with Stat92E (green, single channel B’), Ptc (red, single channel B”), and Zfh1 (blue, single channel B’”) showing that while some Zfh1-positive cells co-labeled for Ptc and Stat92E (red arrowhead), others were only positive for one factor (yellow arrowhead) or for neither (arrow).
C CySC MARCM clones labeled with membrane-targeted CD8-GFP (C’) showing identifiable single cells, some of which contacted the hub (DE-cadherin, blue) with membrane extensions (arrow in C’).
D–F Clonal analysis, GFP (single channels D’–F’) indicates the clone, Vasa (red) labels germ cells and Zfh1 (blue) CySCs and early cyst cells; the hub is indicated by a dotted line. GFP-labeled control clones were generated by the MARCM technique and analyzed at 2 (D) and 14 dpci (E, F). Although clones were small at 2 dpci (D), they varied markedly by 14 dpci (E, F).
G Variation of average size of control clones as a function of time. The data points (boxes) show the mean fraction of labeled CySCs in persisting clones. The black line shows a fit of the neutral drift model to the data using an induction frequency of CySCs at a ratio of one in 10 (i.e., 10%). The dashed orange line represents the predicted clonal evolution if only a single CySC clone was induced with a time-shift of 3 days with the same set of parameters. One may note that the clone sizes observed from multiple independent induction events and from a single induction event converge rapidly. For details of the neutral drift model and the notation, see Supplementary Materials and Methods. n = 83, 74, 73, 81 for 2, 7, 14, 28 dpci, respectively. Error bars denote SEM.
H Comparison of observed (boxes) and predicted (line) frequency of clusters of somatic cell clones. Each cluster is presumed to represent an independent labeling event. The line was generated by a least-squares fit and suggests a labeling efficiency of 11% (q = 0.11). Error bars denote SEM.
I Distribution of persisting clone sizes in wild-type testes. The boxes show experimental data, and lines show the predictions of the model. n = 83, 74, 73, 81 for 2, 7, 14, 28 dpci, respectively. Error bars denote SEM.
The molecular signals governing self-renewal at the testis niche have been well characterized (de Cuevas & Matunis, 2011). GSCs are maintained by Bone Morphogenetic Protein (BMP) signals originating from both the hub and CySCs (Shivdasani & Ingham, 2003; Kawase et al, 2004; Leatherman & Dinardo, 2010). CySCs require at least two signaling inputs, the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) and Hedgehog (Hh) pathways, in order to self-renew (Kiger et al, 2001; Leatherman & Dinardo, 2008; Michel et al, 2012; Amoyel et al, 2013). Ligands for both pathways, Unpaired (Upd) and Hh, respectively, are produced by the hub cells (Forbes et al, 1996; Kiger et al, 2001; Tulina & Matunis, 2001; Dinardo et al, 2011). Two known targets are expressed in CySCs in response to JAK/STAT pathway activation, Zfh1 and Chinmo. Overexpression in CySCs of the JAK Hopscotch (Hop) or of either pathway target results in autonomous hyper-proliferation of CySCs and non-autonomous hyper-proliferation of GSCs, due to BMP production by the CySCs (Leatherman & Dinardo, 2008, 2010; Wang et al, 2008; Flaherty et al, 2010). Conversely, Hh activation only regulates the self-renewal and numbers of the CySCs, without affecting the GSC niche (Amoyel et al, 2013).
Although both stem cells co-exist at the same niche, and although the CySCs are a necessary component of the niche for GSCs, these two populations compete for access to the niche, as revealed by analysis of the mutant phenotype of the JAK/STAT negative feedback regulator Socs36E (Issigonis et al, 2009; Singh et al, 2010). This reduction of GSCs in Socs36E mutants was attributed to increased JAK/STAT signaling in Socs36E mutant CySCs, leading to upregulation of integrin-based adhesion and enabling the mutant cells to displace wild-type GSCs and CySCs from the niche.
Here, we characterize CySC behavior by clonal analysis. We found that the behavior of CySCs was consistent with them being lost and replaced stochastically, as predicted by the neutral competition model. For this study, we made clones homozygous mutant for patched (ptc), which encodes the Hh receptor (Chen & Struhl, 1996); loss of ptc causes constitutive activation of the pathway. We found that ptc mutant CySCs outcompeted both wild-type CySCs and GSCs for niche access. We determined that this phenotype was due to biased competition, skewing normal behavioral dynamics in favor of the mutant cell. We showed that adhesion and JAK/STAT signaling could not cause stem cells to acquire colonizing capabilities. Rather, we showed that simply accelerating proliferation was sufficient to cause a single CySC and its descendants to outcompete wild-type CySCs and GSCs. Furthermore, we established a critical role for the conserved growth regulatory Hippo pathway in regulating competition and self-renewal in CySCs independently of Hh signaling. Thus, we demonstrate that proliferation is the key driver of somatic stem cell behavior and provide a model for how oncogenic mutations can spread throughout a stem cell pool by exploiting a fundamental homeostatic process of stochastic stem cell replacement.
Results
Characterizing the CySC pool
We first attempted to use molecular markers to sub-divide the somatic population near the niche. We reasoned that only a subset of the ∼44 Zfh1-positive cells could constitute the true stem cell pool. We therefore examined whether markers of self-renewal pathways in CySCs—Ptc for Hh and Stat92E for JAK/STAT—were co-expressed. We only found expression of these markers in Zfh1-positive somatic cells located one cell diameter from the hub. Within this group, only a subset co-expressed Ptc and Stat92E (Fig 1B–B’’’, red arrowhead), while others expressed only one or neither (Fig 1B–B’’’, yellow arrowhead and arrow, respectively). This analysis suggests that using the best available molecular markers may not be the most robust method to identify CySCs. Since membrane contact with the niche appears to be the defining feature of stemness in the Drosophila testis (Hardy et al, 1979; de Cuevas & Matunis, 2011), we estimated the actual number of CySCs by generating single-cell control MARCM clones (Lee & Luo, 1999), expressing a membrane-targeted GFP (Fig 1C). We used this clonal method because labeling all somatic cell membranes did not allow us to determine whether an individual cell contacts the hub or not. Only 30.5% of Zfh1-positive clones (29/95 single cell clones) had membrane extensions contacting the hub (Fig 1C’, arrow). Extrapolating this proportion to an average of 43 ± 7 Zfh1-positive cells per testis that we counted in these samples (n = 59), we estimated 13 CySCs per testis, consistent with the 12.6 value that has been previously reported (Hardy et al, 1979). In the genotype we examined, there were 13.2 GSCs (n = 34). In the Drosophila testis, stem cells are actively dividing, and within the somatic lineage, only CySCs divide (Hardy et al, 1979; Inaba et al, 2011). As further confirmation of the number of CySCs, we examined markers of cycling cells, PCNA-GFP to mark cells in S-phase (Thacker et al, 2003) and Cyclin B (CycB) for G2/M. We found 11.2 somatic cells one cell diameter away from the niche undergoing replication that were positive for PCNA (Supplementary Fig S1A–A”’, arrow). In the same testes, 9.2 out of 12.2 total GSCs on average expressed PCNA-GFP, suggesting a 1.3:1 ratio of CySCs to GSCs and by extrapolation a total of ∼15 CySCs. Similarly, in an unrelated genetic background that contained on average 7.9 GSCs, we observed 5.6 GSCs and 5.6 CySCs-expressing CycB (Supplementary Fig S1B–B”’, arrows). Taken together, these data suggest that GSCs and CySCs exist in a ratio close to 1:1.
Two different models have been proposed to explain stem cell behavior in actively cycling homeostatic tissues; in the first, stem cells are invariant and divide asymmetrically to self-renew and are only rarely lost in cases of damage or loss of fitness. In the other, asymmetry is achieved only at the level of the stem cell population. In the latter case, stem cell populations are dynamic, and their clonal make-up changes according to stochastic variations such that some clones are lost entirely while others expand to occupy empty stem cell berths, through a process termed ‘neutral competition’ (Simons & Clevers, 2011).
We tested these models by generating control FRT42D MARCM clones that mis-expressed only membrane CD8-GFP and scored the number of labeled somatic cells contacting the hub. While the membrane labeling of clones allows for direct identification of CySCs, this methodology has two drawbacks. First, CySCs outside the clones (which are unmarked) have to be scored more subjectively by their position relative to the hub. Second, once many cells around the niche are labeled, it becomes difficult to distinguish the membranes of individual cells, resulting in a slight overestimation of the total number of CySCs (∼16–21 obtained by this method versus ∼13 obtained above). Therefore, to circumvent this uncertainty, we monitored both the total number of GFP-labeled and unlabeled cells considered to be contacting the hub and used these values to determine the fraction of labeled CySCs as a percentage in each testis.
At 2 days post-clone induction (dpci), we found few GFP-labeled CySCs, consistent with a low clone induction rate (Fig 1D and H, Supplementary Materials and Methods, see below). To characterize CySC dynamics, we separated testes according to whether they maintained at least one GFP-expressing cell in contact with the hub (termed ‘persisting’) and those in which all GFP-expressing cells had detached from the hub (termed ‘differentiating’). We observed empirically that the mean fraction of labeled CySCs in persisting clones increased steadily as a function of time (Fig 1G), while the number of labeled CySCs in individual clones varied considerably between samples at the same time point, as exemplified by the 14 dpci samples shown in Fig 1E and F. The increased number of labeled CySCs in persisting clones is inconsistent with the model of invariant asymmetric stem cell division as in this scenario this parameter should not change over time. However, the change observed is consistent with CySCs undergoing loss and replacement (Fig 1G).
We next subjected these data to a quantitative analysis, using a parallel approach to that developed to study stem cell dynamics in the murine intestinal crypt (Lopez-Garcia et al, 2010). The assumptions contained in the model are the following: (1) CySCs form a single equipotent population in which any cell has an equal chance of being lost and replaced; (2) in line with the geometry of the testis, the ensemble of CySCs can be approximated as a one-dimensional ‘necklace’ of cells around the niche; (3) as CySCs proliferate, some lose contact with the niche and differentiate, and this is perfectly compensated by the duplication of a neighboring CySC to maintain a constant total number of CySCs. By contrast, an asymmetrical CySC division leaves the number of labeled CySCs unchanged (Fig 1A). As a simplification, we do not take into account GSCs, which are simply regarded as a separate lineage with their own fate behavior. In this modeling scheme, clonal dynamics of the CySC compartment is dependent upon only two parameters—the CySC loss/replacement rate, λ, and the total number of CySCs contacting the hub, N (see Supplementary Materials and Methods). Since CySC divisions that lead to asymmetric fate outcome do not change the number of marked CySCs in a clone, the clonal fate data are insensitive to the CySC division rate.
To implement the modeling scheme, it is important to define the labeling efficiency of the FRT42D MARCM system. From the frequency of unlabeled testes at 2 dpci, we estimated a labeling efficiency of around 10% for each of the 13 CySCs following the heat shock. At this level of induction, we therefore expect that testes will experience multiple induction events, leading to isolated “clusters” of GFP-expressing cells. By comparing the predicted frequency of clusters with direct measurements at 2 dpci, a least-squares fit suggested a labeling efficiency of q = 11% (Fig 1H), consistent with the observed frequency of unlabeled testes. With the labeling efficiency and CySC number defined, only the CySC loss/replacement rate, λ, remained to be determined.
To fix the loss/replacement rate, we compared the predictions of the model with measurements of the average fraction of labeled CySCs in persisting clones using an induction frequency of 10%. Adjusting the loss/replacement rate, we found that the mean fraction of labeled CySCs could be well reproduced by a loss/replacement rate of λ = 0.84 ± 0.05 per day (Fig 1G, compare lines and boxes). However, alongside the mean fraction, the model also predicts the variation in the size distribution of individual clones as a function of time post-induction. Taking the inferred loss/replacement rate and induction frequency, we found that the model provides an excellent prediction of the measured cumulative clone size distribution (Fig 1I, compare lines to boxes), defined as the fraction of testes that have a fraction of labeled CySCs larger than the given value. (We note that N and λ exist in a fixed ratio N2/λ (see Supplementary Materials and Methods), meaning that the fit to a model with a different N would generate quite reasonable agreement with the data. However, for a larger N, we would require a proportionately larger λ, potentially in excess of the CySC division rate.) Taken together, these empirical and modeling data strongly suggest that the ∼13 CySCs in a wild-type testis produce equivalent offspring which have stochastic fates.
Given the potential uncertainty of the membrane labeling method as a means to identify CySCs, we chose to challenge our findings by following a second unbiased (albeit less direct) approach to measuring CySC number and scoring clones. We generated control MARCM clones on two separate chromosome arms (FRT40A and FRT42D) that mis-expressed only nuclear GFP and scored the size of CySC clones relative to the total Zfh1-positive population and the clone recovery rate (percentage of testes with any marked clone positive for Zfh1) at various times after clone induction. We assumed that each CySC contributes equally to the total Zfh1-positive pool and counted the number of Zfh1-positive cells (N) that were labeled with the clone marker and expressed this as a fraction N/43 where 43 was the average number of Zfh1-positive cells found per testis (see above). At 2 dpci, we found few GFP-labeled Zfh1-positive cells, consistent with a low clone induction rate (q = 0.18 for FRT40A and q = 0.3 for FRT42D (Supplementary Fig S2A and B and Supplementary Materials and Methods)). At 14 dpci, individual clones varied considerably between samples at the same time point (Supplementary Fig S2C and D). Using this method, we obtained similar results to those observed for membrane labeling: the mean fraction of labeled CySCs increased as a function of time (Supplementary Fig S2E and F) and the number of testes harboring labeled clones decreased over time (Supplementary Fig S2G and H). Quantitative analysis of nuclear GFP MARCM control clones (Supplementary Fig S2E–J) revealed that they obeyed similar neutral drift dynamics to the membrane CD8-GFP MARCM control clones (Fig 1D–I). Importantly, we were able to infer a CySC loss/replacement rate of around once per day, which is comparable to the loss/replacement rate of 0.84 per day inferred from the earlier modeling scheme (see above). Using two different labeling methods, generating clones on two chromosome arms, scoring CySCs by two independent methods, we reach a similar conclusion: CySCs are lost and replaced stochastically and obey neutral drift dynamics.
ptc mutant CySCs skew neutral drift dynamics and outcompete wild-type CySCs
The dynamics of neutral stem cell competition have been reported in mammalian and Drosophila stem cells (Wallenfang et al, 2006; Clayton et al, 2007; Klein et al, 2010; Lopez-Garcia et al, 2010; Snippert et al, 2010; de Navascues et al, 2012), but mutations that co-opt the homeostatic mechanisms underlying this process for the benefit of the mutant cell have only recently been described (Vermeulen et al, 2013; Snippert et al, 2014). We and others previously showed that Hh signal reception is required for the maintenance of CySC fate. CySCs that are unable to transduce the Hh signal are lost from the niche and differentiate (Michel et al, 2012; Amoyel et al, 2013). Here, we studied the effect of clonal gain of Hh signaling by making clones homozygous mutant for patched (ptc). Cells lacking ptc function can no longer inhibit Smoothened activity and experience sustained ligand-independent Hh signal transduction (Ingham et al, 1991; Chen & Struhl, 1996). We examined FRT42D ptc mutant CD8-GFP MARCM clones as compared to the appropriate control, that is, FRT42D CD8-GFP MARCM control clones. Similar to control, we found few GFP-labeled ptc mutant CySCs at 2 dpci (Fig 2A). In contrast to control clones, ptc mutant clones contained more labeled CySCs on average by 14 dpci and were often seen to take over the entire somatic lineage, presumably by causing the displacement of wild-type CySCs (Fig 2B, compare boxes in Fig 2C to those in Fig 1G). We counted unlabeled CySCs in control and ptc samples and found that there were significantly fewer when ptc mutant clones were present (Fig 2E, P < 0.004). These results indicated that ptc mutant CySCs expanded at the expense of their wild-type neighbors. However, like control clones, the frequency of persistent ptc mutant CySC clones decreased over time (Fig 8E, red line). These results show that ptc clones could differentiate, indicating that they are not locked in a perpetual state of stem cell self-renewal, which is consistent with a prior report (Michel et al, 2012). Indeed, we observed differentiating ptc mutant cyst cells ensheathing spermatogonial cysts similar to control clones (Supplementary Fig S3A and B). This represents a different situation from a previously observed instance of stem cell competition, where stem cells in the ovary that cannot differentiate eventually replaced their wild-type neighbors (Jin et al, 2008). Thus, these data suggest that ptc mutant CySCs have a competitive advantage over wild-type CySCs in effecting stem cell replacement.
Figure 2. Neutral drift dynamics are skewed by ptc mutant clones.

A, B Clonal analysis, GFP (single channels A’, B’) indicates the clone, Vasa (red, single channels A”, B”) labels germ cells and Zfh1 (blue, single channels A’”, B’”) CySCs and early cyst cells; the hub is indicated by a dotted line. GFP-labeled ptc mutant clones were generated by the MARCM technique and analyzed at 2 (A) and 14 dpci (B). Arrow (B–B’”) shows displacement of GSCs by ptc mutant CySCs.
C Variation of average size of ptc mutant clones as a function of time. The data points (boxes) show the mean fraction of labeled CySCs in persisting clones. The black line shows a fit of the neutral drift model, modified to have a bias in favor of the labeled cell, to the data using an induction frequency of 10%. The dashed orange line represents the predicted clonal evolution if only a single CySC clone were induced with a time-shift of 3 days with the same set of parameters. One may note that the clone sizes observed from multiple independent induction events and from a single induction event converge rapidly. For details of the biased drift model and the notation, see Supplementary Materials and Methods. n = 63, 81, 79, 66 for 2, 7, 14, 28 dpci, respectively. Error bars denote SEM.
D Distribution of clone sizes of persistent ptc mutant clones. The boxes show experimental data, and lines show the predictions of the model. Error bars denote SEM.
E Number of unlabeled CySCs at 14 dpci in testes containing either control (green) or ptc mutant (red) clones. Lines show mean and standard deviation. Asterisks denote statistically significant difference from control. n = 73 and 79 for control and ptc mutant, respectively.
F Number of GSCs at 14 dpci when control or ptc CySC clones were present. ptc mutant CySCs displaced wild-type GSCs, leading to a significant decrease in the number of GSCs. Asterisks denote statistically significant difference from control. n = 48 and 49 for control and ptc mutant, respectively. Error bars denote SEM.
Figure 8. Hh and Yki regulate niche competition and self-renewal independently.

A–C yki mutant CySCs did not self-renew (A) and loss of yki suppressed the ptc colonization phenotype (C). Testes with negatively marked yki (A), ptc (B), or ptc yki double mutant (C) clones at 7 dpci. Absence of GFP (single channel A’, B’, C’) marks the clones, Vasa (single channel A”, B”, C”) is red, and Tj (single channel A”’, B’”, C’”) is blue. Arrows point to differentiated mutant cyst cells in (A) and (C).
D ptc mutant clones (arrow) did not display increases in ex-lacZ expression compared to wild type (arrowhead). Clones are labeled by GFP (single channel D’), β-gal is in red (single channel D”), and Zfh1 is in blue (single channel D’”). The hub is outlined with a dotted line in all panels.
E Graph of clone recovery rates over time for the indicated genotypes. n = 61, 21, 88 for control at 2, 7, 14 dpci, respectively; n = 46, 39, 89 for ptc at 2, 7, 14 dpci, respectively; n = 50, 94, 35 for yki at 2, 7, 14 dpci, respectively; n = 40, 59, 64 for ptc yki at 2, 7, 14 dpci, respectively; n = 39, 67 for hpo at 2, 14 dpci, respectively.
F yki mutant GSCs could be recovered at 2 and 7 dpci. yki mutant CySCs could be recovered at 2 dpci but not 7 dpci. n = 61, 21 for control at 2, 7 dpci, respectively. n = 50, 94 for yki at 2, 7 dpci, respectively.
To assess whether the dynamics of ptc mutant clones represent a biasing of the neutral competition process toward persistence, we sought for the simplest revision of the neutral drift model which could accommodate the observed behavior. In particular, we assumed that, following the loss of a CySC (control or ptc mutant) through commitment to differentiation, a neighboring ptc mutant CySC will have a higher chance of replacing it through symmetric cell division than a wild-type neighboring CySC. We also assumed that the competitive advantage of the ptc mutant CySC is sustained since the loss rate of CySCs is not differentially affected by ptc mutation. Once again, using the frequency of unlabeled testes at 2 dpci, we inferred a CySC labeling efficiency of around 10%, similar to the control. Then, taking the loss/replacement rate of wild-type CySCs to be unperturbed from its control value, by adjusting the bias of ptc mutant CySCs away from loss and toward replacement (by around 35%), we found a good agreement between the model dynamics and the experimental data (Fig 2C, compare lines and boxes, and Supplementary Materials and Methods). Significantly, taking these model parameters, comparison of the cumulative clone size distribution revealed an excellent agreement of the model prediction with the data over the range of time points (Fig 2D, compare lines and boxes).
Once again, we repeated this experiment using the alternative labeling and scoring method in which CySC number is estimated by the labeled fraction of Zfh1-expressing cells. We analyzed FRT42D ptc mutant nuclear GFP MARCM clones as compared to FRT42D nuclear GFP MARCM control clones (compare Supplementary Fig S4A and B to Supplementary Fig S2B and D). We found that the number of these ptc mutant CySCs increased faster than control CySCs and that they were lost less frequently (compare boxes in Supplementary Fig S4C and D to those in Supplementary Fig S2F and H), similar to the results obtained for the membrane labeling experiment. Finally, we carried out modeling using the same bias as before (Supplementary Fig S4C–E, lines) and found that the computational output for number and distribution of CySCs as well as clone recovery rate was well matched to the experimental data for FRT42D ptc mutant nuclear GFP MARCM clones. Taken together, the data indicate that the behavior of ptc mutant clones is consistent with a biasing of competition between stem cells.
ptc mutant CySCs outcompete wild-type GSCs
As a readout for the competitive activity of ptc mutant CySCs, we also quantified the number of GSCs (defined as Vasa-positive cells in contact with the niche) in testes with control or ptc mutant CySCs at 14 dpci. Indeed, we found that GSC number was significantly reduced (P < 0.0001) non-autonomously when ptc mutant CySC clones were present (Fig 2F, red bar). At the same time, colonizing CySCs contacted the hub in place of the outcompeted GSCs (Fig 2B, arrow, Supplementary Fig S4B), similar to the phenotype described for Socs36E (Issigonis et al, 2009). GSC loss was only observed once the majority of CySCs were replaced by ptc mutant CySCs (Supplementary Fig S4F), suggesting a sequential outcompetition of first wild-type CySCs and then wild-type GSCs by ptc mutant CySCs. The fact that ptc mutant CySCs had normal levels of factors that mediate GSC extended niche function, that is, Stat92E and Zfh1 (Leatherman & Dinardo, 2010) (Fig 3A, Supplementary Fig S5, arrows, for Stat92E; Fig 2B, Supplementary Fig S4B for Zfh1), strongly suggests that GSC loss is not due to lack of appropriate support from ptc mutant CySCs. Thus, gain of Hh signaling results in niche colonization by the mutant cell, as a consequence of the displacement of resident wild-type CySCs and GSCs at the niche.
Figure 3. Clonal overactivation of the Hh pathway, but not JAK/STAT, causes niche competition phenotypes.

A No increase in Stat92E staining (red, single channel in A”) was seen in a ptc mutant CySC (green, arrow, single channel in A’) compared to neighboring wild-type CySCs (arrowhead) at 2 dpci. Somatic cells were labeled with Tj (blue, single channel in A’”). See also Supplementary Fig S5 for Stat92E staining in clones at 7 and 14 dpci.
B–G MARCM clones at 14 dpci, with single channels showing the clone marker GFP in (B’–F’), Vasa in red and Zfh1 (B, C) or Tj (D–F) in blue. Control clones (B, C) showed variation in the number of cells labeled. Overexpression of CiAct (D) or RNAi against ptc (E) recapitulated the ptc mutant phenotype (compare with Fig 2B), but overexpression of Hop did not (F). Hop overexpression activated JAK/STAT signaling (G), as seen by stabilization of Stat92E protein (blue, single channel in G’) in the clones (green, arrows).
H Number of GSCs at 14 dpci when CySC clones of the indicated genotype were induced. Hop overexpression did not affect GSC number, while CiAct and ptc RNAi-expressing clones caused loss of GSCs, similar to ptc mutant clones (see Fig 2F). Asterisks denote statistically significant change from control. n = 15, 24, 27, 8 for control, UAS-Hop, UAS-CiAct, UAS-ptc RNAi, respectively. Error bars denote SEM.
I Number of GSCs at 14 dpci when control or ptc mutant CySCs were present, showing an enhancement of GSC loss when ptc mutant clones were induced in a background lacking one copy of the Stat92E gene. Asterisks denote statistically significant change from the ptc mutant clones alone. n = 48 (control), 36 (control; Stat92E/+), 49 (ptc), 20 (ptc; Stat92E/+). Error bars denote SEM.
JAK/STAT signaling, adhesion and cell competition factors are not causal to niche competition
Several possibilities could explain niche colonization by ptc mutant CySCs. We ruled out the trivial explanation that the niche size was altered in testes with ptc mutant clones (Supplementary Table S1). We next tested whether an increase in integrin-based adhesion downstream of Stat92E, as proposed for Socs36E mutants (Issigonis et al, 2009), caused ptc mutant CySCs to anchor more securely to the niche. We previously found no epistatic relationship between Hh and STAT signaling in the testis (Amoyel et al, 2013). Consistent with this, we found that Stat92E levels were unchanged in ptc mutant CySCs (Fig 3A–A’’’, arrow, Supplementary Fig S5). We used the MARCM technique to assess whether the clonal overexpression of various factors could induce niche competition. As expected, control clones that only overexpressed GFP had variable clone sizes (Fig 3B and C). By contrast, clonal overexpression of either a constitutively active form of the Hh signal transducer Cubitus interruptus (CiAct) (Price & Kalderon, 1999) or of an RNAi hairpin against ptc recapitulated the ptc mutant phenotype (Fig 3D and E) and caused a statistically significant reduction of GSCs (Fig 3H, P < 0.0001 for both CiAct and ptc RNAi), thus validating our technique. Surprisingly, clonal hyper-activation of Stat92E by mis-expression of Hop did not cause CySC clones to compete with either wild-type CySCs or with GSCs (Fig 3F and H), despite clearly elevated levels of Stat92E in the clone (Fig 3G, arrows), a well-established readout of Stat92E activity (Chen et al, 2002). We reasoned that if the ptc mutant phenotype was due to elevated Stat92E levels in CySCs, we could suppress ptc-dependent GSC loss by removing a copy of Stat92E. The number of GSCs in Stat92E/+ heterozygotes was indistinguishable from wild type (Fig 3I, dark green bar). However, GSC loss was actually enhanced when ptc mutant clones were induced in a Stat92E/+ heterozygous background (Fig 3I, red bar, P < 0.008), presumably due to the role of Stat92E in GSC-hub adhesion (Leatherman & Dinardo, 2010).
We also examined whether the ptc mutant phenotype could be ascribed to changes in cell-matrix (integrin) or cell–cell (cadherin) adhesion. We did not detect changes in βPS-integrin in ptc clones (Fig 4A, arrow), in contrast to the observations reported for Socs36E mutants (Issigonis et al, 2009). Furthermore, clonal overexpression of βPS-integrin, or a dominant-active form of Talin (TalinH), which strengthens integrin adhesion (Tanentzapf & Brown, 2006), neither recapitulated the ptc phenotype nor induced competition with CySCs and GSCs (Fig 4C and H). Importantly, we found that rhea, which encodes the Drosophila Talin, was dispensable for CySC self-renewal (Fig 4E–G). DE-cadherin levels did not change in ptc mutant clones (Fig 4B, arrow). Moreover, clonal mis-expression of DE-cadherin also did not cause niche colonization (Fig 4D and H). Furthermore, competition caused by ptc mutant clones was not altered by reducing the genetic dose of α-Catenin, which connects DE-cadherin to the cytoskeleton (Sarpal et al, 2012) (Fig 4I). Although one mutant allele of rhea partially suppressed the ptc phenotype (Fig 4I, P < 0.65 for rhea1 and P < 0.051 for rhea6-66), this is likely to be an indirect effect of loosening the tethering of the hub to the muscle sheath and allowing more stem cells to surround the hub (Tanentzapf et al, 2007). Consistent with this, there were more GSCs in testes from rhea/+ heterozygous animals (Fig 4I). These data strongly suggest that increased adhesion does not skew neutral drift dynamics in CySCs.
Figure 4. Increased adhesion is not causal to niche competition.

A, B ptc mutant clones did not upregulate adhesion molecules. No change in βPS-integrin (A, red, single channel in A’) or in DE-cadherin expression (B, blue, single channel in B’) was seen at the hub in testes with ptc mutant clones (green). Vasa labels germ cells in red (B), Tj labels somatic cells in blue (A). The hub is indicated with a dotted line.
C, D GFP-positive MARCM clones (green, single channels in C’,D’) overexpressing βPS-integrin (C) or DE-cadherin (D) did not outcompete neighboring wild-type CySCs or GSCs. Vasa labels germ cells in red, Tj labels somatic cells in blue. The hub is indicated with a dotted line.
E, F Control (E) and rhea mutant (F) MARCM clones showing marked CySCs which contacted the hub at 7 dpci (arrows). Vasa labels germ cells in red, Tj labels somatic cells in blue. The hub is indicated with a dotted line.
G CySC clone recovery rates at 2 (blue bars) and 7 (red bars) dpci for control (left) and rhea mutant (right). The presence of rhea mutant clones at the niche at the 7-day time point indicates that rhea was not required in CySCs for self-renewal. n = 38 and 24 for control at 2 and 7 dpci, respectively, and n = 9 and 49 for rhea1 at 2 and 7 dpci, respectively.
H Number of GSCs present when CySC clones of the indicated genotype were generated at 14 dpci. Overexpression of βPS-integrin, TalinH or DE-cadherin did not affect GSC numbers. n = 15, 19, 25, 17 for control, UAS-βPS-integrin, UAS-TalinH or UAS-DE-cadherin, respectively. Error bars denote SEM.
I Number of GSCs when ptc mutant CySC clones were present along with a single mutant copy of the indicated genes at 14 dpci. Reduction of α-cat had no effect on the ptc phenotype, while one rhea allele partly suppressed GSC loss. n = 48 (control); 21 (control; rhea1/+); 49 (ptc); 26 (ptc; α-cat/+); 19 (ptc; rhea6-66/+); 35 (ptc; rhea1/+). Error bars denote SEM.
An alternative explanation for the ptc phenotype is that ptc mutant CySCs induce death in neighboring wild-type cells, akin to classical cell competition in which more robust cells kill and take the place of weaker cells (Amoyel & Bach, 2014). A key process in cell competition is ribosomal function, which in turn is dependent on optimal levels of the cellular growth regulator dMyc and of ribosomal subunits, encoded by Minute genes (M) (Morata & Ripoll, 1975; de la Cova et al, 2004; Moreno & Basler, 2004). Clonal overexpression of dMyc, which causes cell competition in imaginal discs (de la Cova et al, 2004), did not cause niche colonization or loss of GSCs (Supplementary Fig S6A and B). Similarly, testes from a M/+ animal harboring wild-type clones (labeled M+), which normally predominate in a M/+ heterozygous background, contained a normal complement of GSCs (Supplementary Fig S6B). Finally, we found no evidence of cell death in testes with ptc mutant clones (Supplementary Fig S6C and D), and removing a copy of the pro-apoptotic gene hid (which suppresses dMyc-dependent cell competition) did not suppress ptc-dependent competition (Supplementary Fig S6E, red bar).
ptc mutant CySCs proliferate faster than controls
Having ruled out increased JAK/STAT signaling or adhesion as causal factors in niche competition, we reasoned that proliferation might be a driving force of clone dominance within the stem cell pool. We therefore tested whether ptc mutant CySCs had an altered rate of proliferation relative to control clones and, in doing so, might outcompete wild-type CySCs in the race to replace neighbors. Labeling with 5-ethynyl-2′-deoxyuridine (EdU) revealed that ptc mutant CySCs had a higher S-phase index than control clones (Fig 5A, B and E, P < 0.0001). The E2f-responsive reporter PCNA-GFP is a marker of S-phase and was normally expressed at higher levels in GSCs than in CySCs (Supplementary Fig S1A’). However, in ptc mutant CySCs, PCNA-GFP was upregulated to the level observed in GSCs (Fig 5D, arrowhead, quantified in Fig 5F). ptc mutant CySC clones also had increased M phase (Michel et al, 2012), suggesting that ptc clones accelerate proliferation as opposed to shortening only one phase of the cell cycle. We examined a protein-trap reporter for string (stg, the cdc25 homolog in Drosophila; Edgar & O’Farrell, 1989). Stg-GFP was upregulated in ptc mutant cells (Fig 5C, arrowheads).
Figure 5. ptc mutant CySCs proliferate faster than controls.

A, B There was an increase in the S-phase index in CySCs mutant for ptc. Quantification of S-phase in control (A) or ptc mutant (B) clones. Clones expressing GFP (green, single channel A’, B’) were labeled with Tj (red, single channel A”, B”) and EdU (blue, single channel A”’, B’”). Triply labeled cells (yellow arrowheads) were counted as a ratio of total cells double positive for GFP and Tj, with quantification shown in (E).
C stg-GFP (green, single channel C’) was upregulated in ptc mutant CySCs (yellow arrowheads). Zfh1 (red, single channel C”) labels CySCs, and their offspring and clones are identified by loss of the βgal marker (blue, single channel C’”).
D PCNA-GFP (green, single channel D’) was upregulated in ptc mutant clones. Clones are labeled by loss of βgal (blue, single channel D”’). Zfh1 (red, single channel D”) marks CySCs and their offspring. Arrow shows control CySC, and arrowhead shows a ptc mutant CySC.
E S-phase index. See legend of (A) above. Asterisks denote statistically significant change from control. Error bars denote SEM.
F Quantification of PCNA-GFP fluorescence intensity in control or ptc mutant CySCs. n = 11 for both genotypes. An asterisk denotes statistically significant change from control. Error bars denote SEM.
Increased proliferation downstream of ptc is necessary and sufficient for colonizing behavior
We next addressed if the competitive behavior of ptc mutant CySCs depended on their ability to increase their proliferation rate. To accomplish this, we removed one copy of stg and counted the number of labeled CySCs and of GSCs at the niche. In a stg/+ heterozygous background, the number of ptc mutant CySCs was significantly reduced (Fig 6A, P < 0.034), suggesting that ptc mutant CySCs have a reduced competitive advantage when stg is limiting. In addition, in a stg/+ background, the outcompetition of GSCs by ptc mutant CySCs was significantly suppressed (Fig 6B, red bar, P < 0.008). We note that the number of GSCs was not changed in stg/+ heterozygotes when control clones were present (Fig 6B, dark green bar). These data indicate that increased proliferation downstream of Ptc is necessary for niche competition in the Drosophila testis and that CySC–CySC and CySC–GSC competitive interactions are related, making GSC number a good readout for CySC competitiveness.
Figure 6. Increased proliferation downstream of ptc is necessary and sufficient for colonizing behavior.

A, B Loss of one copy of stg suppressed the ptc mutant phenotype at 14 dpci. Graph showing number of labeled CySCs in the indicated genotypes (A). Lines in (A) show mean and standard deviation. n = 36 (control), 59 (ptc), 31 (ptc; stg/+). Graph showing the number of GSCs when CySC clones were present in the indicated genotype (B). Asterisks denote statistically significant change from the ptc mutant clones alone (A, B). n = 48 (control), 10 (control; stg/+), 49 (ptc), 17 (ptc; stg/+). Error bars in (B) denote SEM.
C Graph showing the number of GSCs when CySC clones were present in the indicated genotypes at 14 dpci. An asterisk indicates statistically significant suppression of the ptc mutant phenotype. n = 48 (control), 25 (control; Akt/+), 21 (control; InR/+), 30 (control; S6k/+), 19 (control; cdk2/+), 21 (control, E2f/+), 49 (ptc), 21 (ptc; Akt/+), 30 (ptc; InR/+), 35 (ptc; S6k/+), 28 (ptc; cdk2/+), 30 (ptc, E2f/+). Error bars denote SEM.
D Graph showing the total number of labeled cells within control clones (blue line) or clones overexpressing UAS-CycE + UAS-Stg (red line) at 48, 72, and 96 h pci. n = 21, 21, and 26 for control at 48, 72, and 96 h pci, respectively. n = 6, 12, and 15 for UAS-CycE+UAS-Stg at 48, 72 and 96 h pci, respectively.
E Clonal overexpression of CycE and Stg caused CySC overproliferation and GSC loss. Clones are labeled by GFP expression (green, single channel E’), Vasa (red, single channel E”) marks germ cells, and Tj (blue, single channel E’“) marks the somatic lineage. The hub is indicated by a dotted line.
F Quantification of GSC loss in the presence of CycE+Stg overexpressing CySC clones. Asterisks denote statistically significant difference from control. n = 15 and 22 for control and UAS-CycE, UAS-Stg, respectively. Error bars denote SEM.
To corroborate the hypothesis that increased proliferation is necessary for niche competition by CySCs and to determine which pathways are normally active in CySCs, we examined other cellular growth and proliferation factors for their ability to rescue the ptc mutant phenotype when reduced. Removing one copy of the gene E2f, which encodes an S-phase regulator (Duronio et al, 1995), partially suppressed the loss of GSCs; similar genetic interactions were found with cdk2, which encodes a cyclin-dependent kinase (Lehner & O’Farrell, 1990) (Fig 6C, P < 0.04, and P < 0.02, respectively). We note that GSC number was not changed in E2f/+, cdk2/+ or in any of the heterozygous backgrounds tested below (Fig 6C). In Drosophila, cellular growth and proliferation are genetically separable (Neufeld et al, 1998), so we also tested whether increased cellular growth was required for CySC colonization. Removal of one copy of the gene encoding the Drosophila Insulin receptor, InR, or genes encoding its effectors Akt1 and S6k, did not suppress niche colonization by ptc mutant clones (Fig 6C) (Chen et al, 1996; Montagne et al, 1999; Verdu et al, 1999). In fact, clonal mis-expression of Drosophila Phosphoinositide 3-kinase (PI3K) Dp110, or clonal loss of PI3K pathway inhibitors Tsc1 or Pten, was incompatible with CySC fate (Supplementary Fig S7 and Supplementary Table S2) (Leevers et al, 1996; Goberdhan et al, 1999; Potter et al, 2001; Tapon et al, 2001). Consistently, we also recovered fewer dMyc-expressing CySC clones at 14 dpci compared to control (Supplementary Table S2). Thus, cell cycle progression is essential for CySCs to gain an advantage over their neighbors at the niche, while excessive activation of cellular growth pathways like PI3K and dMyc is detrimental to CySC function.
We next tested whether increasing proliferation was sufficient to cause niche competition by expressing the G1/S-phase promoting factor CyclinE (CycE) and G2/M-phase promoting factor Stg together in a clonal fashion. In imaginal discs, clonal overexpression of these factors together led to marked acceleration of the cell cycle and increased cell number (Neufeld et al, 1998). We found that CycE+Stg overexpressing clones with at least one labeled CySC grew at a faster rate than control clones, indicating that CycE+Stg overexpression also led to cell cycle acceleration in the testis (Fig 6D). Strikingly, CycE+Stg overexpressing clones outcompeted both wild-type CySCs and GSCs at the niche (Fig 6E and F, P < 0.004), in a manner reminiscent of ptc mutant clones. Combined, these data indicate that proliferation downstream of ptc is necessary and sufficient to induce competition at the niche. Thus, altering the rate of cell division skews the stochastic process of stem cell loss and replacement at the niche in favor of the faster proliferating CySCs, and disrupting the normal homeostatic balance between GSCs and CySCs, in favor of the latter.
The Hippo pathway regulates proliferation, self-renewal, and niche competition independently of Hh
As a proof of concept for the central role of proliferation in niche competition, we examined a universal regulator of proliferation, the Hippo (Hpo) pathway (Pan, 2010) using clonal assays. Hpo restrains the activity of the transcriptional co-activator Yorkie (Yki), the Drosophila homolog of Yes-associated protein (YAP), which is oncogenic in flies and mice (Dong et al, 2007). We noted that this pathway was active in the soma, as seen by expression of the pathway target expanded (ex)-lacZ (Hamaratoglu et al, 2006) (Fig 7A). Next, we generated hpo mutant clones and measured the number of mutant CySCs at several time points. We note that FRT42D CD8-GFP hpo mutant MARCM clones were induced at rates comparable to FRT42D CD8-GFP control MARCM clones (compare Fig 7B to Fig 1D). Strikingly, hpo mutant clones displayed overproliferation and colonized the niche at the expense of wild-type stem cells (Fig 7C and F). Importantly, hpo mutant cyst cells differentiated normally and were readily observed ensheathing spermatogonial cysts (Supplementary Fig S3C).
Figure 7. hpo mutant CySCs also skew neutral drift dynamics and outcompete GSCs.

A ex-lacZ (green, single channel A’) expression in the testis was observed in the somatic lineage (Zfh1, blue, single channel A’“) near the hub (DE-cadherin, red, single channel A”).
B, C Clonal analysis, GFP (single channels, B’, C’) indicates the clone, Vasa (red) labels germ cells and Zfh1 (blue) CySCs and early cyst cells; the hub is indicated by a dotted line. GFP-labeled hpo mutant clones were generated by the MARCM technique and analyzed at 2 (B) and 14 dpci (C). Arrow (C, C’) shows displacement of wild-type GSCs by hpo mutant CySCs.
D Variation of average size of hpo mutant clones as a function of time. The data points (boxes) show the mean fraction of labeled CySCs in persisting clones. The black line shows a fit of the neutral drift model, modified to have a bias in favor of the labeled cell, to the data using an induction frequency of 10%. The dashed orange line represents the predicted clonal evolution if only a single CySC clone were induced with a time-shift of 3 days with the same set of parameters. For details of the biased drift model and the notation, see Supplementary Materials and Methods. n = 69, 64, 71, 43 for 2, 7, 14, 28 dpci, respectively. Error bars denote SEM.
E Distribution of clone sizes of persistent hpo mutant clones. The boxes show experimental data, and lines show the predictions of the model. Error bars denote SEM.
F Graph showing the number of GSCs at 14 dpci when CySC clones of the indicated genotype were present. Asterisks denote statistically significant change for the comparisons indicated. n = 26 (control), 36 (ptc), 47 (hpo), 15 (hpo; stg/+). Error bars denote SEM.
G, H YkiAct-overexpressing clones outcompeted both CySCs and GSCs at 14 dpci. Graph in (G) shows the number of GSCs when clones overexpressing YkiAct were present. Asterisks denote statistically significant change for the comparisons indicated. n = 15 (control), 19 (UAS-YkiAct). Error bars denote SEM. Clones are green (single channel H’), Vasa is red (single channel H”), and Tj is blue (single channel H”’). The hub is indicated by a dotted line.
We applied the same quantitative analysis to hpo mutant clones as described above for ptc mutant clones. Noting that the labeling efficiency of the CySC was comparable to that of the control and ptc mutant (at around 10%), we used the same strategy to analyze the clonal fate data. In doing so, we found that the behavior of hpo mutant clones was consistent with a bias in neutral drift in favor of the mutant cell (Fig 7D and E, compare lines and boxes), quantitatively similar to the trend we found for ptc mutant clones (compare Fig 7D and E with Fig 2C and D). Indeed, within error bars, we could discern no distinction between the bias for ptc and hpo mutants. Furthermore, hpo mutant CySCs displaced GSCs from the niche (Fig 7F, P < 0.0001 hpo versus control), similar to the competitive behavior of ptc mutant clones (Fig 7F, P < 0.0001 ptc versus control). Like ptc-dependent niche colonization, the loss of GSCs caused by hpo mutant CySCs could be suppressed by removing one copy of stg (Fig 7F, dark red bar, P < 0.0001 hpo versus hpo; stg/+).
We next tested the role of the Hpo pathway effector Yki in niche competition. Clonal mis-expression of an activated form of Yki (YkiAct (Oh & Irvine, 2008)) resulted in CySC clones that out-competed wild-type CySCs and GSCs at the niche (Fig 7G and H, P < 0.0014). Consistent with an essential role of yki in CySCs, we found that yki was required autonomously for self-renewal in CySCs but not in GSCs (Fig 8A, E and F), the latter consistent with a prior report (Sun et al, 2008). Finally, we addressed whether Hh and Hpo, two proliferative pathways in CySCs, were epistatic. To test this, we generated clones that were mutant for both ptc and yki, with the expectation that loss of yki would suppress the competitiveness in ptc mutant CySCs. Indeed, CySCs lacking ptc and yki did not overproliferate and colonize the niche (Fig 8B, C and E, compare red to purple line), indicating that Hpo is epistatic to Hh signaling in the testis. However, we observed no change in expression of the Yki target gene ex-lacZ in ptc mutant clones (Fig 8D, arrow), suggesting no direct link between these pathways in this tissue. These data establish Yki as a central regulator of somatic stem cell fate in the testis and suggest a parallel requirement for the Hh and Hpo pathways in CySC proliferation, through independent or convergent control of cell cycle progression genes.
Discussion
In this study we characterized the behavior of somatic CySCs in the Drosophila testis and explored the molecular mechanisms that regulate their ability to compete with their neighbors for limited space at the niche. We found that single stem cell clones bias stem cell replacement dynamics in their favor, leading to non-neutral competition, when they had increases in Hh signaling, Yki activity or in the rate of proliferation, but not when JAK/STAT signaling or adhesion were dys-regulated. Furthermore, we found that the dynamics of CySCs were well-described by a model in which they were continually and stochastically lost and replaced, leading to neutral drift dynamics and a consolidation of clonal diversity.
This observation contrasts with the dynamics of GSC offspring fate choices, where oriented divisions and mother centromere retention determine which cells remain as stem cells and which are thrust out of the niche to differentiate (Yamashita et al, 2003, 2007; Sheng & Matunis, 2011). However, careful analysis of GSC dynamics has suggested that they also undergo neutral competition, albeit at a slower loss/replacement rate than CySCs (Wallenfang et al, 2006; Sheng & Matunis, 2011; Salzmann et al, 2013). Thus, within the same stem cell niche, two markedly different strategies for self-renewal are in use, exemplified by the requirement for yki in CySC self-renewal, but not in GSC self-renewal (this study and Sun et al, 2008). This is particularly surprising as the two stem cell populations are by necessity linked, in that they need to produce offspring in the correct ratio, as well as the fact that CySCs support GSC self-renewal through BMP production (Leatherman & Dinardo, 2010). It has been hypothesized that the careful choice of stem cell retention in the GSC pool is a requirement of their role in preserving the genetic integrity of the species (Yuan & Yamashita, 2010). CySCs are under no such constraint, and moreover, need to proliferate twice as fast in order to produce two cyst cells for every germ cyst (Inaba et al, 2011). Thus it may be that the functional imperatives of the tissue (e.g., careful replication of DNA versus rapid production of offspring) determine which type of self-renewal strategy a stem cell adopts.
Characterizing the testis stem cell niche
Our study revealed an unexpected ratio of CySCs to GSCs, close to 1:1 and different from the 2:1 ratio described by Hardy et al. However we note that both studies find the same number of CySCs (approximately 13), and that the difference resides in the number of GSCs. Indeed, Hardy et al find a ratio of 1.3 CySCs:1 GSC in larval testes which increases to 1.8:1 in young adults, due entirely to a drop in the number of GSCs (Hardy et al, 1979). This may be a function of the genetic background used by these authors, as we established our 1:1 ratio through three different experiments in distinct genetic stocks. Although the analysis of the data is consistent with neutral competition between 13 equipotent CySCs, by the nature of the neutral competition model, we cannot rule out the possibility that the stem cell compartment is heterogeneous with cells moving reversibly between states in which they become primed for duplication or loss, as recently defined in the mouse intestinal crypt (Ritsma et al, 2014). In this case, the effective number of CySCs may be smaller than the observed figure of N = 13, while the true loss/replacement rate, λ, might be proportionately adjusted to a lower value such that the ratio N2/λ remains constant.
Mechanisms of niche competition by CySCs
Our results also show that the predominant force driving niche colonization by CySCs is proliferation. How proliferation causes stem cells to replace neighbors more efficiently is not established by this study. However, we hypothesize that in such a competitive situation, the rate of stem cell loss is not altered but the overproliferating mutants simply produce more offspring, which are in the right place to fill a vacant seat at the niche. It remains possible that a mechanism of active displacement is involved in CySC dominance (i.e., the colonizing stem cells crowd out the wild-type ones), and live-imaging of competing clones might distinguish between passive replacement and active displacement.
A related issue is how CySCs outcompete GSCs. We found that GSC loss is only observed after most of the CySC pool is comprised of colonizing mutant CySCs (Supplementary Fig S4F). We therefore favor the model that competition among CySCs for niche space precedes that between CySCs and GSCs. It is unclear whether the numerous offspring of the competitive CySCs are passively replacing GSCs that have spontaneously left a vacancy at the niche, or whether colonizing CySCs actively push the GSCs out of the niche. The latter scenario is reminiscent of competition among GSCs in the Drosophila ovary, where the contact area between the GSC and niche depended on DE-cadherin. GSCs that elevated cadherins adhered better to the niche and caused the physical displacement of neighbors (Jin et al, 2008; Tian et al, 2012). We explored the contribution of integrin- and cadherin-based adhesion and found that neither affected the competitiveness of CySCs. Moreover, we found that integrin binding was entirely dispensable for CySC self-renewal, unlike cadherin (Voog et al, 2008). Importantly, clonal gain of integrin or cadherin did not lead to niche colonization, indicating that they are not instructive for CySC maintenance. Moreover, we found no role for JAK/STAT signaling in inducing competition at the niche. The fact that neither Stat92E nor integrin was causal to colonization in clonal assays is surprising because both were ascribed critical roles in CySC-dependent niche competition (Issigonis et al, 2009). The reasons for the difference in results by our group and the previous study are not entirely clear. However, we note that gain of Stat92E activity in CySCs in an otherwise wild-type background leads to expansion (not loss) of GSCs because JAK/STAT signaling in CySCs enables their extended niche function to support GSC self-renewal (Leatherman & Dinardo, 2008, 2010). The latter niche role is specific to JAK/STAT signaling in CySCs and cannot be fulfilled by Hh signaling, another CySC self-renewal pathway (Amoyel et al, 2013). Moreover, our clonal assays (as opposed to lineage mis-expression) are able to recapitulate the constant jostling for space at the niche that normally occurs. Regardless, our findings establish that competition and self-renewal are two facets of the same homeostatic process (i.e., proliferation) and that colonizing stem cells have not acquired a new cellular property, but are simply better at self-renewing.
Our study exemplifies how corrupting the naturally occurring process of neutral competition endows a stem cell with greater competitiveness, enabling it to gain dominance within a tissue. Such behavior may be relevant to the early steps of oncogenesis driven by tumor-initiating cells, which have stem cell-like properties (Reya et al, 2001), as in the case of carcinoma, glioma and leukemia caused by sustained Hh signaling (Clement et al, 2007; Zhao et al, 2009; Youssef et al, 2012). The process described here of biasing neutral drift by stem cells harboring oncogenic mutations and the mechanism underlying it appear to be conserved (Vermeulen et al, 2013; Snippert et al, 2014). Taken together, these findings may explain observations such as field cancerization, in which a molecular lesion spreads through a tissue, causing multiple foci of the primary tumor (Vanharanta & Massague, 2012).
Materials and Methods
Fly stocks and genotypes are described in Supplementary Materials and Methods. For ptc mutants, ptcS2 was used in all experiments shown, but similar results were obtained with ptcIIw. hpoKC202 phenotypes were confirmed using hpo42-47.
Freshly eclosed adult males were aged for 1 day and then heat shocked for 1 h at 37°C to induce clones and raised at 25°C until the appropriate time for dissection. For self-renewal assays, CySCs were scored as Zfh1-positive or Tj-positive cells one cell diameter away from the hub, and GSCs as Vasa-positive cells in contact with the hub. For control, ptc or hpo CySCs, the method of counting is detailed in the text.
Dissections and immunohistochemistry were performed as previously described (Flaherty et al, 2010). Primary antibodies used were rabbit anti-GFP (1:500, Invitrogen), mouse anti-GFP (1:500, Invitrogen), chicken anti-β-galactosidase (1:250, Immunology Consultants Lab), goat anti-Vasa (1:400, Santa Cruz), rabbit anti-Zfh1 (1:5,000, gift of Ruth Lehmann), guinea pig anti-Zfh1 (1:1,000, gift of James Skeath), guinea pig anti-Tj (1:3,000, gift of Dorothea Godt), rabbit anti-Stat92E (1:1,000), mouse anti-Ptc (1:100, DSHB), rat anti-DE cadherin (1:50, DSHB), mouse anti-βPS-integrin (1:20, DSHB), rabbit anti-cleaved caspase 3 (1:50, Cell Signaling).
For 5-ethynyl-2′-deoxyuridine (EdU, Invitrogen) labeling, samples were incubated for 30 min before fixation in Ringer’s medium containing 10 μM EdU. Testes were fixed and processed normally for antibody labeling and then treated per manufacturer’s instructions.
For statistical tests, we used the GraphPad Prism software. To compare two samples, we used the Mann–Whitney U-test to determine significance; for multiple conditions, we used the Kruskal–Wallis test and the Sidak’s multiple comparisons test for post hoc analysis.
The mathematical model is described in Supplementary Materials and Methods.
Acknowledgments
We thank R. Lehmann, F. Schöck, L. Johnston, D. Kalderon, U. Tepass, J. Treisman, S. Grewal, Y. Yamashita, T. Harris, B. Ohlstein, L. Buttitta, D. Godt, Bloomington, and DSHB for antibodies and reagents. We thank members of the Bach laboratory for fruitful discussions. We are extremely grateful to Esteban Mazzoni, Andrew Tomlinson, and Gary Struhl for their generosity in the aftermath of Superstorm Sandy. BDS acknowledges the support of the Wellcome Trust (Grant Number 098357/Z/12/Z). Work in the Bach laboratory is supported by grants from the NIH (R01-GM085075-05 and R01-GM085075-05S1) and NYSTEM (C028132 and C024284).
Author contributions
MA designed, carried out, and analyzed the experiments. EAB contributed to experimental design and analysis. BDS contributed to experimental design and provided the mathematical analysis. MA, BDS, and EAB wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Amoyel M, Bach EA. Cell competition: how to eliminate your neighbours. Development. 2014;141:988–1000. doi: 10.1242/dev.079129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoyel M, Sanny J, Burel M, Bach EA. Hedgehog is required for CySC self-renewal but does not contribute to the GSC niche in the Drosophila testis. Development. 2013;140:56–65. doi: 10.1242/dev.086413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Jack J, Garofalo RS. The Drosophila insulin receptor is required for normal growth. Endocrinology. 1996;137:846–856. doi: 10.1210/endo.137.3.8603594. [DOI] [PubMed] [Google Scholar]
- Chen Y, Struhl G. Dual roles for patched in sequestering and transducing Hedgehog. Cell. 1996;87:553–563. doi: 10.1016/s0092-8674(00)81374-4. [DOI] [PubMed] [Google Scholar]
- Chen HW, Chen X, Oh SW, Marinissen MJ, Gutkind JS, Hou SX. mom identifies a receptor for the Drosophila JAK/STAT signal transduction pathway and encodes a protein distantly related to the mammalian cytokine receptor family. Genes Dev. 2002;16:388–398. doi: 10.1101/gad.955202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Tiyaboonchai A, Yamashita YM, Hunt AJ. Asymmetric division of cyst stem cells in Drosophila testis is ensured by anaphase spindle repositioning. Development. 2011;138:831–837. doi: 10.1242/dev.057901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton E, Doupe DP, Klein AM, Winton DJ, Simons BD, Jones PH. A single type of progenitor cell maintains normal epidermis. Nature. 2007;446:185–189. doi: 10.1038/nature05574. [DOI] [PubMed] [Google Scholar]
- Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165–172. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–116. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- de Cuevas M, Matunis EL. The stem cell niche: lessons from the Drosophila testis. Development. 2011;138:2861–2869. doi: 10.1242/dev.056242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinardo S, Okegbe T, Wingert L, Freilich S, Terry N. Lines and bowl affect the specification of cyst stem cells and niche cells in the Drosophila testis. Development. 2011;138:1687–1696. doi: 10.1242/dev.057364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupe DP, Alcolea MP, Roshan A, Zhang G, Klein AM, Simons BD, Jones PH. A single progenitor population switches behavior to maintain and repair esophageal epithelium. Science. 2012;337:1091–1093. doi: 10.1126/science.1218835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duronio RJ, O’Farrell PH, Xie JE, Brook A, Dyson N. The transcription factor E2F is required for S phase during Drosophila embryogenesis. Genes Dev. 1995;9:1445–1455. doi: 10.1101/gad.9.12.1445. [DOI] [PubMed] [Google Scholar]
- Edgar BA, O’Farrell PH. Genetic control of cell division patterns in the Drosophila embryo. Cell. 1989;57:177–187. doi: 10.1016/0092-8674(89)90183-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty MS, Salis P, Evans CJ, Ekas LA, Marouf A, Zavadil J, Banerjee U, Bach EA. Chinmo is a functional effector of the JAK/STAT pathway that regulates eye development, tumor formation and stem cell self-renewal in Drosophila. Dev Cell. 2010;18:556–568. doi: 10.1016/j.devcel.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes AJ, Lin H, Ingham PW, Spradling AC. Hedgehog is required for the proliferation and specification of ovarian somatic cells prior to egg chamber formation in Drosophila. Development. 1996;122:1125–1135. doi: 10.1242/dev.122.4.1125. [DOI] [PubMed] [Google Scholar]
- Goberdhan DC, Paricio N, Goodman EC, Mlodzik M, Wilson C. Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes Dev. 1999;13:3244–3258. doi: 10.1101/gad.13.24.3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, Jafar-Nejad H, Halder G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol. 2006;8:27–36. doi: 10.1038/ncb1339. [DOI] [PubMed] [Google Scholar]
- Hardy RW, Tokuyasu KT, Lindsley DL, Garavito M. The germinal proliferation center in the testis of Drosophila melanogaster. J Ultrastruct Res. 1979;69:180–190. doi: 10.1016/s0022-5320(79)90108-4. [DOI] [PubMed] [Google Scholar]
- Inaba M, Yuan H, Yamashita YM. String (Cdc25) regulates stem cell maintenance, proliferation and aging in Drosophila testis. Development. 2011;138:5079–5086. doi: 10.1242/dev.072579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW, Taylor AM, Nakano Y. Role of the Drosophila patched gene in positional signalling. Nature. 1991;353:184–187. doi: 10.1038/353184a0. [DOI] [PubMed] [Google Scholar]
- Issigonis M, Tulina N, de Cuevas M, Brawley C, Sandler L, Matunis E. JAK-STAT signal inhibition regulates competition in the Drosophila testis stem cell niche. Science. 2009;326:153–156. doi: 10.1126/science.1176817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Kirilly D, Weng C, Kawase E, Song X, Smith S, Schwartz J, Xie T. Differentiation-defective stem cells outcompete normal stem cells for niche occupancy in the Drosophila ovary. Cell Stem Cell. 2008;2:39–49. doi: 10.1016/j.stem.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase E, Wong MD, Ding BC, Xie T. Gbb/Bmp signaling is essential for maintaining germline stem cells and for repressing bam transcription in the Drosophila testis. Development. 2004;131:1365–1375. doi: 10.1242/dev.01025. [DOI] [PubMed] [Google Scholar]
- Kiger AA, Jones DL, Schulz C, Rogers MB, Fuller MT. Stem cell self-renewal specified by JAK-STAT activation in response to a support cell cue. Science. 2001;294:2542–2545. doi: 10.1126/science.1066707. [DOI] [PubMed] [Google Scholar]
- Klein AM, Nakagawa T, Ichikawa R, Yoshida S, Simons BD. Mouse germ line stem cells undergo rapid and stochastic turnover. Cell Stem Cell. 2010;7:214–224. doi: 10.1016/j.stem.2010.05.017. [DOI] [PubMed] [Google Scholar]
- Leatherman JL, Dinardo S. Zfh-1 controls somatic stem cell self-renewal in the Drosophila testis and nonautonomously influences germline stem cell self-renewal. Cell Stem Cell. 2008;3:44–54. doi: 10.1016/j.stem.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatherman JL, Dinardo S. Germline self-renewal requires cyst stem cells and stat regulates niche adhesion in Drosophila testes. Nat Cell Biol. 2010;12:806–811. doi: 10.1038/ncb2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD. The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J. 1996;15:6584–6594. [PMC free article] [PubMed] [Google Scholar]
- Lehner CF, O’Farrell PH. Drosophila cdc2 homologs: a functional homolog is coexpressed with a cognate variant. EMBO J. 1990;9:3573–3581. doi: 10.1002/j.1460-2075.1990.tb07568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Garcia C, Klein AM, Simons BD, Winton DJ. Intestinal stem cell replacement follows a pattern of neutral drift. Science. 2010;330:822–825. doi: 10.1126/science.1196236. [DOI] [PubMed] [Google Scholar]
- Margolis J, Spradling A. Identification and behavior of epithelial stem cells in the Drosophila ovary. Development. 1995;121:3797–3807. doi: 10.1242/dev.121.11.3797. [DOI] [PubMed] [Google Scholar]
- Michel M, Kupinski AP, Raabe I, Bokel C. Hh signalling is essential for somatic stem cell maintenance in the Drosophila testis niche. Development. 2012;139:2663–2669. doi: 10.1242/dev.075242. [DOI] [PubMed] [Google Scholar]
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: a regulator of cell size. Science. 1999;285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- Morata G, Ripoll P. Minutes: mutants of drosophila autonomously affecting cell division rate. Dev Biol. 1975;42:211–221. doi: 10.1016/0012-1606(75)90330-9. [DOI] [PubMed] [Google Scholar]
- Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–129. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- de Navascues J, Perdigoto CN, Bian Y, Schneider MH, Bardin AJ, Martinez-Arias A, Simons BD. Drosophila midgut homeostasis involves neutral competition between symmetrically dividing intestinal stem cells. EMBO J. 2012;31:2473–2485. doi: 10.1038/emboj.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA. Coordination of growth and cell division in the Drosophila wing. Cell. 1998;93:1183–1193. doi: 10.1016/s0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- Nystul T, Spradling A. An epithelial niche in the Drosophila ovary undergoes long-range stem cell replacement. Cell Stem Cell. 2007;1:277–285. doi: 10.1016/j.stem.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Oh H, Irvine KD. In vivo regulation of Yorkie phosphorylation and localization. Development. 2008;135:1081–1088. doi: 10.1242/dev.015255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter CJ, Huang H, Xu T. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell. 2001;105:357–368. doi: 10.1016/s0092-8674(01)00333-6. [DOI] [PubMed] [Google Scholar]
- Price MA, Kalderon D. Proteolysis of cubitus interruptus in Drosophila requires phosphorylation by protein kinase A. Development. 1999;126:4331–4339. doi: 10.1242/dev.126.19.4331. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Ritsma L, Ellenbroek SI, Zomer A, Snippert HJ, de Sauvage FJ, Simons BD, Clevers H, van Rheenen J. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature. 2014;507:362–365. doi: 10.1038/nature12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzmann V, Inaba M, Cheng J, Yamashita YM. Lineage tracing quantification reveals symmetric stem cell division in Drosophila male germline stem cells. Cell Mol Bioeng. 2013;6:441–448. doi: 10.1007/s12195-013-0295-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarpal R, Pellikka M, Patel RR, Hui FY, Godt D, Tepass U. Mutational analysis supports a core role for Drosophila alpha-catenin in adherens junction function. J Cell Sci. 2012;125:233–245. doi: 10.1242/jcs.096644. [DOI] [PubMed] [Google Scholar]
- Sheng XR, Matunis E. Live imaging of the Drosophila spermatogonial stem cell niche reveals novel mechanisms regulating germline stem cell output. Development. 2011;138:3367–3376. doi: 10.1242/dev.065797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivdasani AA, Ingham PW. Regulation of stem cell maintenance and transit amplifying cell proliferation by tgf-beta signaling in Drosophila spermatogenesis. Curr Biol. 2003;13:2065–2072. doi: 10.1016/j.cub.2003.10.063. [DOI] [PubMed] [Google Scholar]
- Simons BD, Clevers H. Strategies for homeostatic stem cell self-renewal in adult tissues. Cell. 2011;145:851–862. doi: 10.1016/j.cell.2011.05.033. [DOI] [PubMed] [Google Scholar]
- Singh SR, Zheng Z, Wang H, Oh SW, Chen X, Hou SX. Competitiveness for the niche and mutual dependence of the germline and somatic stem cells in the Drosophila testis are regulated by the JAK/STAT signaling. J Cell Physiol. 2010;223:500–510. doi: 10.1002/jcp.22073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, Clevers H. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- Snippert HJ, Schepers AG, van Es JH, Simons BD, Clevers H. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep. 2014;15:62–69. doi: 10.1002/embr.201337799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Zhao S, Wang Z. Genes of Hippo signaling network act unconventionally in the control of germline proliferation in Drosophila. Dev Dyn. 2008;237:270–275. doi: 10.1002/dvdy.21411. [DOI] [PubMed] [Google Scholar]
- Tanentzapf G, Brown NH. An interaction between integrin and the talin FERM domain mediates integrin activation but not linkage to the cytoskeleton. Nat Cell Biol. 2006;8:601–606. doi: 10.1038/ncb1411. [DOI] [PubMed] [Google Scholar]
- Tanentzapf G, Devenport D, Godt D, Brown NH. Integrin-dependent anchoring of a stem-cell niche. Nat Cell Biol. 2007;9:1413–1418. doi: 10.1038/ncb1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N, Ito N, Dickson BJ, Treisman JE, Hariharan IK. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell. 2001;105:345–355. doi: 10.1016/s0092-8674(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Thacker SA, Bonnette PC, Duronio RJ. The contribution of E2F-regulated transcription to Drosophila PCNA gene function. Curr Biol. 2003;13:53–58. doi: 10.1016/s0960-9822(02)01400-8. [DOI] [PubMed] [Google Scholar]
- Tian JP, Jin Z, Xie T. Mathematical model for two germline stem cells competing for niche occupancy. Bull Math Biol. 2012;74:1207–1225. doi: 10.1007/s11538-011-9713-x. [DOI] [PubMed] [Google Scholar]
- Tulina N, Matunis E. Control of stem cell self-renewal in Drosophila spermatogenesis by JAK-STAT signaling. Science. 2001;294:2546–2549. doi: 10.1126/science.1066700. [DOI] [PubMed] [Google Scholar]
- Vanharanta S, Massague J. Field cancerization: something new under the sun. Cell. 2012;149:1179–1181. doi: 10.1016/j.cell.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Verdu J, Buratovich MA, Wilder EL, Birnbaum MJ. Cell-autonomous regulation of cell and organ growth in Drosophila by Akt/PKB. Nat Cell Biol. 1999;1:500–506. doi: 10.1038/70293. [DOI] [PubMed] [Google Scholar]
- Vermeulen L, Morrissey E, van der Heijden M, Nicholson AM, Sottoriva A, Buczacki S, Kemp R, Tavare S, Winton DJ. Defining stem cell dynamics in models of intestinal tumor initiation. Science. 2013;342:995–998. doi: 10.1126/science.1243148. [DOI] [PubMed] [Google Scholar]
- Voog J, D’Alterio C, Jones DL. Multipotent somatic stem cells contribute to the stem cell niche in the Drosophila testis. Nature. 2008;454:1132–1136. doi: 10.1038/nature07173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallenfang MR, Nayak R, DiNardo S. Dynamics of the male germline stem cell population during aging of Drosophila melanogaster. Aging Cell. 2006;5:297–304. doi: 10.1111/j.1474-9726.2006.00221.x. [DOI] [PubMed] [Google Scholar]
- Wang L, Li Z, Cai Y. The JAK/STAT pathway positively regulates DPP signaling in the Drosophila germline stem cell niche. J Cell Biol. 2008;180:721–728. doi: 10.1083/jcb.200711022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Spradling AC. Decapentaplegic is essential for the maintenance and division of germline stem cells in the Drosophila ovary. Cell. 1998;94:251–260. doi: 10.1016/s0092-8674(00)81424-5. [DOI] [PubMed] [Google Scholar]
- Xie T, Spradling AC. A niche maintaining germ line stem cells in the Drosophila ovary. Science. 2000;290:328–330. doi: 10.1126/science.290.5490.328. [DOI] [PubMed] [Google Scholar]
- Yamashita YM, Jones DL, Fuller MT. Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science. 2003;301:1547–1550. doi: 10.1126/science.1087795. [DOI] [PubMed] [Google Scholar]
- Yamashita YM, Mahowald AP, Perlin JR, Fuller MT. Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science. 2007;315:518–521. doi: 10.1126/science.1134910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef KK, Lapouge G, Bouvree K, Rorive S, Brohee S, Appelstein O, Larsimont JC, Sukumaran V, Van de Sande B, Pucci D, Dekoninck S, Berthe JV, Aerts S, Salmon I, del Marmol V, Blanpain C. Adult interfollicular tumour-initiating cells are reprogrammed into an embryonic hair follicle progenitor-like fate during basal cell carcinoma initiation. Nat Cell Biol. 2012;14:1282–1294. doi: 10.1038/ncb2628. [DOI] [PubMed] [Google Scholar]
- Yuan H, Yamashita YM. Germline stem cells: stems of the next generation. Curr Opin Cell Biol. 2010;22:730–736. doi: 10.1016/j.ceb.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kalderon D. Hedgehog acts as a somatic stem cell factor in the Drosophila ovary. Nature. 2001;410:599–604. doi: 10.1038/35069099. [DOI] [PubMed] [Google Scholar]
- Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, Munchhof M, VanArsdale T, Beachy PA, Reya T. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–779. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.