

Abstract
The mechanisms of intramembrane proteases are incompletely understood due to the lack of structural data on substrate complexes. To gain insight into substrate binding by rhomboid proteases, we have synthesised a series of novel peptidyl-chloromethylketone (CMK) inhibitors and analysed their interactions with Escherichia coli rhomboid GlpG enzymologically and structurally. We show that peptidyl-CMKs derived from the natural rhomboid substrate TatA from bacterium Providencia stuartii bind GlpG in a substrate-like manner, and their co-crystal structures with GlpG reveal the S1 to S4 subsites of the protease. The S1 subsite is prominent and merges into the ‘water retention site’, suggesting intimate interplay between substrate binding, specificity and catalysis. Unexpectedly, the S4 subsite is plastically formed by residues of the L1 loop, an important but hitherto enigmatic feature of the rhomboid fold. We propose that the homologous region of members of the wider rhomboid-like protein superfamily may have similar substrate or client-protein binding function. Finally, using molecular dynamics, we generate a model of the Michaelis complex of the substrate bound in the active site of GlpG.
Keywords: intramembrane protease, rhomboid family, rhomboid protease, structure, substrate recognition
Introduction
Cleavage of transmembrane domains (TMDs) by intramembrane proteases has emerged as an important and evolutionarily widespread signalling and quality control mechanism with medical significance (Brown et al, 2000; Lemberg, 2011), but a full understanding of the biological roles and design of pharmacological interventions against intramembrane proteases requires a greater knowledge of their mechanism and structure. Intramembrane proteases are very different from the classical water soluble proteases, since they evolved independently and operate in a distinct biophysical environment—at the interface of lipid membrane and aqueous solvent (Strisovsky, 2013). Although the crystal structures of prokaryotic homologues of all four catalytic types of intramembrane proteases have been solved (Wang et al, 2006; Feng et al, 2007; Li et al, 2013; Manolaridis et al, 2013), mechanistic understanding is limited by the lack of structures of enzyme–substrate complexes.
Rhomboids are serine proteases—probably the best characterised intramembrane proteases as regards structure and mechanism. Rhomboid proteases are widely conserved and regulate many biological processes including intercellular signalling, mitochondrial dynamics, invasion of eukaryotic parasites and membrane protein quality control (Lemberg, 2013). In addition, the recently discovered rhomboid-like proteins that share a similar scaffold, but are devoid of enzymatic activity, have emerged as important regulators of membrane protein quality control (Greenblatt et al, 2011; Zettl et al, 2011) and trafficking (Adrain et al, 2012). Non-catalytic rhomboid-like proteins regulate growth factor signalling (Zettl et al, 2011), inflammatory signalling via tumour necrosis factor in macrophages (Adrain et al, 2012) and NK-cell signalling (Liu et al, 2013), which illustrates their wide medical importance. In contrast to the advances in the biology of the non-protease rhomboid-family proteins, their mechanistic understanding lags behind. The only current source of structural information about rhomboid-family proteins are the bacterial rhomboid proteases.
The structures of bacterial rhomboid proteases published over the last 8 years have provided the first glimpses into the molecular architecture of an intramembrane protease. However, the mechanism of action and the structural basis of substrate specificity of rhomboids remain unresolved, largely due to the absence of structural analyses of rhomboid–substrate complexes. The recently published structures of GlpG bound to various small, mechanism-based inhibitors (Vinothkumar et al, 2010, 2013; Xue & Ha, 2012; Vosyka et al, 2013) have served as models for speculations on substrate binding, but their utility in this respect is limited since the inhibitors are relatively small and structurally very different from peptide or protein substrates.
Here, we report crystal structures of a rhomboid intramembrane protease in complex with substrate-derived peptides, providing the first direct structural view of rhomboid specificity and catalytic mechanism. We show that tetrapeptidyl-chloromethylketone inhibitors bind the Escherichia coli rhomboid protease GlpG in a way that mimics the substrate, which allows us to map the specificity determining pockets of GlpG with confidence. Unexpectedly, the S4 subsite (which binds to the P4 residue of the substrate) is formed by the residues from the L1 loop, a conspicuous but enigmatic structural feature of rhomboid proteases (Wang et al, 2007; Bondar et al, 2009; Baker & Urban, 2012). Using site-directed mutagenesis, quantitative enzymatic assays and structural analyses, we demonstrate the plasticity of the S4 subsite. Furthermore, our work has implications for the recently discovered proteolytically inactive members of the rhomboid-like family (such as iRhoms or Derlins). It suggests that their domains topologically corresponding to the L1 loop of rhomboids may have client-binding roles. Finally, using molecular modelling and dynamics, we generate an extended model of our complex structure comprising the P4 to P3′ fragment of a bound substrate, allowing us to speculate about the mode of interaction of substrate’s transmembrane domain with rhomboid.
Results
The inhibitory properties of peptidyl-chloromethylketones
One of the problems complicating structural analyses of rhomboid–substrate complexes is the relatively low affinity of rhomboids for their substrates (Dickey et al, 2013). To overcome this hurdle and gain insight into rhomboid substrate binding, we developed mechanism-based irreversible inhibitors modified with a peptide derived from a natural rhomboid substrate. The currently used rhomboid inhibitors, isocoumarins, phosphonofluoridates and monocyclic β-lactams (Vinothkumar et al, 2010, 2013; Pierrat et al, 2011; Xue & Ha, 2012; Xue et al, 2012), were unsuitable as warheads because the stereochemical similarity of peptidyl conjugates of isocoumarins and β-lactams to the acyl enzyme intermediate would be limited, and phosphonofluoridates have proven difficult to synthesise in the desired sequence diversity. We therefore turned our attention to peptidyl-chloromethylketones (CMKs) (Fig 1A), whose complexes with serine proteases resemble the tetrahedral transition state intermediate (Mac Sweeney et al, 2000; Malthouse, 2007) and which are readily synthesisable. The commercially available CMKs TLCK (N-α-tosyl-L-lysine chloromethylketone) and TPCK (N-α-tosyl-L-phenylalanine chloromethylketone) had shown only weak inhibition of YqgP and Drosophila rhomboid 1 (Urban et al, 2001; Urban & Wolfe, 2005), but we reasoned that this could have been due to their unsuitable P1 residues (Lys or Phe), since P1 residues with large side chains are not tolerated in substrates by several rhomboids including GlpG (Strisovsky et al, 2009; Vinothkumar et al, 2010).
Figure 1. Tetrapeptidyl-chloromethylketones are mechanism-based inhibitors of rhomboid proteases.
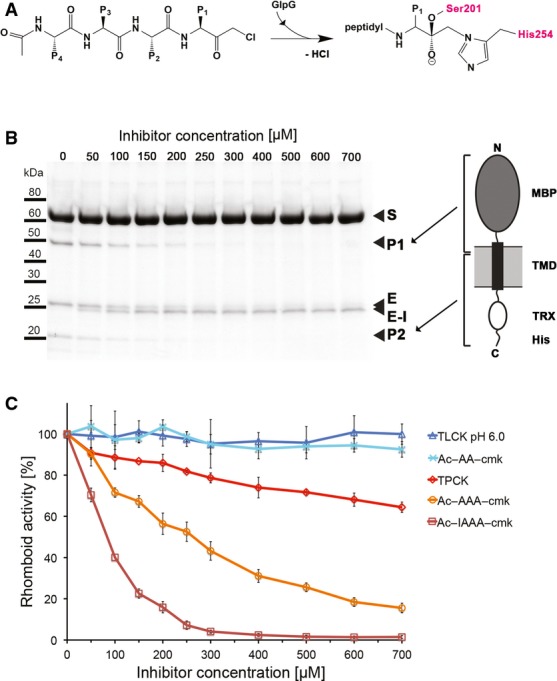
A Scheme of a tetrapeptidyl-chloromethylketone and mechanism of its reaction with the catalytic dyad of GlpG. In the final adduct, the inhibitor has lost chlorine and is covalently bound to serine 201 and histidine 254.
B SDS gel showing the inhibition of GlpG by increasing concentrations of Ac-IAAA-cmk. The identity of cleavage products P1 and P2 is illustrated by the schematic drawing of chimeric TatA that was used as substrate (right panel). Reaction of the inhibitor with catalytic residues links TMDs 4 and 6 of GlpG, resulting in a faster migrating band in SDS-PAGE (left panel). MBP, maltose-binding protein; TRX, thioredoxin; S, substrate; P1, product 1; P2, product 2, E, enzyme; E-I, enzyme inhibitor complex.
C The inhibition properties of chloromethylketones depend on the length and sequence of their peptidyl chain. All compounds were pre-incubated with GlpG for 180 min and reacted with the TatA substrate for 30 min as described in Materials and Methods. The assays were performed in triplicate, and data points show average ± standard deviation.
We have first examined the inhibitory properties of tetrapeptidyl-CMK Ac-IleAlaAlaAla-COCH2Cl (abbreviated as Ac-IAAA-cmk henceforth) based on the well-characterised bacterial rhomboid substrate TatA (Stevenson et al, 2007; Strisovsky et al, 2009). Like all other peptidyl-CMKs used in this study, this compound was stable in aqueous solution for more than 4 h (Supplementary Fig S1) and was soluble in rhomboid assay buffer up to 1 mM concentration (data not shown), allowing robust inhibition measurements. The compound Ac-IAAA-cmk inhibited GlpG in a concentration- and time-dependent manner (Fig 1B and Supplementary Fig S2A), and mass-spectrometric analysis indicated that it formed a stoichiometric (1:1) complex with the enzyme, which was dependent on the catalytic residues Ser201 and His254 (Supplementary Fig S2B). Upon reaction of Ac-IAAA-cmk with wild-type (wt) GlpG, but not with its S201A and H254A mutants, a faster migrating species on SDS-PAGE arose (Fig 1B and Supplementary Fig S2B). A similar effect has been observed recently upon disulphide cross-linking of TMDs 2 and 5 in GlpG (Xue & Ha, 2013), which suggested that Ac-IAAA-cmk may be cross-linking two TMDs of GlpG. The mass shift of GlpG in the presence of Ac-IAAA-cmk was consistent with the formation of the inhibitor–enzyme complex and elimination of a leaving group of approximately 36 Da (consistent with the molecular weight of HCl). This behaviour was analogous to how CMKs react with classical serine proteases, and we concluded that Ac-IAAA-cmk acted as a mechanism-based inhibitor of GlpG, forming a covalent adduct with the catalytic dyad residues, thus cross-linking TMDs 4 and 6. Furthermore, N-terminal truncation analysis of Ac-IAAA-cmk revealed that the inhibitory potency markedly decreased with progressive truncation of peptidyl chain of the inhibitor (Fig 1C).
Tetrapeptidyl-chloromethylketone inhibitors bind GlpG in a substrate-like manner
To assess whether our peptidyl-CMKs bound to rhomboid in a manner similar to the parent substrate, we analysed the sensitivity of the substrate and inhibitors to identical amino acid changes. We first investigated the subsite preferences of GlpG in the context of the TatA substrate in vitro by conducting a complete positional scanning mutagenesis of its P5 to P1 region. The P1 position was the most restrictive one, where GlpG strongly preferred small amino acids with non-branched side chain, such as Ala or Cys (Fig 2A and Supplementary Fig S3); the second most restrictive position was P4 with preference for hydrophobic residues. Positions P5, P3 and P2 were much less restrictive, with P2 accepting almost any amino acid with little impact on cleavage efficiency. Interestingly, aspartate inhibited cleavage profoundly anywhere between P1 to P4 positions, and glycine was not tolerated well at P1, P3 and P4 positions. To verify these results in biological membranes, we introduced some of the strongest inhibitory mutations in the context of full-length TatA into a chimeric substrate construct based on fusions with maltose-binding protein and thioredoxin (Strisovsky et al, 2009) and tested the cleavability of the mutants by endogenous GlpG in vivo. Consistently, mutations in the P4 position (I5S or I5G), the P3 position (A6D) and the P1 position (A8G or A8V) led to a dramatic decrease in substrate cleavage to nearly undetectable levels, as documented by Western blotting (Fig 2B), confirming our in vitro inhibition data.
Figure 2. Tetrapeptidyl-chloromethylketones bind GlpG in a substrate-like manner.

A Specificity matrix of GlpG preferences on TatA variants in vitro. Preferences for TatA positions P5–P1 (residues 4–8) are displayed in shades of grey. Substrates showing increased cleavage are additionally marked with black dots. GlpG preferences are most stringent for positions P1 and P4 of TatA. The assays have been done in duplicates and representative source data are shown in Supplementary Fig S3.
B In vivo cleavage efficiency of TatA variants with mutations not tolerated in vitro. Consistent with the in vitro assay, substrates with mutations T4W (P5 position), I5G or I5S (P4 position), A6D (P3 position) and A8G or A8V (P1 position) are refractory to cleavage or show severely inhibited cleavage by GlpG in biological membranes.
C Correlation of effects of amino acid changes in inhibitors with corresponding mutations in substrates. Amino acids that are not tolerated in TatA by GlpG in vitro and in vivo cause a loss of inhibitory property in the respective inhibitors Ac-GATA-cmk, Ac-SATA-cmk, Ac-IATG-cmk, Ac-IATV-cmk and Ac-IDTA-cmk. The parent compound Ac-IATA-cmk, having the same P1–P4 sequence as wild-type TatA, or its variant Ac-ISKA-cmk harbouring mutations innocuous in the substrate, inhibit GlpG efficiently. The assays have been done in independent triplicates and plotted as average ± standard deviation. Representative source data are shown in Supplementary Fig S4.
Having defined the positional sequence preferences of GlpG in a substrate, we determined whether the peptidyl-CMK inhibitors showed the same specificity, implying a similar binding mechanism. We focussed on the amino acid changes in positions P4, P3 and P1 of TatA that strongly impaired substrate cleavage by GlpG both in vitro (Fig 2A) and in vivo (Fig 2B): I5S, I5G, A6D, A8V and A8G. These amino acid changes were introduced into the TatA-derived parent compound Ac-IATA-cmk, and inhibitory properties of the resulting compounds were compared at a range of concentrations and fixed pre-incubation time. While all the amino acid changes that impaired cleavage of mutant TatA substrates (I5S, I5G, A6D, A8V and A8G) also profoundly worsened the inhibitory properties of the variant peptidyl-CMKs, those amino acid changes that did not negatively affect cleavage of mutant substrate (T7A and A6S/T7K) had no impact on the inhibitory properties of the respective CMK derivatives (Figs 1C and 2C, and Supplementary Fig S4). This demonstrates that TatA-derived peptidyl-CMKs bind GlpG in a substrate-like manner and can hence be used as substrate mimetics in crystallographic experiments.
The GlpG:Ac-IATA-cmk complex structure reveals substrate interactions in the active site
The experiments described above provided us with validated tools for structural characterisation of rhomboid–substrate interaction. We co-crystallised Ac-IATA-cmk with the transmembrane core of the wild-type GlpG rhomboid protease and solved the complex structure at 2.1 Å resolution (data collection and refinement statistics in Supplementary Table S1). The electron density for the whole inhibitor was clearly defined and allowed unambiguous model building (Fig 3A). The inhibitor is anchored in the active site by two covalent bonds to the catalytic dyad residues S201 and H254, confirming that the CMK warhead reacts as expected. The peptidyl part of the inhibitor fills the active site lying wedged between loops 5 (L5) and 3 (L3), forming a parallel β-sheet with the latter (Fig 3B). The carbonyl oxygen of the CMK warhead forms a weak hydrogen bond to the side chain amido group of N154, but not to the main chain amides of S201 or L200, unlike previously observed in isocoumarin (ISM) and diisopropylfluorophosphonate (DFP) inhibitor complexes (Vinothkumar et al, 2010; Xue & Ha, 2012). This minor difference could be a consequence of the covalent binding of the CMK to both the catalytic serine and histidine, which might slightly distort the carbonyl oxygen from the position it would adopt in the natural (singly bonded) tetrahedral intermediate (Mac Sweeney et al, 2000). Nevertheless, the position of the P1 carbonyl oxygen is similar to the position of the ISM benzoyl carbonyl (Vinothkumar et al, 2010) and DFP phosphonyl oxygens (Xue & Ha, 2012) (Fig 3C), suggesting that the double binding of the CMK warhead to the catalytic dyad is unlikely to affect the conformation of the tetrapeptide ligand in the active site significantly.
Figure 3. Crystal structure of GlpG complexed to Ac-IATA-cmk reveals the mode of substrate binding to GlpG.

A Overall structure of the GlpG:Ac-IATA-cmk complex. Ac-IATA-cmk (yellow) is covalently bound to the catalytic histidine and serine (pink). An Fo–Fc simulated annealing omit map, calculated at 2.1 Å and contoured at 3 σ, is shown 2 Å around Ac-IATA-cmk. Crystallographic statistics is shown in Supplementary Table S1.
B Interactions between Ac-IATA-cmk and GlpG. GlpG residues forming hydrogen bonds with Ac-IATA-cmk are coloured green. These residues are additionally engaged in van der Waals (vdW) contacts; residues making vdW contacts only are depicted in blue. All interactions were calculated using the program Ligplot.
C Superposition of the GlpG:Ac-IATA-cmk complex with the isocoumarin (ISM) and diisopropylfluorophosphonate (DFP) complexes (PDB-IDs 2XOV and 4H1D, respectively). The side chain of the P1 alanine superimposes well with one of the DFP isopropyl groups and points into the S1 subsite. The ISM ring points away from the P1 alanine, but still into the cavity. The oxyanion position is occupied by the superimposing DFP phosphonyl and the ISM benzoyl carbonyl oxygens. The P1 alanine carbonyl oxygen of Ac-IATA-cmk points slightly away from them and forms a hydrogen bond with N154 (see panel B).
D, E Ac-IATA-cmk bound in the active site of GlpG. Shown are views from two different angles. Positions of the S1–S4 subsites are indicated. A dashed line contours the water-filled S1 subsite. Water molecules are depicted as red spheres. ISM, 7-amino-4-chloro-3-methoxy-isocoumarin; DFP, diisopropyl fluorophosphonate.
The peptide ligand is further stabilised in the active site by hydrogen bonds of its backbone with the backbone carbonyl and amido groups of residues S248/A250 of the L5 loop, and residues G198/W196 of the L3 loop (Fig 3B). Side chain and main chain atoms in each position of the ligand are also engaged in van der Waals interactions with residues of the L3 loop (P1 → G199, P3 → F197), the L5 loop (P2 → M249) and the L1 loop. The terminal P4 isoleucine of the ligand has the right orientation and distance to be considered to interact with the aromatic ring of F146 of the L1 loop via a CH–π interaction (Fig 3B), a weak hydrogen bond with a dominant dispersive character (Brandl et al, 2001; Plevin et al, 2010). These numerous interactions run along the entire length of the peptide, and, although relatively weak individually, they collectively contribute to the productive positioning of the peptide in the active site in a significant way. This may explain why N-terminal truncations of Ac-IAAA-cmk led to a dramatic progressive decrease in inhibitory potency (Fig 1C).
Since we observed weak sequence preferences also at the P5 position of the substrate (Fig 2A), we solved the GlpG complex with the pentapeptide Ac-TIATA-cmk to get insight into their structural basis. However, no additional electron density for the P5 threonine could be observed in this structure, and the overall orientation of the P1–P4 residues was the same as in Ac-IATA-cmk complex (Supplementary Fig S5). These findings indicate that substrate residues beyond P4 are unlikely to interact with GlpG significantly and are completely solvent-exposed. This is consistent with the observation that only hydrophobic amino acids are not tolerated well in the P5 position of the substrate (Fig 2A).
Substrate-binding subsites in GlpG
The structure of Ac-IATA-cmk complex with GlpG reveals substrate interactions in the active site of a rhomboid protease, allowing us to correlate them to the observed amino acid preferences in the TatA substrate from which Ac-IATA-cmk is derived (Fig 2A). GlpG shows a strict requirement for a small P1 residue, strongly preferring alanine and less well accepting cysteine and serine (Fig 2A). The side chain of the P1 alanine in Ac-IATA-cmk is bound into a well-formed S1 subsite, corresponding to the one proposed earlier (Vinothkumar et al, 2010) (Fig 3C). The S1 subsite is the proximal part of a deeper cavity, whose distal part has a strongly hydrophilic character with negative surface electrostatic potential (Supplementary Fig S6) and contains three conspicuous conserved water molecules present in all structures of GlpG from different crystallisation conditions and space groups (Wang et al, 2006; Ben-Shem et al, 2007; Vinothkumar, 2011). It was recently proposed that this region constitutes a ‘water retention site’ in GlpG that facilitates channelling of water molecules from the aqueous environment into the body of the hydrophobic protease to confer catalytic efficiency (Zhou et al, 2012; Fig 3D and E). The mechanistic implications of its proximity to the S1 subsite will be discussed later.
In contrast to the P1 position, P2 and P3 positions in TatA are relatively insensitive to residue changes (Fig 2A). Consistent with this, both S2 and S3 subsites are large and open enough to accommodate residues of any size. While the S2 subsite is half-open to the periplasm, S3 subsite resembles a mere notch in the rim of the active site of GlpG, through which the side chain of the P3 alanine of Ac-IATA-cmk points towards Q189 (Fig 3D and E). The P4 isoleucine of the bound peptide interacts with the aromatic ring of F146, possibly via a CH–π bond. This interaction defines the S4 subsite as a recessed area on the periplasmic face of GlpG, the borders and bottom of which are delineated mainly by residues of the L1 loop with some contribution from the side chain of W196 in the L3 loop. This patch is unusual because it is fully solvent-exposed, yet strongly hydrophobic in nature (Fig 4), which suggests functional importance. Indeed, the character of the S4 subsite provides a structural explanation of the preference for large and hydrophobic residues and the intolerance for polar residues in the P4 position of TatA (Fig 2A).
Figure 4. A patch of hydrophobic residues from the L1 loop forms the S4 subsite of GlpG.

A Compensatory effect of mutations in the S4 subsite of GlpG and the P4 position of TatA. Mutation of the S4 subsite residue F146 to the smaller hydrophobic residues alanine or isoleucine is nearly or completely inactivating only for substrate variants with small- to medium-sized side chains (A, S, C, T, V, I) in the P4 position. This attenuating effect of the GlpG S4 subsite mutants can be compensated by a mutation of the TatA P4 residue to a residue with a bigger side chain. Hence, while wild-type (F146) GlpG cleaves the TatA substrate with a large P4 residue (such as I5W) very poorly, the activity can be fully recovered by replacing the bulky phenylalanine 146 in the S4 subsite of the enzyme by a small hydrophobic side chain (such as F146A). The assays have been conducted three times independently, and representative data are shown (source data in Supplementary Fig S7).
B The S4 subsite in the complex structures GlpG:Ac-IATA-cmk, GlpG:Ac-FATA-cmk and GlpG_F146I:Ac-FATA-cmk. Crystallographic statistics are shown in Supplementary Table S1. Upper panels: close-up view of the S4 subsite with the surface of GlpG coloured according to the Kyte-Doolittle hydrophobicity scale (Kyte & Doolittle, 1982). The S4 subsite is a surface-exposed hydrophobic patch formed by residues from the L1 loop. Lower panels: residues making vdW interactions with the bound tetrapeptide are shown as contact surfaces. For residues W196, F197 and G198 of the L3 loop, only main chain atoms (mc) are engaged in interactions. Residues F146, M120, M144 and I146 of the L1 loop make vdW contacts with their side chains. The isoleucine in the P4 position of Ac-IATA forms a CH-π interaction with the aromatic side chain of F146.
The S4 subsite is plastically formed by residues of the L1 loop
As P4 residue crucially contributes to substrate recognition by several rhomboids (Strisovsky et al, 2009), strongly influencing mainly the kcat of the reaction (Dickey et al, 2013), we examined the functional and structural properties of S4 subsite in greater detail. The mutation of F146 to alanine was reported to inactivate GlpG without substantially affecting its thermodynamic stability (Baker & Urban, 2012), which was previously difficult to explain. Since F146 interacts with the P4 residue side chain of the substrate, we hypothesised that mutations in F146 could actually affect the P4 specificity of GlpG. To test this hypothesis, we engineered complementary enzyme and substrate mutants by introducing hydrophobic residues of different side chain volumes to position 146 of GlpG (F146A and F146I) and by testing their activity against all 20 possible mutations in the P4 position of TatA substrate. Indeed, the F146A mutant was not inactive as previously reported (Baker & Urban, 2012), but it rather showed a shift in specificity for the P4 residue. TatA variants with smaller residues in P4 position (e.g. A, C, V) were cleaved less efficiently by both the F146A and F146I mutants than by wt GlpG, while TatA variants with larger hydrophobic side chains in P4 position (such as M, F, W) were cleaved significantly better by F146A and F146I mutants than by wt GlpG (Fig 4A and Supplementary Fig S7).
To understand the properties of S4 subsite structurally, we determined the structures of wt GlpG and its F146I mutant complexed to Ac-FATA-cmk (2.9 and 2.55 Å resolution, respectively, Supplementary Table S1) and compared the ligand-binding mode to the parent structure of GlpG and Ac-IATA-cmk complex. Interestingly, the P4 residue of the ligand binds GlpG in a slightly different way in the three complexes (Fig 4B), illustrating the plasticity of S4 subsite. In wt GlpG, the isoleucine of Ac-IATA-cmk interacts with the main chain atoms of W196 of the L3 loop and the side chain of F146 (Fig 4B), while the ring of the P4 phenylalanine of Ac-FATA-cmk is accommodated additionally by the side chain of M120 contributing to the hydrophobic patch that constitutes the S4 subsite (Fig 4B). In the F146I mutant of GlpG, the P4 phenylalanine points down into a well-formed, hydrophobic pocket and engages in contacts with the main chain atoms of F197 and G198 of L3 loop and the side chains of I146 and M144 of L1 loop (Fig 4B). Our structural analyses therefore reveal a function for the L1 loop in rhomboid specificity determination: the S4 subsite is plastically formed by the side chains of three L1 loop residues, aided by the main chain atoms of L3 loop. This finding is consistent with the observations that mutations at the L1-L3 loop interface often lead to a significant decrease in GlpG activity (Baker & Urban, 2012).
Structural changes upon inhibitor binding—implications for rhomboid mechanism
The previously published inhibitor-bound complex structures of GlpG (Vinothkumar et al, 2010, 2013; Xue & Ha, 2012; Xue et al, 2012) were useful first approximations for uncovering the structural changes involved in GlpG catalysis, but the small size and chemical dissimilarity of the inhibitors to a polypeptide limited their use as models for substrate binding. The present structures of GlpG with substrate-derived peptides resemble the tetrahedral intermediate and the acylenzyme, thus allowing us to characterise more accurately structural changes during catalysis.
Alignment of the unliganded and Ac-IATA-cmk complex structures of GlpG (Fig 5A and B) reveals that only minor TMD movements occur in the complex. TMD6 is slightly turned inwards in the ligand-bound state, but this may be the consequence of the double binding of the CMK warhead to both H254 and S201 (Mac Sweeney et al, 2000). The lateral movement of TMD5, thought to be required for substrate access (Baker et al, 2007), is negligibly small in the Ac-IATA-cmk complex structure. However, since our ligands include neither the TMD of the substrate nor the prime-side residues, which would probably co-localise with the top of TMD5 in the enzyme–substrate complex, we cannot exclude the possibility of larger TMD5 movements in other phases of the catalytic cycle of rhomboid. The most dramatic secondary structure changes involve the L5 loop: it caps the active site in the apoenzyme while swinging upwards and shifting laterally upon binding of Ac-IATA-cmk (Fig 5A and B).
Figure 5. Binding of Ac-IATA-cmk to GlpG induces displacement of the L5 loop and side chain rotamer changes in TMD2 and 5.
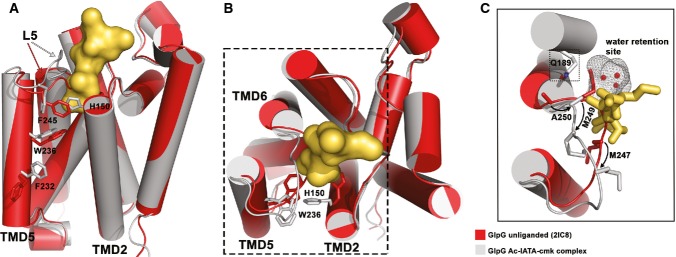
A Structural alignment of unliganded GlpG and the GlpG:Ac-IATA-cmk complex, side view. Unliganded GlpG (PDB-ID 2IC8) is coloured in red, the complex in grey/yellow. Ac-IATA-cmk is represented as contact surface. Residues with different side chain rotamers are shown for TMD2, TMD5 and the L5 loop.
B Top view. F232 and F245 are omitted for clarity.
C Displacement of the L5 loop causes a positional shift for M247, M249 and A250. Both methionines form the S2 subsite in the complex. A250 adopts the position in the complex that was occupied by M249 in the apoenzyme. The presence of the shorter side chain of alanine between Q189 and the ‘water retention site’ might play a role in facilitating the relay of water molecules for the reaction (see Discussion).
In addition to secondary structure changes, we detect several pronounced rotamer changes in residues of TMD2, TMD5 and L5 loop, which may indicate the importance of these residues for the catalytic mechanism. The movement of the L5 loop inflicts a positional change on the side chains of M247 and M249 (Fig 5C), having profound impact on S1 and S2 subsite formation and potentially also on catalysis (see Discussion). Upon binding of Ac-IATA-cmk, M249 shifts and becomes engaged in van der Waals interactions with the methyl group of threonine in the P2 position of the substrate, while the original position of M249 in the unliganded enzyme is adopted by A250 in the complex structure (Fig 5C). Methionine 247 fills the centre of the active site in the apoenzyme, while in the complex structure, it moves to the entrance of the active site, where it confines the S2 subsite together with H150. In the apoenzyme, the side chain of H150 fills the space that corresponds to the S2 cavity, swinging far out from this position upon binding of Ac-IATA-cmk. If H150 stayed in its original position, it would sterically clash with the side chain of the P2 threonine (Fig 5A and B), suggesting that the role of H150 in catalysis may be more dynamic than previously thought.
Several other conspicuous rotamer changes occur in the Ac-IATA-cmk complex. The L5 residue F245 obstructs the entrance to the active site at the level of the catalytic dyad residues in the apoenzyme, while in the complex structure, it has rotated to the side (Fig 5A). Given the position of F245 and the fact that F245A mutation results in a modest enhancement of proteolytic activity (Baker & Urban, 2012), it is suggestive that rotation of F245 may be required for substrate entry into the active site. The indole ring of W236 of TMD5 has rotated 180° in the complex when compared to the apoenzyme, thus allowing the formation of an internal cavity thought to represent the S2′ subsite (Vinothkumar et al, 2010, 2013) (Fig 5A and B). It is noteworthy that this cavity forms even in the absence of prime-side residues in our complex or in complexes with small molecular inhibitors, isocoumarins and β-lactams (Vinothkumar et al, 2010, 2013; Xue & Ha, 2012). Finally, residue F232 of TMD5 is also found in a different conformation in the complex structure than in the apoenzyme, closing the gap to TMD2 residue W157 (Fig 5A). Since the F232A mutation has been shown to result in increased enzymatic activity (Baker & Urban, 2012), it is possible that F232 directly or indirectly participates in substrate binding.
Molecular dynamics reveals active site interactions of the substrate in the Michaelis complex
Besides revealing the substrate-binding subsites on GlpG, crystal structures of the peptidyl-CMK complexes enabled us to investigate rhomboid mechanism in closer detail. We used the complex structures, molecular modelling and molecular dynamics (MD) to create a model of the Michaelis complex of rhomboid protease and the substrate spanning the P4 to P3′ subsites. The model was validated by monitoring (i) the root-mean-square deviation (RMSD) of protein and substrate backbone (Supplementary Fig S8A) and (ii) hydrogen bonds (H-bonds) at the non-prime side of the substrate during the MD run. Throughout MD simulations, H-bonds between the L3/L5 loop and the substrate backbone, as present in the crystal structure (Fig 3B), were retained (Supplementary Fig S8B). Furthermore, we observed (i) the formation of H-bonds between the catalytic dyad residues, (ii) the scissile bond carbonyl carbon and the S201 side chain oxygen coming into close spatial proximity compatible with nucleophilic attack, and (iii) formation of H-bonds between the P1 carbonyl oxygen and residues thought to form the oxyanion hole (Supplementary Fig S8B). The interactions (iii) involved mainly the H-bonds by the N154 side chain nitrogen and by the S201 main chain amide. The former H-bond was stable, while the latter one was transient, and the previously observed H-bond to L200 main chain amide (Vinothkumar et al, 2010) could not be detected. During MD simulations, H150 transiently flipped back into the position it adopts in the unliganded enzyme (data not shown), suggesting that H150 (and maybe also L200) may hydrogen-bond to the negatively charged oxyanion that forms in the tetrahedral intermediate (but is absent from the Michaelis complex). Overall, the carbonyl oxygen of the P1 residue adopts a similar orientation in our MD simulations as found in the complex structure with diisopropylfuorophosphonate (DFP), deemed to mimic the tetrahedral intermediate (Xue & Ha, 2012) (Supplementary Fig S8C). This finding makes us confident that our MD model of the Michaelis complex (Fig 6A) is realistic, allowing us to examine the interactions of the prime-side residues with GlpG and estimate the likely exit position of the unwound C-terminus of the substrate from the body of GlpG.
Figure 6. Molecular dynamics-based model of the Michaelis complex and possible interaction modes of substrate transmembrane domain.

A The molecular dynamics model for the active site bound unwound part of TatA comprising positions P4-P3′. The P3′ residue exits the active site of GlpG between W236 (TMD5) and F153 (TMD2).
B A cartoon model of the full transmembrane TatA substrate interacting with GlpG. The substrate continues by the N-terminus of its helical transmembrane domain from the point of where its P3′ residue ‘exits’ the active site of GlpG in the Michaelis complex model. This arrangement suggests three principally different orientations of TatA TMD that are shown in the illustrative cartoon in different colours. Detailed views of the boxed areas around TatA P4-P3′ segments are shown in Supplementary Fig S8D and E.
The MD model of the Michaelis complex reveals the likely interactions of the P2′ residue, which is important for substrate recognition by P. stuartii AarA and E. coli GlpG rhomboids (Strisovsky et al, 2009; Dickey et al, 2013). The major ensemble (92%) of conformations of the P2′ phenyl of TatA (Supplementary Fig S8D and E) snugly fits into the previously proposed S2′ subsite (Vinothkumar et al, 2010, 2013). The ‘back wall’ of the subsite is formed by residues of TMD4 deeply buried within the core of the enzyme (Supplementary Fig S8E). The bulk of this interaction interface is provided by Y205, assisted by V204, M208 and A233, all of which make van der Waals contacts to the P2′ residue of the substrate. Phenylalanine 245, located at the tip of L5 loop, constitutes the ‘roof’ above the S1′ and S2′ subsites, making van der Waals contacts with the P1′ and P2′ residues (Supplementary Fig S8E). Amino acids F153 and W157 of TMD2 and W236 of TMD5 form the outer rim of the active site cavity that opens to the lipid bilayer, making van der Waals contacts to the P2′ residue as well as to the glycine in P3′ position (Supplementary Fig S8E). This arrangement suggests that F153, W157 and W236 could directly interact with the substrate as opposed to having just an indirect ‘gating’ role in limiting the mobility of TMD5, as proposed earlier (Baker et al, 2007).
Our data indicate that the full extent of the enzyme–substrate interactions in the active site of GlpG comprises a stretch of seven consecutive residues of the substrate in an extended conformation, from the P4 to P3′ position (I5 to G11 in TatA) (Fig 6A). The P3′ glycine marks the end of the unwound part of the TatA substrate, suggesting that its transmembrane helical part begins just after the helix-destabilising proline in P4′. The P3′ glycine exits the active site of GlpG within or just above the plane of the Cα atoms of residues W236 and F153. It was recently reported that intramolecular disulphide cross-linking of a W236C/F153C mutant of GlpG via 1,2-ethanediyl bismethanethiosulfonate (M2M) does not impair enzyme activity (Xue & Ha, 2013), suggesting that substrate accesses the active site above these residues (above the M2M cross-link). That report is compatible with our MD simulations, since the Cα–Cα distance between W236 and F153 is 12.5 ± 0.6 Å, which matches the calculated distance of 13 Å between the Cα atoms of the M2M-cross-linked cysteine pair mutant, calculated from the respective MD model (Fig 6A).
In conclusion, our crystallographic, biochemical and molecular dynamics data reveal for the first time substrate interactions in the active site of an intramembrane protease, explain the observed substrate specificity of rhomboid proteases structurally and reveal a role in substrate binding for the hitherto enigmatic conserved element of the rhomboid fold—the L1 loop. Besides providing new insights into intramembrane protease mechanism, our work raises testable mechanistic hypotheses that, if confirmed, could facilitate development of selective rhomboid inhibitors.
Discussion
Understanding of the mechanism and specificity of intramembrane proteases would be significantly advanced by high-resolution structural characterisation of substrate binding, but it has long been an unattained goal. Rhomboids, the most structurally characterised intramembrane proteases, have so far been co-crystallised only with small molecular mechanism-based inhibitors (Vinothkumar et al, 2010, 2013; Xue & Ha, 2012; Xue et al, 2012; Vosyka et al, 2013) useful for only indirect inferences about mechanism and specificity (Vinothkumar et al, 2010, 2013). We have developed a new series of peptidic chloromethylketone inhibitors based on a natural bacterial rhomboid substrate sequence (Providencia stuartii TatA) (Stevenson et al, 2007) and solved X-ray structures of their complexes with GlpG, thus providing the first structural insight into substrate binding to rhomboids. We reveal the subsites for the P1–P4 residues that had been demonstrated to be crucial for substrate recognition and efficient catalysis (Strisovsky et al, 2009; Dickey et al, 2013). Furthermore, we show that the S4 subsite is formed by residues of the highly conserved but previously enigmatic L1 loop, leading us to propose that the domain topologically equivalent to the L1 loop may have evolved for client-protein recruitment in rhomboid-like pseudoproteases.
Rhomboid substrate binding—the unwound, the destabilising and the helical
The peptidyl-CMKs used in this study exhibit identical specificity requirements to natural substrates, validating their ability to provide mechanistic insight. We can now use our data in combination with previous structural and biochemical work to propose a plausible working model of the enzyme–substrate complex. Our work shows that the non-helical P4 to P3′ segment of the substrate is in contact with the active site cleft of GlpG. The importance of the P4, P1 and P2′ positions in the substrate (Strisovsky et al, 2009) was recently confirmed by showing that they determine the kcat of rhomboid cleavage (Dickey et al, 2013). These residues have only a negligible impact on KM (Dickey et al, 2013), suggesting that they do not make a major contribution to the overall binding energy between a full transmembrane substrate and the enzyme. This in turn implies that the overall interaction area of rhomboid–substrate complex is significantly larger than the segment containing the P4 to P2′ residues, and the majority of overall binding energy of the substrate is probably contributed by the part of its TMD directly contacting the enzyme. The mode of binding of substrate TMD is unknown, but our structures and MD models provide a solid framework to reflect on it.
To propose a structure-based conceptual model of a full transmembrane substrate complex with GlpG, we took advantage of the recent solution NMR structure of E. coli TatA (Rodriguez et al, 2013). A homology model of P. stuartii TatA that we generated shows that the region spanning residues P13 (P4′ position) to F27 is α-helical and about 22 Å long. The estimated hydrophobic thickness of GlpG molecule from the point of exit of the P3′ residue to the cytoplasmic boundary of the membrane is about 13 Å (Fig 6B), and manual docking of P. stuartii TatA TMD region P13 (P4′ position) to F27 into a representative structure of the Michaelis complex model suggests that the TatA TMD would ‘stick out’ of the membrane.
Such hydrophobic mismatch would be energetically unfavourable, and different ways of alleviating it can be envisaged, for example (i) tilting of substrate TMD in the membrane or (ii) minimising the solvent-exposed hydrophobic surface area of substrate TMD by its interaction with GlpG. In the first scenario (i), a tilted but straight TMD of the substrate (Fig 6B) would have virtually no interaction interface with the transmembrane region of GlpG (unless GlpG is also tilted in the membrane accordingly) and might therefore be less likely. However, a tilted orientation with a kinked α-helix would still allow some interaction with the transmembrane region of GlpG, making it perhaps more likely (Fig 6B). In the second scenario (ii), a slight ‘inward’ curving of the substrate transmembrane helix that would allow its alignment and interaction with TMD2 of GlpG (which is also slightly bent) might provide a larger interaction interface and shield much of the ‘mismatched’ TMD from the solvent (Fig 6B). Indeed, such a mechanism has been described in cases where positive mismatch is bigger than 4 Å (Lewis & Engelman, 1983). Interestingly, introducing transmembrane helix-destabilising residues at several positions along the TMD of an artificial rhomboid substrate increases its cleavage efficiency by GlpG (Akiyama & Maegawa, 2007; Moin & Urban, 2012), but this effect has been difficult to explain (Ha, 2009). Now, our conceptual models of the complex where substrate TMD is kinked or bent (Fig 6B) would both be consistent with and explain these observations.
Structural changes in rhomboid accompanying substrate binding
Crystal structures of model intramembrane proteases suggest that substrate access to their catalytic residues may be conformationally regulated (Strisovsky, 2013). Based on the alternative conformation of one molecule in the asymmetric unit of a crystal structure of GlpG (Wu et al, 2006), substrate access to rhomboid protease had been suggested to be governed by a ‘gating’ mechanism. In analogy to the translocon (Van den Berg et al, 2004), this mechanism should involve a large dislocation of TMD5 to make the core of the enzyme accessible laterally from the lipid bilayer (Wu et al, 2006; Baker et al, 2007). Mutations in residue pairs W236A/F153A and F232A/W157A, designed to weaken the contacts between TMD2 and 5, increased enzymatic activity, supposedly by opening the TMD5 gate (Baker et al, 2007), which was further supported by enzymatic and thermodynamic studies (Baker & Urban, 2012; Moin & Urban, 2012). In contrast, other authors showed that preventing large lateral movement of TMD5 by chemically cross-linking TMDs 2 ad 5 in a W236C/F153C mutant does not abrogate the activity of GlpG. This suggests that a ‘gating’ movement of TMD5 may not actually be required for substrate binding, and it leaves the mechanism of substrate access to rhomboid controversial.
Our structures of the peptidyl-CMK complexes show that the L5 loop has to be displaced significantly to allow binding of substrate to the active site, but we do not observe any significant movement of the adjoining TMD5. Since our peptide ligands comprise only the non-prime-side residues and capture the reaction at the stage of the tetrahedral or acylenzyme intermediate, we explored rhomboid–substrate interactions at the prime side and possible involvement of TMD5 by molecular modelling and dynamics. The results show that a large lateral movement of TMD5 is not required for the formation of the acylenzyme nor the Michaelis complex with the P4 to P3′ segment of the substrate. Our data are thus compatible with the published cross-linking data suggesting that major movements of TMD5 are not required for substrate access (Xue & Ha, 2013). We cannot formally exclude the possibility of a large TMD5 movement in the earlier phases of a transmembrane substrate binding. However, the positions of residues W236 and F153, which we observe in the Michaelis complex model (Fig 6A and Supplementary Fig S8E), suggest that they may directly interact with the substrate, rather than just acting as ‘openers’ of the TMD5 gate. These results collectively imply that the lateral gate opening analogy with the translocon (Wu et al, 2006; Baker et al, 2007) may not be entirely correct and that substrate access mechanism to rhomboid merits further investigation.
Several other conspicuous movements of side chains accompany ligand binding, among which H150 is worth highlighting. Histidine 150 flips out completely from its position in the unliganded enzyme to make space for the P2 residue of the ligand, which can be almost any amino acid type (Fig 2A). In this conformation, however, the side chain of H150 cannot make a hydrogen bond to the carbonyl oxygen of the substrate. This dislocation of H150 could well be partly due to the chloromethylketone warhead binding to the catalytic dyad and slightly distorting the carbonyl oxygen (Fig 3C; Mac Sweeney et al, 2000). Indeed, our MD simulations of the Michaelis complex suggest that the side chain of H150 can occasionally flip to its original position (M. Lepšík, S. Zoll, K. Strisovsky, unpublished observations), although this may be less likely in substrates with larger P2 residues. Interestingly, the side chain of H150 occupies a similar position in the crystal structure of GlpG complex with 2-phenylethyl 2-(4-azanyl-2-methanoyl-phenyl) ethanoate (Vosyka et al, 2013) as it does in our Ac-IATA-cmk complex, but it is covalently bound to the inhibitor. In summary, these observations collectively indicate that the role of H150 in catalysis may be more dynamic than previously thought and may extend beyond oxyanion hole formation.
Water access to the catalytic site—a key open question
To better understand intramembrane proteolysis, one of the key aspects to consider is the mechanism of water supply to the catalytic site immersed in the hydrophobic environment of the lipid bilayer. It was recently proposed, based on molecular dynamics and mutagenesis data, that GlpG employs a specific mechanism to channel water molecules from bulk solution to an internal ‘water retention site’ near the catalytic dyad (Zhou et al, 2012). Our structural data are consistent with this concept and offer a plausible mechanistic interpretation based on several observations. First, the ‘water retention site’ forms a continuous cavity with the S1 subsite of GlpG. Although the whole cavity is quite large, only alanine and to lower extent also cysteine or serine are accepted in the P1 position of the substrate. One explanation could be that the strongly negative electrostatic potential of this cavity (Supplementary Fig S6) disfavours binding of negatively charged residues and residues with longer aliphatic side chains than that of alanine. Polar natural amino acids other than serine are likely to be either too large to be accommodated (K, R, H) or might engage in hydrogen bonds to the water molecules inside the retention site, thus perturbing the described dynamic hydrogen bonding network (Zhou et al, 2012). Such interference could result in (i) structural destabilisation of the enzyme–substrate complex or (ii) impaired catalysis as water molecules may not effectively access the catalytic site to be used in the deacylation step. The latter mechanism is experimentally testable, since one would predict that a substrate with a P1 residue of a suitable character larger than an alanine could be trapped at the acyl-enzyme stage, bound to the catalytic serine. However, given the structural restrains of the cavity and the structural properties of genetically encoded amino acids, testing this hypothesis might require the use of unnatural amino acids. Our structural analyses also rationalise why glycine is poorly tolerated in the P1 position of a substrate and the corresponding peptidyl-CMK. The poor tolerance cannot be due to steric hindrance because glycine has no side chain, but it can be caused by a higher degree of rotational freedom endowed by glycine, which could prevent optimal alignment of the ligand’s polypeptide chain for hydrogen bonding to the L3 loop backbone in a parallel β-strand and productive exposure of the scissile bond to the catalytic residues.
A second observation relates to glutamine 189 that had been proposed to channel water molecules to the water retention site (besides S185, H141 and S181). The side chain of the P3 residue of the substrate/inhibitor points directly at Q189 (Fig 3D). We can thus speculate that substitution of the P3 alanine in Ac-IATA-cmk by a residue that can either sterically interfere with Q189 (e.g. W in Fig 2A) or form direct or water-mediated hydrogen bonds with Q189 (e.g. D, E, N in Fig 2A) could result in a loss of proteolytic activity due to the interference with water channelling into the retention site. Third, residue M249 from the L5 loop protrudes right in between Q189 and water molecules in the water retention site, again potentially interfering with water channelling to the water retention site. Upon ligand binding, the L5 loop is displaced, and the position of M249 side chain is adopted by the side chain of A250, which may ‘unblock’ the pathway from Q189 to the water retention site (Fig 5C). Although necessarily speculative, the mechanism of water access control supported by the above observations deserves further investigation, also because if proven correct it could represent a unique rhomboid-specific mechanism exploitable in the design of selective rhomboid inhibitors.
L1 loop—a prominent feature of the rhomboid fold—binds substrate
We find that the S4 subsite of GlpG is, unexpectedly, formed by a patch of hydrophobic but solvent-exposed residues from the L1 loop. This interaction surface is plastic, and substitution of the P4 residue requires adjustment of residues in the S4 subsite to maximise the number of van der Waals contacts and preserve catalytic efficiency (Fig 4). Notably, the only other structurally characterised rhomboid, GlpG from Haemophillus influenzae (Lemieux et al, 2007), contains a similar solvent-exposed hydrophobic patch formed mainly by L61 (EcGlpG equivalent F146), V59 (EcGlpG eq. M144) and M35 (EcGlpG eq. M120) (Supplementary Fig S9A), allowing for substrate interactions comparable to the ones observed in the S4 subsite of EcGlpG (Supplementary Fig S9B). In fact, most GlpG homologues harbour hydrophobic residues at the positions corresponding to F146, M144 and M120 of EcGlpG (Supplementary Fig S9C), suggesting that this specificity feature is more widely conserved.
Given how large and diverse the rhomboid protease family is [less than 15% of sequence identity in the conserved region (Koonin et al, 2003)], it is expected that substrate specificity and S4 subsite preferences may differ among phylogenetic clusters of rhomboids. Nevertheless, some key features of rhomboid architecture are likely to be used for a similar purpose even in distant homologues. It has been recently suggested that rhomboids are dimeric (Sampathkumar et al, 2012), and that natural substrates induce dimer-dependent allosteric activation of the enzyme (Arutyunova et al, 2014). The molecular details of the dimerisation interface and the basis for the allosteric regulation are unknown (Strisovsky & Freeman, 2014), but it is attractive to speculate that either of them may involve the L1 loop. Notably, this region of rhomboid architecture, topologically corresponding to the L1 loop, is present in Derlins, and has expanded in size and been conserved in iRhoms (called iRhom homology domain) (Lemberg & Freeman, 2007). Taking the implications of our work evolutionarily further, we speculate that the L1 loop region may have evolved for the interaction with client proteins also in iRhoms and other proteins of the rhomboid-like superfamily (Freeman, 2014).
Materials and Methods
Chemical synthesis
Peptidyl-chloromethylketone inhibitors were prepared by coupling of the protected N-α-acetyl-peptide fragment and the corresponding chloromethylketone derived from the C-terminal (P1) amino acid synthesised analogously with previously described methods (Thomson & Denniss, 1973; Owen & Voorheis, 1976; Jahreis et al, 1984; Hauske et al, 2009). Acidolabile tert-butyl type groups were used for protection of side chain functionalities. The resulting peptidyl-chlomethylketones were then deprotected by trifluoroacetic acid and purified by reversed-phase HPLC. Identity of all compounds was confirmed by mass spectrometry on Waters Micromass ZQ ESCi multimode ionisation mass-spectrometer, using ESI-ionisation method (ESI-MS) and NMR (Bruker AV-400 MHz, data collected at room temperature). Stability of the compounds in aqueous buffers was analysed by reversed-phase HPLC with UV and ESI-MS detection (Supplementary Fig S1), and their solubility was checked using Millipore low-binding hydrophilic centrifugal filters and HPLC with UV detection. Full experimental details on chemical synthesis and analytical characterisation of all synthesised compounds are included in Supplementary Information.
Protein expression and purification
Recombinant GlpG core domain for crystallography was expressed, solubilised in n-decyl-β-D-maltoside (DM, Anatrace) and purified essentially as described (Wang et al, 2006; Vinothkumar et al, 2010) with minor modifications detailed in the Supplementary Information. For purification of full-length GlpG used in inhibition assays, n-dodecyl-β-D-maltoside (DDM, Anatrace) was used instead of DM. Imidazole from the Ni-NTA elution buffer was removed by dialysis into the rhomboid reaction buffer (50 mM Tris (pH 7.4), 100 mM NaCl, 25 mM EDTA, 10% (v/v) glycerol and 0.05% (w/v) DDM). Purification of GlpG mutants (S201A, H254A, F146I and F146A) was performed in the same way. The recombinant chimeric substrate based on TatA TMD was expressed in glpG knock-out E. coli and purified by Ni-NTA and amylose affinity chromatography as described (Strisovsky et al, 2009).
Rhomboid activity assays
To analyse sequence preferences of GlpG, the panel of P. stuartii TatA mutants in positions 4–8 (Strisovsky et al, 2009) was PCR-amplified and in vitro-transcribed and translated in the presence of radioactive [35S]-L-Met as described (Strisovsky et al, 2009) with minor modifications detailed in the Supplementary Information. All mutant TatA variants were used at equimolar concentrations as judged by autoradiography. The substrates were exposed to purified recombinant full-length GlpG (20 ng/μl) in 16-μl reactions in a buffer containing 50 mM HEPES pH 7.4, 0.5 M NaCl, 10% (v/v) glycerol, 5 mM EDTA and 0.05% (w/v) DDM. After 40 min incubation at 37°C, the reactions were stopped by transfer on ice and addition of SDS-PAGE sample buffer. Reaction products were separated on 12% BisTris-MES SDS-PAGE (NuPAGE, Invitrogen), and substrate conversion was analysed by radiography and densitometry as described (Strisovsky et al, 2009) using ImageQuant 8.0 software (GE Healthcare).
For evaluating GlpG activity in vivo, recombinant chimeric MBP-TatAtmd-Trx substrates (Strisovsky et al, 2009) were expressed in the wild-type E. coli MC4100 encoding endogenous GlpG and in its glpG::tet mutant derivative at 37°C under conditions specified in the Supplementary Information, and 3 h after induction, substrate cleavage was analysed by Western blotting.
Inhibition assays
For inhibition assays, the purified MBP-TatAtmd-Trx fusion protein encompassing amino acids 1–50 of P. stuartii TatA (Strisovsky et al, 2009) was used as substrate. Purified full-length GlpG (5.4 μM) was preincubated with peptidyl-chloromethylketone inhibitors at different concentrations (50–700 μM) for 3 h at 37°C in reaction buffer containing 50 mM Tris (pH 7.4), 100 mM NaCl, 25 mM EDTA, 10 % (v/v) glycerol and 0.05 % (v/v) DDM. The cleavage reaction was started by adding substrate in fivefold molar excess over the enzyme, and let proceed for 30 min at 37°C, after which it was stopped by the addition of SDS-PAGE sample buffer and transfer on ice. Reaction products were resolved by 4–20% Tris-Glycine SDS-PAGE (Bio-Rad) and Coomassie stained (Instant Blue, Expedeon, UK). Substrate conversion was quantified densitometrically from the scanned stained gels using the ImageQuant 8.0 software (GE Healthcare).
Crystallisation and structure solution
For co-crystallisation, N-terminally truncated GlpG core domain was complexed with chloromethylketone inhibitors overnight. Excess inhibitor was then removed using desalting columns packed with Sephadex G-25 (PD-10, GE Healthcare), and the completion of complex formation was confirmed by MALDI-MS. The complex was concentrated to 6 mg/ml, mixed with crystallisation buffer in a 1:1 ratio and crystallised by the sitting drop method at 20°C. Crystal diffraction was measured at 100 K using synchrotron radiation at BESSY (Berlin, Germany) and ESRF (Grenoble, France), and structures were solved using molecular replacement. For detailed crystallisation, freezing and measurement conditions and for details on structure solution and refinement, see Supplementary Information. Figures were generated with PyMol (Schrodinger, 2012).
Methods for plasmids and mutagenesis and modelling of the Michaelis complex are fully described in Supplementary Information.
Accession codes
The coordinates of the X-ray structures presented in this paper have been deposited with the Protein Data Bank under identifiers 4QO2, 4QO0 and 4QNZ.
Acknowledgments
We thank Jana Horáková and Martin Hubálek for mass spectrometry, Radko Souček for amino acid analysis and LC-MS, Zdeněk Voburka for N-terminal sequencing, Tobias Kloepper for help with rhomboid phylogeny and sequence alignment, Kateřina Švehlová for technical assistance, Vinothkumar Kutti Ragunath and Matthew Freeman for comments on the manuscript, and the beamline staff at the BESSY in Berlin and the ESRF in Grenoble for beamtime and support. Research in KS’s lab is supported by Czech Science Foundation (project no. P305/11/1886), Ministry of Education, Youth and Sports of the Czech Republic (projects no. LK11206 and LO1302), EMBO Installation Grant (project no. 2329), Marie Curie Career Integration Grant (project no. 304154) and the National Subvention for Development of Research Organisations (RVO: 61388963) to the Institute of Organic Chemistry and Biochemistry (IOCB). SZ was supported by a post-doctoral fellowship from IOCB, JS was supported by a PhD grant project GA UK no. 232313 from Charles University in Prague, and ML by a Czech Science Foundation grant (project no. P208/12/G016).
Author contributions
SZ designed, conducted and evaluated all X-ray crystallographic experiments and inhibition assays and co-wrote the paper. SS and PM designed and carried out all chemical syntheses. JS and JB characterised the specificity of GlpG and its mutants, LP contributed key reagents, and ML performed all molecular dynamics simulations. KS conceived and led the project, designed experiments and evaluated the data, and KS and SZ wrote the manuscript with input from all co-authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Adrain C, Zettl M, Christova Y, Taylor N, Freeman M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science. 2012;335:225–228. doi: 10.1126/science.1214400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama Y, Maegawa S. Sequence features of substrates required for cleavage by GlpG, an Escherichia coli rhomboid protease. Mol Microbiol. 2007;64:1028–1037. doi: 10.1111/j.1365-2958.2007.05715.x. [DOI] [PubMed] [Google Scholar]
- Arutyunova E, Panwar P, Skiba PM, Gale N, Mak MW, Lemieux MJ. Allosteric regulation of rhomboid intramembrane proteolysis. EMBO J. 2014;33:1869–1881. doi: 10.15252/embj.201488149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RP, Young K, Feng L, Shi Y, Urban S. Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc Natl Acad Sci USA. 2007;104:8257–8262. doi: 10.1073/pnas.0700814104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RP, Urban S. Architectural and thermodynamic principles underlying intramembrane protease function. Nat Chem Biol. 2012;8:759–768. doi: 10.1038/nchembio.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shem A, Fass D, Bibi E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc Natl Acad Sci USA. 2007;104:462–466. doi: 10.1073/pnas.0609773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondar AN, del Val C, White SH. Rhomboid protease dynamics and lipid interactions. Structure. 2009;17:395–405. doi: 10.1016/j.str.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl M, Weiss MS, Jabs A, Suhnel J, Hilgenfeld R. C-H..pi-interactions in proteins. J Mol Biol. 2001;307:357–377. doi: 10.1006/jmbi.2000.4473. [DOI] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- Dickey SW, Baker RP, Cho S, Urban S. Proteolysis inside the membrane is a rate-governed reaction not driven by substrate affinity. Cell. 2013;155:1270–1281. doi: 10.1016/j.cell.2013.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Yan H, Wu Z, Yan N, Wang Z, Jeffrey PD, Shi Y. Structure of a Site-2 Protease Family Intramembrane Metalloprotease. Science. 2007;318:1608–1612. doi: 10.1126/science.1150755. [DOI] [PubMed] [Google Scholar]
- Freeman M. The rhomboid-like superfamily: molecular mechanisms and biological roles. Annu Rev Cell Dev Biol. 2014 doi: 10.1146/annurev-cellbio-100913-012944. doi: 10.1146/annurev-cellbio-100913-012944. [DOI] [PubMed] [Google Scholar]
- Greenblatt EJ, Olzmann JA, Kopito RR. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant alpha-1 antitrypsin from the endoplasmic reticulum. Nat Struct Mol Biol. 2011;18:1147–1152. doi: 10.1038/nsmb.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha Y. Structure and mechanism of intramembrane protease. Semin Cell Dev Biol. 2009;20:240–250. doi: 10.1016/j.semcdb.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauske P, Meltzer M, Ottmann C, Krojer T, Clausen T, Ehrmann M, Kaiser M. Selectivity profiling of DegP substrates and inhibitors. Bioorg Med Chem. 2009;17:2920–2924. doi: 10.1016/j.bmc.2009.01.073. [DOI] [PubMed] [Google Scholar]
- Jahreis G, Smalla K, Fittkau S. Thermitase – eine thermostabile Serinprotease. II. Synthese von substratanalogen Peptidchlormethylketonen als potentielle irreversible Enzyminhibitoren. J Prakt Chem. 1984;326:41–47. [Google Scholar]
- Koonin EV, Makarova KS, Rogozin IB, Davidovic L, Letellier MC, Pellegrini L. The rhomboids: a nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol. 2003;4:R19. doi: 10.1186/gb-2003-4-3-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lemberg MK, Freeman M. Functional and evolutionary implications of enhanced genomic analysis of rhomboid intramembrane proteases. Genome Res. 2007;17:1634–1646. doi: 10.1101/gr.6425307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg MK. Intramembrane proteolysis in regulated protein trafficking. Traffic. 2011;12:1109–1118. doi: 10.1111/j.1600-0854.2011.01219.x. [DOI] [PubMed] [Google Scholar]
- Lemberg MK. Sampling the membrane: function of rhomboid-family proteins. Trends Cell Biol. 2013;23:210–217. doi: 10.1016/j.tcb.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Lemieux MJ, Fischer SJ, Cherney MM, Bateman KS, James MN. The crystal structure of the rhomboid peptidase from Haemophilus influenzae provides insight into intramembrane proteolysis. Proc Natl Acad Sci USA. 2007;104:750–754. doi: 10.1073/pnas.0609981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BA, Engelman DM. Bacteriorhodopsin remains dispersed in fluid phospholipid bilayers over a wide range of bilayer thicknesses. J Mol Biol. 1983;166:203–210. doi: 10.1016/s0022-2836(83)80006-0. [DOI] [PubMed] [Google Scholar]
- Li X, Dang S, Yan C, Gong X, Wang J, Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493:56–61. doi: 10.1038/nature11801. [DOI] [PubMed] [Google Scholar]
- Liu J, Liu S, Xia M, Xu S, Wang C, Bao Y, Jiang M, Wu Y, Xu T, Cao X. Rhomboid domain-containing protein 3 is a negative regulator of TLR3-triggered natural killer cell activation. Proc Natl Acad Sci USA. 2013;110:7814–7819. doi: 10.1073/pnas.1220466110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Sweeney A, Birrane G, Walsh MA, O’Connell T, Malthouse JP, Higgins TM. Crystal structure of delta-chymotrypsin bound to a peptidyl chloromethyl ketone inhibitor. Acta Crystallogr D Biol Crystallogr. 2000;56:280–286. doi: 10.1107/s0907444999016583. [DOI] [PubMed] [Google Scholar]
- Malthouse JP. 13C- and 1H-NMR studies of oxyanion and tetrahedral intermediate stabilization by the serine proteinases: optimizing inhibitor warhead specificity and potency by studying the inhibition of the serine proteinases by peptide-derived chloromethane and glyoxal inhibitors. Biochem Soc Trans. 2007;35:566–570. doi: 10.1042/BST0350566. [DOI] [PubMed] [Google Scholar]
- Manolaridis I, Kulkarni K, Dodd RB, Ogasawara S, Zhang Z, Bineva G, O’Reilly N, Hanrahan SJ, Thompson AJ, Cronin N, Iwata S, Barford D. Mechanism of farnesylated CAAX protein processing by the intramembrane protease Rce1. Nature. 2013;504:301–305. doi: 10.1038/nature12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moin SM, Urban S. Membrane immersion allows rhomboid proteases to achieve specificity by reading transmembrane segment dynamics. ELife. 2012;1:e00173. doi: 10.7554/eLife.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MJ, Voorheis HP. Active-site-directed inhibition of the plasma-membrane carrier transporting short-chain, neutral amino acids into trypanosoma brucei. Eur J Biochem. 1976;62:619–624. doi: 10.1111/j.1432-1033.1976.tb10197.x. [DOI] [PubMed] [Google Scholar]
- Pierrat OA, Strisovsky K, Christova Y, Large J, Ansell K, Bouloc N, Smiljanic E, Freeman M. Monocyclic beta-lactams are selective, mechanism-based inhibitors of rhomboid intramembrane proteases. ACS Chem Biol. 2011;6:325–335. doi: 10.1021/cb100314y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plevin MJ, Bryce DL, Boisbouvier J. Direct detection of CH/pi interactions in proteins. Nat Chem. 2010;2:466–471. doi: 10.1038/nchem.650. [DOI] [PubMed] [Google Scholar]
- Rodriguez F, Rouse SL, Tait CE, Harmer J, De Riso A, Timmel CR, Sansom MS, Berks BC, Schnell JR. Structural model for the protein-translocating element of the twin-arginine transport system. Proc Natl Acad Sci USA. 2013;110:E1092–E1101. doi: 10.1073/pnas.1219486110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampathkumar P, Mak MW, Fischer-Witholt SJ, Guigard E, Kay CM, Lemieux MJ. Oligomeric state study of prokaryotic rhomboid proteases. Biochim Biophys Acta. 2012;1818:3090–3097. doi: 10.1016/j.bbamem.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Schrodinger LLC. 2012. The PyMOL Molecular Graphics System, Version 1.5.0.4.
- Stevenson LG, Strisovsky K, Clemmer KM, Bhatt S, Freeman M, Rather PN. Rhomboid protease AarA mediates quorum-sensing in Providencia stuartii by activating TatA of the twin-arginine translocase. Proc Natl Acad Sci USA. 2007;104:1003–1008. doi: 10.1073/pnas.0608140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strisovsky K, Sharpe HJ, Freeman M. Sequence-specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol Cell. 2009;36:1048–1059. doi: 10.1016/j.molcel.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strisovsky K. Structural and mechanistic principles of intramembrane proteolysis–lessons from rhomboids. FEBS J. 2013;280:1579–1603. doi: 10.1111/febs.12199. [DOI] [PubMed] [Google Scholar]
- Strisovsky K, Freeman M. Sharpening rhomboid specificity by dimerisation and allostery. EMBO J. 2014;33:1847–1848. doi: 10.15252/embj.201489373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson A, Denniss IS. The reaction of active-site inhibitors with elastase using a new assay substrate. Eur J Biochem. 1973;38:1–5. doi: 10.1111/j.1432-1033.1973.tb03024.x. [DOI] [PubMed] [Google Scholar]
- Urban S, Lee JR, Freeman M. Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell. 2001;107:173–182. doi: 10.1016/s0092-8674(01)00525-6. [DOI] [PubMed] [Google Scholar]
- Urban S, Wolfe MS. Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity. Proc Natl Acad Sci USA. 2005;102:1883–1888. doi: 10.1073/pnas.0408306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berg B, Clemons WM, Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- Vinothkumar KR, Strisovsky K, Andreeva A, Christova Y, Verhelst S, Freeman M. The structural basis for catalysis and substrate specificity of a rhomboid protease. EMBO J. 2010;29:3797–3809. doi: 10.1038/emboj.2010.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar KR. Structure of rhomboid protease in a lipid environment. J Mol Biol. 2011;407:232–247. doi: 10.1016/j.jmb.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar KR, Pierrat OA, Large JM, Freeman M. Structure of rhomboid protease in complex with beta-lactam inhibitors defines the S2′ cavity. Structure. 2013;21:1051–1058. doi: 10.1016/j.str.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosyka O, Vinothkumar KR, Wolf EV, Brouwer AJ, Liskamp RM, Verhelst SH. Activity-based probes for rhomboid proteases discovered in a mass spectrometry-based assay. Proc Natl Acad Sci USA. 2013;110:2472–2477. doi: 10.1073/pnas.1215076110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- Wang Y, Maegawa S, Akiyama Y, Ha Y. The role of L1 loop in the mechanism of rhomboid intramembrane protease GlpG. J Mol Biol. 2007;374:1104–1113. doi: 10.1016/j.jmb.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Yan N, Feng L, Oberstein A, Yan H, Baker RP, Gu L, Jeffrey PD, Urban S, Shi Y. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat Struct Mol Biol. 2006;13:1084–1091. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- Xue Y, Chowdhury S, Liu X, Akiyama Y, Ellman J, Ha Y. Conformational change in rhomboid protease GlpG induced by inhibitor binding to its S’ subsites. Biochemistry. 2012;51:3723–3731. doi: 10.1021/bi300368b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ha Y. Catalytic mechanism of rhomboid protease GlpG probed by 3,4-dichloroisocoumarin and diisopropyl fluorophosphonate. J Biol Chem. 2012;287:3099–3107. doi: 10.1074/jbc.M111.310482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ha Y. Large lateral movement of transmembrane helix S5 is not required for substrate access to the active site of rhomboid intramembrane protease. J Biol Chem. 2013;288:16645–16654. doi: 10.1074/jbc.M112.438127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zettl M, Adrain C, Strisovsky K, Lastun V, Freeman M. Rhomboid family pseudoproteases use the ER quality control machinery to regulate intercellular signaling. Cell. 2011;145:79–91. doi: 10.1016/j.cell.2011.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Moin SM, Urban S, Zhang Y. An internal water-retention site in the rhomboid intramembrane protease GlpG ensures catalytic efficiency. Structure. 2012;20:1255–1263. doi: 10.1016/j.str.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.