

Abstract
The Mre11–Rad50–Nbs1 (MRN) complex plays important roles in sensing DNA damage, as well as in resecting and tethering DNA ends, and thus participates in double-strand break repair. An earlier structure of Mre11 bound to a short duplex DNA molecule suggested that each Mre11 in a dimer recognizes one DNA duplex to bridge two DNA ends at a short distance. Here, we provide an alternative DNA recognition model based on the structures of Methanococcus jannaschii Mre11 (MjMre11) bound to longer DNA molecules, which may more accurately reflect a broken chromosome. An extended stretch of B-form DNA asymmetrically runs across the whole dimer, with each end of this DNA molecule being recognized by an individual Mre11 monomer. DNA binding induces rigid-body rotation of the Mre11 dimer, which could facilitate melting of the DNA end and its juxtaposition to an active site of Mre11. The identified Mre11 interface binding DNA duplex ends is structurally conserved and shown to functionally contribute to efficient resection, non-homologous end joining, and tolerance to DNA-damaging agents when other resection enzymes are absent. Together, the structural, biochemical, and genetic findings presented here offer new insights into how Mre11 recognizes damaged DNA and facilitates DNA repair.
Keywords: conserved basic region, crystal structure, DNA end recognition, DSB repair, Mre11-DNA
Introduction
DNA double-strand breaks (DSBs) are primarily repaired by homology-directed repair (HDR) or non-homologous and alternative end-joining mechanisms. The Mre11–Rad50–Nbs1 (Xrs2 in Saccharomyces cerevisiae) (MRN/X) complex repairs DSBs by sensing, resecting, and tethering damaged sites (Paull, 2010; Stracker & Petrini, 2011). In addition, the MRN complex propagates damage signals via the Ataxia telangiectasia-mutated (ATM) kinase to initiate DNA damage response. The MR complex is conserved in prokaryotes, archaea, and eukaryotes, whereas Nbs1/Xrs2 is confined to eukaryotes.
Mre11 consists of nuclease and capping domains (Hopfner et al, 2001; Park et al, 2011; Schiller et al, 2012). In vitro, Mre11 exhibits 3′–5′ exonuclease activity on blunt and 3′ recessed DNA as well as endonuclease activity on single-strand hairpin loops in the presence of Mn2+ or Mg2+ ion (Paull & Gellert, 1998; Trujillo & Sung, 2001; Williams et al, 2008). Rad50 is a structural maintenance of chromosome (SMC) family member that binds and hydrolyzes ATP to regulate the nuclease activities of Mre11 (Alani et al, 1990; Paull & Gellert, 1999; Hopfner et al, 2000; Moncalian et al, 2004; Chen et al, 2005; Williams et al, 2011). DSB recognition and end resection by Mre11 is crucial as inactivation of Mre11 endonuclease activities leads to early embryonic lethality in mice and acute clastogen sensitivity in fission yeast (Arthur et al, 2004; Buis et al, 2008; Williams et al, 2008). Hypomorphic mutation of Mre11 also leads to the cancer-causing ATLD (ataxia telangiectasia-like disorder), resulting in cell cycle checkpoint defects, genome instability, and ionizing radiation (IR) hypersensitivity (Stewart et al, 1999; Giannini et al, 2002).
The structure of the MR complex can be divided into three parts: the globular head domain, the zinc-hook domain, and the extended coiled-coil domain that separates the head and hook domains (Hopfner et al, 2001; de Jager et al, 2001; Moreno-Herrero et al, 2005). The head domain formed by the Mre11 dimer and the two nucleotide-binding domains (NBD) of Rad50 plays a central role in damage recognition and resection. ATP-dependent conformational changes in the MR complex regulates the decision between DNA tethering, ATM signaling, and end resection and are believed to control repair pathway choices (Deshpande et al, 2014). In the presence of ATP, the two NBDs of the Rad50 subunits are engaged on top of the nuclease domain of Mre11 and sandwich the two ATP molecules. As a result, the head region of the MR complex forms a closed structure in which the Mre11 active site is blocked by Rad50; DNA is believed to occupy the central groove on top of the Rad50 dimer (Lim et al, 2011; Möckel et al, 2012). Upon ATP hydrolysis, the Rad50 dimer is disengaged to expose the active site of Mre11, which can then perform binding and resection of DNA ends (Lammens et al, 2011; Lim et al, 2011). In addition to end resection, Mre11 alone can contribute to DNA end joining in the presence of ligase in vitro, although the efficiency is lower than that of the ATP-bound MR complex (Paull & Gellert, 2000; Deshpande et al, 2014).
The MR complex recognizes DNA via its head domain, and both Mre11 and Rad50 independently bind to DNA (Hopfner et al, 2000; Paull & Gellert, 2000; de Jager et al, 2001; Moreno-Herrero et al, 2005). Although it is not clear how Rad50 recognizes DNA damage, two structures of the Pyrococcus furiosus (Pf) Mre11–DNA complex have provided insight on how Mre11 binds to DNA lesions. In one structure (referred to as Mre11–synaptic DNA), an Mre11 dimer interacts with two short [7 base pairs (bp) with 1-base overhangs on each end] double-stranded (ds) DNA molecules, with individual Mre11 molecule binding each DNA end, explaining how Mre11 bridges two DNA ends at a short distance (Williams et al, 2008). In this structure, the Mre11 nuclease domain is involved in DNA recognition. In the other structure (Mre11–branched DNA), a branched DNA with a hairpin (8 bp with a 3-base overhang) is recognized by an Mre11 dimer, reflecting the role of Mre11 in repair of collapsed replication forks via its endonuclease activity: A short DNA duplex binds the nuclease domain of one Mre11, and the ssDNA overhang interacts with the capping domain of another Mre11 (Williams et al, 2008).
However, damaged DNA is typically longer than the DNA used in crystallization, and therefore, the proposed model for DNA alignment and joining by Mre11 requires further investigation. For instance, a short DNA could be extended toward another Mre11, which would allow an Mre11 dimer to interact with only one elongated DNA. Also, if an Mre11 dimer was to bind entirely to a duplex region within a longer DNA, it could not interact with an ssDNA overhang. Additionally, previous PfMre11–DNA structures showed that the bound DNA is too distantly located from the active site: The closest end is about 10 Å away from the metal binding site of PfMre11 (Williams et al, 2008). To explain the cleavage mechanism, it has been proposed that the movement of the capping domain contributes to direct the DNA ends to the active site. To date, however, no experiment has been conducted to test this idea.
Re-examination of the Mre11 structure using a longer DNA will help us to understand more accurately the three dimensional arrangements of Mre11 and DNA in a physiological setting. Here, we report crystal structures of Methanococcus jannaschii Mre11 (MjMre11) and DNA molecules 17 or 22 bp long with 4–5 base overhangs (Fig 1A). The MjMre11–DNA structures reveal that the whole Mre11 dimer binds a B-form DNA with each Mre11 monomer recognizing the corresponding end of DNA as well as the middle region. The DNA-binding mode of MjMre11 is substantially different from those previously observed for PfMre11. Both structural and biochemical features suggested that the dynamics of the Mre11 quaternary structure are important for DNA sensing and cleavage. We also identified a novel DNA-binding interface that is important for repair of DSBs in budding yeasts. These findings provide new insights regarding how the Mre11 complex recognizes, resects, and tethers DNA ends during repair of DNA DSBs.
Figure 1. Schematic diagram of the overall structure of the DNA–MjMre11 complex.

A DNA substrates used in the co-crystallization with MjMre11. The disordered region is shown in red. Top and bottom strands are marked as “non-template strand” and “template strand”, respectively.
B Schematic ribbon diagram of the asymmetric unit of the MjMre11–DNA1 complex. The overall structure of the asymmetric unit of the MjMre11–DNA2 complex is virtually identical. The DNA-binding Mre11 A and B are shown in cyan and pink, respectively. The Mre11 C/D dimer and E/F dimer are shown in green and gold, respectively. For a close-up view, see Supplementary Fig S2.
C Overall structure of the MjMre11–DNA1 complex. Mre11 A and B are shown in cyan and pink, respectively. DNA is shown in yellow (template strand) and orange (non-template strand). Mg2+ ions are shown as two red spheres. Two recognition loops (RL) that wedge into the central minor groove are shown in dark blue at the center. Two RLs that bind the duplex ends are shown in deep purple. See Supplementary Fig S3A for comparison with the MjMre11–DNA2 interaction.
D An orthogonal view of (C).
E Overall structure of the PfMre11-synaptic DNA complex (PDB: 3DSC). The figure is shown in the same orientation as that of (C) by superimposing Mre11 A on MjMre11.
F Overall structure of the PfMre11-branched DNA complex (PDB: 3DSD).
Results
Overall structure of the DNA-bound MjMre11 complex
To obtain Mre11–DNA crystals, we used MjMre11 core (residues 1–313) and two different DNA substrates: a 22-bp dsDNA with one blunt end and 4-base 3′ and 5′ overhangs on the other end (DNA1), or a 17-bp dsDNA with one blunt end and a 5-base 3′ overhang on the other end (DNA2) (Fig 1A). For convenience, throughout the text, we refer to each strand as the template or non-template strand. We determined the structures of both MjMre11–DNA complexes in the presence of Mg2+ at 3.55 Å (Mre11–DNA1) and 3.59 Å (Mre11–DNA2) (see electron density maps in Supplementary Fig S1A–D; Supplementary Table S1). The nuclease activity of Mre11 is much weaker in the presence of Mg2+ than in the presence of Mn2+, so the addition of Mg2+ to the buffer aided in crystallization of the wild-type Mre11–DNA complex (Hopkins & Paull, 2008; Cannon et al, 2013).
Both crystals contain three Mre11 dimers and one DNA molecule in the asymmetric unit, but only one Mre11 dimer (A/B) is involved in interaction with DNA (Fig 1B, Supplementary Fig S2). MjMre11 is dimerized via four-helix bundle formation, with helices α2 and α3 derived from each nuclease domain. In the asymmetric unit, the DNA-bound Mre11 dimer interacts with other two Mre11 dimers (referred to here as Mre11 C/D and Mre11 E/F). In the inter-dimer interaction within the asymmetric unit, Mre11 C of a second dimer binds both Mre11 A and B via its capping domain and the C-terminal tail; the extended C-terminal tail (residues 304–312) of Mre11 C interacts with two helices (α1 and α2) of Mre11 A in a perpendicular orientation, whereas strand β15 and loop β15–β16 of the capping domain of Mre11 C binds to loop β3–α3 of Mre11 B via H-bonds and ion-pairs (Supplementary Fig S2A). The crystallographic symmetry-related Mre11 A binds to the interface of Mre11 C and D in the same manner. In another dimer–dimer interaction, helices α5 and α4 of Mre11F bind to loop α1–β2 and loop β12–β13 of Mre11 A, respectively (Supplementary Fig S2B). DNA1 (or DNA2) does not make any contact to symmetry-related DNA1 or Mre11.
The six Mre11 monomers are similar with overall root-mean-square deviation (rmsd) value of 0.4–1.0 Å for 311 Cα atoms. However, the quaternary structures of three Mre11 dimers are noticeably different (see below). For unclear reasons, electron density for the Mg2+ ions is obvious only in the Mre11–DNA1 structure although both Mre11–DNA1 and Mre11–DNA2 complexes were crystallized in the presence of 1 mM MgCl2. The coordination geometry of Mg2+ to MjMre11 is similar to that of Mn2+ (Hopfner et al, 2001; Lim et al, 2011). We observed additional density near the active site of MjMre11 C, which is only present in the Mre11–DNA1 complex (Supplementary Fig S1D). Although the size of the electron density is similar to that of a mononucleotide, we cannot precisely assign the source of this density due to the limited resolution of the structure.
In the Mre11–DNA1 structure, the three to ten terminal nucleotides are disordered, but the central 16 nucleotides were well ordered to be modeled (Supplementary Fig S1A). In the Mre11–DNA2 structure, three or four terminal nucleotides are not visible on either side, and only the central 14 nucleotides are modeled (Fig 1A). Thus, despite the differences in the sequence and structure of DNA molecules used in crystallization, the structures of both Mre11–DNA complexes are similar (Supplementary Fig S3A). However, some interactions in Mre11–DNA1 are not observed in Mre11–DNA2 due to DNA length as described below (Supplementary Fig S3A–C). Thus, we will primarily focus on describing the Mre11–DNA1 structure.
The B-form DNA runs across the Mre11 dimer and is orientated more toward one Mre11 molecule (Mre11 B). The DNA is tilted by about 30° relative to the interface axis between the two Mre11 molecules (Fig 1C and D). Mre11 A and B interact asymmetrically with DNA through their nuclease domains. DNA recognition by Mre11 is illustrated by the 14-base duplex that spans the Mre11 dimer. Mre11 A recognizes one end (3′ end of the template strand) and the middle region of DNA. Mre11 B binds the opposite end (5′ end of the template strand) and the middle region of DNA. The two recognition loops (RLs, dark blue) wedged into the minor groove in the center of DNA and cause distortion of the middle region, and one RL (deep purple) from each nuclease domain interacts with each DNA end (Fig 1C and D). The capping domains of both Mre11 subunits are distant from DNA, although the capping domain from Mre11 B is closer (5.5 Å between Lys229 and the phosphate group) to the minor groove. These features clearly distinguish MjMre11–DNA binding from the mode of DNA recognition by PfMre11, in which the two DNA molecules bind to a PfMre11 dimer with the axes of two DNA molecules offset by one dsDNA width (synaptic DNA) or with one capping domain interacting exclusively with the ssDNA overhang (branched DNA) (Fig 1E and F). In both PfMre11–DNA structures, no duplex extension is observed as a result of the short length of the DNA used for crystallization.
DNA recognition by MjMre11
The DNA-binding sites can be divided into two groups (Fig 2A and B; Supplementary Fig S3B and C); first, the middle region of DNA binds the center of the dimeric interface. Second, both DNA ends interact with loop β6–α4 where basic residues are clustered (Fig 2C). Each end of the template strand is more closely juxtaposed with the active sites of Mre11 A and B.
Figure 2. Close-up view of DNA recognition by Mre11.

A Close-up view of MjMre11–DNA interaction. DNA-interacting residues from Mre11 A and Mre11 B are shown in cyan and pink, respectively. See also Supplementary Fig S3C and D for cartoon representations of the MjMre11–DNA1 and Mre11–DNA2 interfaces, respectively. Hydrogen bonds and ion-pairs are shown as dotted lines. An interaction between Ser131 (Mre11B) and the 5′ end of template strand is observed only in Mre11–DNA1.
B An orthogonal view of (A).
C Surface representation of the MjMre11 dimer bound to DNA1 (yellow and orange). Positively and negatively charged regions are shown in blue and red, respectively. A cluster of basic residues including Lys129, Lys130, and Lys132 on each Mre11 molecule is marked with a dashed circle.
D Close-up view of the PfMre11-synaptic DNA interaction (3DSC). The figure is drawn in the same orientation as that of (A).
E Close-up view of the PfMre11-branched DNA complex (3DSD).
F Structural comparison of DNA1 (yellow and orange) and B-form DNA (blue).
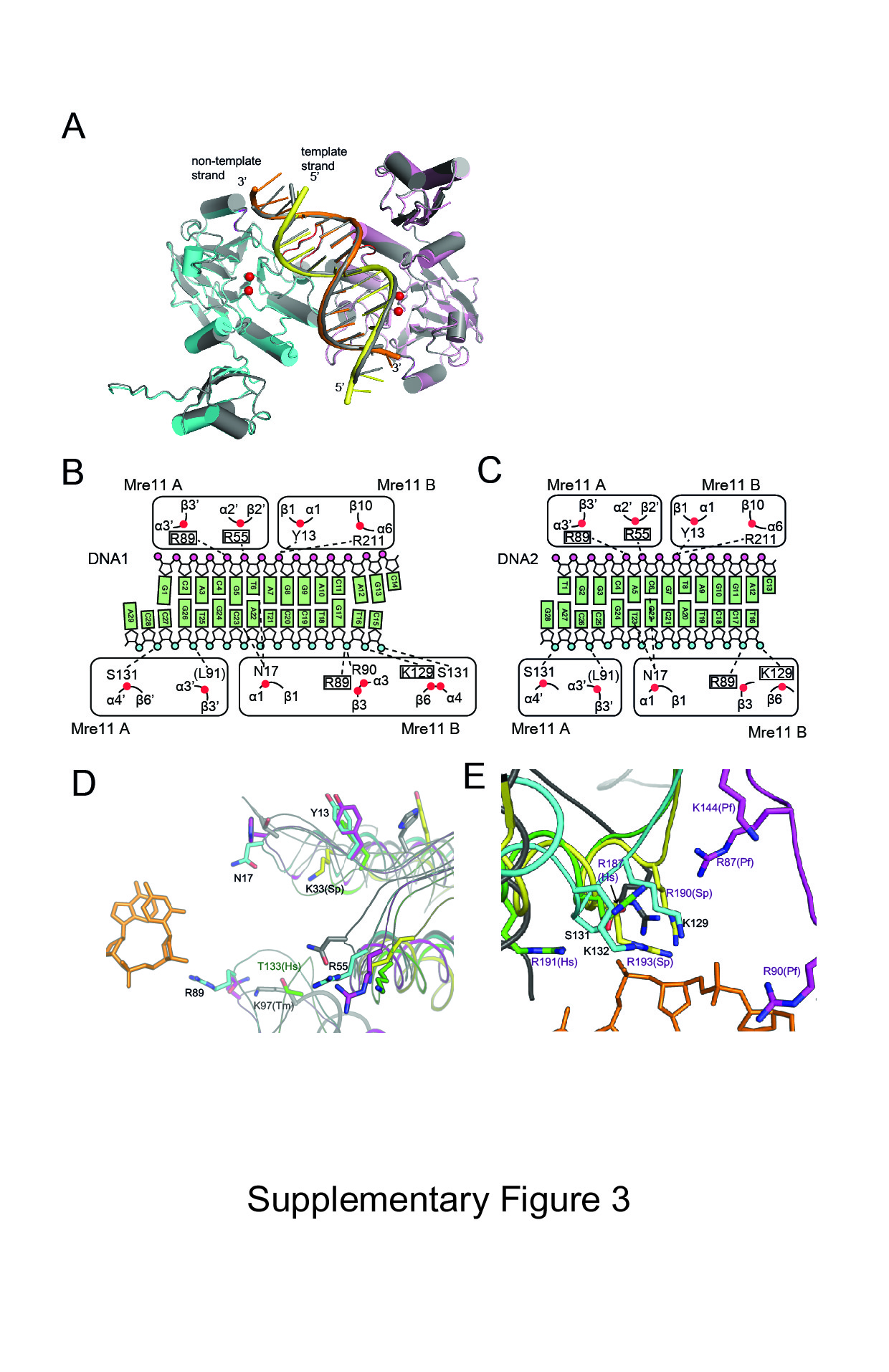
In the middle region, DNA recognition by Mre11 is achieved through RLs β1–α1, β2–α2, and β3–α3. This region of interaction is observed in both DNA1 and DNA2. Arg55 from Mre11 A (β2–α2) interacts with phosphate oxygen of T6, whereas Arg55 from Mre11 B binds to the main chain carbonyl group of Arg55 from Mre11 A and stabilizes the four-helix bundle. Arg89 and Arg90 from Mre11 B (β3–α3) bind phosphate oxygens of G17 and T16 of the template strand at the major groove, whereas Arg89 and Arg90 from Mre11 A interact with G5 of the non-template strand at the minor groove. Asn17 (loop β1–α1 from Mre11 B) is inserted into the minor groove of DNA, where it interacts with both T6 and C23 (Fig 2B, Supplementary Fig S3B and C). Arg14 and Asp19 from Mre11 B stabilize Asn17 (Mre11 A) through a hydrogen-bond network, which is further supported by Arg90 from Mre11 A. A cluster of these residues participates in wedging the two strands. By contrast, Asn17 from Mre11 A is more than 4 Å away from T18 of the template strand. Thus, residues from the Mre11 A and B interact asymmetrically with the phosphate groups in the middle of the DNA. DNA recognition by Mre11 in the middle region is conserved to the MjMre11–DNA structures; Tyr13, Asn17, Arg55, and Arg89 are equivalent to Tyr13, His17, Arg55, and Gln89 of DNA–PfMre11 (Fig 2D and E; Supplementary Fig S3D). However, Arg89 is not conserved in yeast and human Mre11 (Fig 3A); the loop containing the equivalent residue is disordered in fission yeast Mre11, and the equivalent residue (Thr133) in human Mre11 is more than 8 Å apart from Arg89 of MjMre11 (Supplementary Fig S3D).
Figure 3. In vitro analyses of Mre11 mutants.

A Structure-based sequence alignment of MjMre11 orthologues, generated using the Clustar Omega software (http://www.ebi.ac.uk/Tools/msa/clustalo/). Only regions of the aligned sequences near the mutated residues (DNA-interacting and capping domain) are shown. Secondary structure is shown on top of the alignment. Cyan squares indicate residues that interact with DNA in MjMre11. Strictly conserved residues are marked with a red box, and highly conserved residues are marked with a yellow box. MjMre11, M. jannaschii, UniProt accession number Q58719; PfMre11, P. furiosus, Q8U1N9; TmMre11, Thermotoga maritima, Q9X1X0; HsMre11, Homo sapiens, P49959; ScMre11, Saccharomyces cerevisiae, P32829; SpMre11, Schizosaccharomyces pombe, Q09683.
B A schematic diagram of the mutated residues in the MjMre11–DNA1 complex.
C DNA-binding analysis of wild-type and mutant MjMre11 (Arg55, Arg89, Lys129, Lys132, I302R and I302Y) proteins using the TP124/580 substrate. The molar ratios of protein:DNA were 50:1, 250:1, and 750:1. Reactions containing buffer (10 mM BTP-HCl, 50 mM NaCl, 5 mM DTT, 5% glycerol, pH 7.5) were incubated at 37°C for 30 min. Reaction products were resolved on 6% native PAGE gels. SDS–PAGE gel at the bottom shows that equal amounts of various Mre11 proteins were used in the reaction.
D Nuclease activities of wild-type and mutant (Arg55, Arg89, Lys129, Lys132, I302R and I302Y) MjMre11 proteins toward the DAR134 substrate. Reaction mixtures containing 20 nM 32P-labeled DNA substrate and MjMre11 (200 nM or 600 nM) were incubated at 55°C for 30 min. Standard molecular marker size is shown on the left. Quantitation of substrate cleavage is shown on right; the percentage of the DNA substrate remaining after the reaction was calculated from images collected using a phosphorimager. Error bars are calculated from at least three independent experiments. SDS–PAGE gel at the bottom shows that equal amounts of various Mre11 proteins were used in the reaction.
E Analysis of assembly of wild-type and mutant MjMre11 dimers using gel-filtration chromatography. Gel-filtration analysis using a buffer containing 20 mM BTP-HCl (pH 7.0), 200 mM NaCl, 5% glycerol, and 5 mM 2-mercaptoethanol showed that the mutant MjMre11 proteins formed dimers.
In both ends of DNA1, Lys129 and Ser131 (loop β6–α4) from Mre11 A and B bind or are closely juxtaposed (Lys129/ Mre11 A) to phosphate groups (Fig 2A and B); Lys129 (Nε) from Mre11 A and B is 4.3 and 2.8 Å away from the phosphate group of the 3′ and 5′ end of templates strand, respectively. Lys132 (Nε) from Mre11 A and B is located 3.9 Å from the phosphate group of the 3′ end and 6.3 Å from the phosphate group of the 5′ end of the template strand. Although these residues are not conserved at the sequence level, they are structurally conserved (Fig 3A, Supplementary Fig S4). Superposition reveals that Arg87, Arg90, and Lys144 of PfMre11 are near this region, and Arg196 of TmMre11 is close to Lys132 of MjMre11 (Supplementary Fig S3E). In addition, Arg190 and Arg193 in fission yeast Mre11, and Arg188 and Arg191 in human Mre11 are located in this basic loop. Thus, this region contains a basic cluster in all known Mre11 structures.
Mre11 distorts DNA conformation. Although the deformation of DNA is not as substantial as that induced by some DNA repair enzymes or transcription factors (Rice & Correll, 2008), the phosphate oxygen atoms of several nucleotides are markedly shifted relative to ideal B-form DNA (Fig 2F). This conformational change of the DNA is particularly noticeable at the central minor groove and at both ends. At the center, distances between the phosphate groups where RL β3–α3 of Mre11 A and RL β1–α1 of Mre11 B are wedged at the minor groove are 13.5 Å (11.8 Å for B-form DNA) and 12.4 Å, respectively. The terminal ends are deviated by approximately 6 Å. The short duplex DNA of the PfMre11–DNA complex (3DSC) also exhibited 2.4 Å increase in the minor groove width by wedging of His17 (Williams et al, 2008). The increased minor groove width by Mre11 is opposite to the compression of the minor groove by p53 (Cho et al, 1994).
Mutational analyses
To elucidate the significance of the DNA-interacting residues identified from current Mre11–DNA structures, we mutated residues at the DNA-binding interfaces and assessed their DNA-binding and nuclease activities (Fig 3A and B). We used a duplex containing five phosphorothioate bonds at the 3′ end of the top strand (TP124/580; Hopkins & Paull, 2008) for the exonuclease assay and a hairpin-structured DNA substrates (DAR134; Paull & Gellert, 1998) for the endonuclease assay. We first examined the roles of Arg55 and Arg89 because these residues are involved in DNA binding in both the MjMre11–DNA and PfMre11–DNA structures. Both R55S and R89S mutants did not interact with DNA (TP124/580) under 1:250 (DNA:protein) ratio (Fig 3C). The R55S mutant exhibited significantly decreased exo- and endonuclease activities (Fig 3D, lane 4 and 5, Supplementary Fig S5). This mutant forms a stable dimer, suggesting that the diminished DNA binding and cleavage is unlikely to be due to perturbation of dimerization (Fig 3E). The R89S mutant also exhibited substantially reduced nuclease activities, suggesting that this residue is important for DNA processing (Fig 3D, lane 6 and 7, Supplementary Fig S5).
We next examined the effect of mutating two residues, Lys129 and Lys132, which form an interface with both DNA ends. The K129A and K132D mutants failed to bind DNA under 1:250 (DNA:protein) ratio. Both K129A and K132D mutants also exhibited moderately or significantly decreased exo- and endonuclease activity toward TP124/580 and DAR134, respectively (Fig 3D, lanes 8–11; Supplementary Fig S5, lanes 8–11): The K129A and K132D mutants cleaved approximately 20 and 50% of DAR134 under 1:30 ratio (substrate:Mre11), whereas the wild-type Mre11 cleaved 85% of a substrate.
Quaternary structural changes of Mre11
Because the closest phosphate oxygen of the bound DNA is found far from the active site (∼10 Å) in MjMre11–DNA, we expect that Mre11 undergoes conformational changes in order to melt and place the DNA end in or near the active site. Previously, movement of the capping domain has been proposed to bring the DNA end to the active site of PfMre11 (Williams et al, 2008).
To examine the conformational changes of MjMre11, we compared the structures of the three MjMre11 dimers in the asymmetric unit. The most striking difference between these structures is the subunit arrangement of three dimers (rmsd 0.8–1.6 Å) (Fig 4A). The angle between α2 and α2′ of the four-helix bundle interface of the DNA-bound dimer is wider by 7° to 10° than those of the DNA-free MjMre11 dimers. The largest difference is observed between the DNA-bound Mre11 A/B (cyan, pink) and the DNA-free Mre11 E/F (gray) as shown in Fig 4A. Thus, DNA binding induces the rigid-body rotation of the two Mre11 molecules; consequently, the two capping domains become closer, such that the Mre11 dimer more tightly accommodates the substrate DNA. The four-helix bundle of the Rad50-bound Mre11 dimer (PDB 3AV0; Lim et al, 2011) exhibited a narrower angle (by 4°) than that of DNA-free dimer (E/F), further supporting the idea that the Mre11 dimer is dynamic. Within the Mre11 monomer, the capping domain was slightly shifted (up to 2 Å) relative to the nuclease domain.
Figure 4. DNA binding-induced conformational change of the MjMre11 dimer.

A Structural comparison between the DNA-bound MjMre11 dimer (A/B, cyan and pink) and DNA-free MjMre11 dimer (E/F, gray). The angle between helices α2 and α2′ of the four-helix bundle at the dimeric interface becomes larger in the presence of DNA, which shifts the two capping domains of the dimer closer to the DNA.
B Close-up view of the dimeric interface, showing interface residues mutated in this study. For sequences in this interface (α2 and α3), see Fig 3A.
C Cross-linking analysis of dimeric interface mutants. The K59C/E94C mutant successfully cross-linked in the presence of H2O2 and shifted to the dimer position on native PAGE gels (lane 2, 3), whereas the V58C/L99C mutant failed to form a covalent link (lane 4, 5).
D Nuclease activities of mutant Mre11 mutant proteins at the four-helix bundle in the reduced (5 mM DTT) and oxidized states (4 mM H2O2). The activities of V58C/L99C and K59C/E94C were examined using the DAR134 substrate. Quantitation of substrate cleavage is shown.
E Nuclease activities of the wild-type and mutant Mre11 (V58C/L99C and K59C/E94C) proteins at the four-helix bundle in the reduced (DTT) and oxidized states (H2O2) toward the TP124/580 substrate.
Cross-linking of the dimeric interface increases the nuclease activity
In the Mre11–DNA structure, Mre11 A primarily holds one end of DNA (3′ end of template strand), whereas Mre11 B grabs the opposite end (5′ end of template strand) (Fig 2A and B). Because the rigid-body rotation of each Mre11 is the most significant structural change of the Mre11 dimer upon DNA binding, we hypothesized that rotation of the Mre11 might twist and melt the DNA end, which is subsequently guided to the active site. For this dynamic motion to occur, formation of the Mre11 dimer is essential. To test this idea, we cross-linked the interface of the four-helix bundle by introducing double-Cys mutations. In the oxidized state, these Cys residues could introduce a disulfide bond in the oxidized state, thereby stabilizing the Mre11 dimer. Disulfide-bond formation at the dimeric interface might also affect the quaternary structure of the Mre11 dimer relative to the reduced Mre11 mutant. In one mutant, Val58 (α2) and Leu99 (α3) were replaced with Cys, and in another mutant, Lys59 (α2) and Glu94 (α3) were mutated to Cys. The distance between the Cβ atoms of Val58 and Leu99 is 4.2 Å, whereas the distance between the Cβ atoms of Lys59 and Glu94 is 2.7 Å (Fig 4B). Mobility-shift analysis confirmed that the K59C/E94C mutant formed a disulfide, whereas the V58C/L99C mutant did not form a disulfide link in the presence of an oxidizing agent (Fig 4C).
The V58C/L99C mutant exhibited reduced endonuclease activity toward DAR134 substrate in the reduced and oxidized states, suggesting that the double mutation affected the dimeric interface of Mre11 (Fig 4D, lane 4, 5, 10 and 11). The V58C/L99C mutant exhibited slightly reduced or similar exonuclease activity toward TP124/580 in the reduced and oxidized states, respectively (Fig 4E). The K59C/E94C mutant also exhibited reduced endo- and exonuclease activities in the reduced state (Fig 4D, lane 6 and 7, Fig 4E, lane 6 and 7). However, disulfide-bond formation between Cys59 and Cys94 under oxidized conditions resulted in comparable or even elevated nuclease activities relative to the reduced K59C/E94C Mre11 mutant or wild-type Mre11. The disulfide-bond effect of the K59C/E94C mutant was especially noticeable in regard to endonuclease activity toward DAR134. This result together with the quaternary structure change in the DNA-bound Mre11 dimer suggests that stabilization and dynamics of the Mre11 dimer are important for the nuclease activity of Mre11.
Rigid-body rotation of Mre11, as shown in the structural analysis, can shift the capping domain toward a substrate DNA by 10° (Fig 4A). The previously reported structure of the PfMre11–branched DNA complex also revealed an interaction between the capping domain and ssDNA (Williams et al, 2008). Therefore, we next attempted to determine whether movement of the capping domain could affect the nuclease activity of Mre11. To this end, we replaced Ile302 at the capping domain with Arg or Tyr (Fig 3C and D, Supplementary Fig S5). The I302R and I302Y mutants exhibited reduced endonuclease activities toward DAR134 (Fig 3D, lane 12–15). The I302R mutant cleaved TP124/580 as efficiently as WT. The I302Y mutant showed moderately reduced nuclease activity toward TP124/580 (Supplementary Fig S5, lanes 12–15). These results suggest that movement of the capping domain is important for the nuclease activity of Mre11.
In vivo analysis of the Mre11 mutants
To elucidate the roles of the newly discovered DNA-binding residues and their contributions to Mre11 functions in vivo, we generated budding yeast strains that expressed mre11 derivatives with mutations in the conserved DNA-binding sites and then examined their capacities for resection, classical non-homologous end joining (NHEJ), and conferring tolerance to camptothecin (CPT) and phleomycin (PHL)-induced DNA damage.
To test the integrity of NHEJ function, yeast strains deleted for HML and HMR but expressing a galactose-inducible HO endonuclease were engineered to express mre11 mutant derivatives in which Lys62 (Arg55 in MjMre11) and Arg184 (Lys132) were replaced with alanine (Moore & Haber, 1996; Figs 3A, B and 5A, Supplementary Fig S4). Upon addition of galactose, HO produced a DSB at the MAT locus that depends on NHEJ for repair. Deletion of MRE11 caused a severe NHEJ defect when HO was expressed for a short interval (1 h) or expressed persistently (Lee et al, 2002). We found that mre11–K62A and mre11–R184A exhibited a moderate (3- to 4-fold) but clear NHEJ defect under both short-term and persistent HO expression (Fig 5B and C). Strains expressing the nuclease-deficient mre11–H125N allele remained fully competent to perform NHEJ of the HO-induced break (Fig 5B and C). Analysis of the repair junctions among survivors of persistent HO expression confirmed that most of these repair events involved deletion or insertion of a few nucleotides, a hallmark of NHEJ (Moore & Haber, 1996; Supplementary Table S2). The distribution of junction types in mutant mre11 strains was largely indistinguishable from that in the wild type, even though junctions identified in the mre11–62A mutant contained more insertions than deletions (Supplementary Table S2). The results suggest that the conserved DNA-binding site is important for efficient NHEJ following HO-induced DSB.
Figure 5. Analysis of mre11 mutants in vivo.

A Schematic illustration of the NHEJ assay.
B, C NHEJ proficiency of yeast mutants was determined by measuring their survival rate upon induction of an HO break at the MAT locus. Deletion of HML and HMR forced repair of this DSB to occur by NHEJ only. Survival rate was calculated by dividing the number of colonies on YEP-galactose or YEPD following addition of galactose, by the number of colonies on YEPD on which HO was not induced. Survival of SLY1 (MRE11+, JKM139 derivatives), mre11Δ, mre11-K62A, mre11-K184A, and nuclease-deficient mre11-H125N (B) after expression of HO endonuclease for 1 h or (C) continuously by plating onto YEP-galactose plates is shown. Each point represents the average of at least three independent experiments ± SD.
D Sensitivity to DNA-damaging agents. Fivefold serial dilutions of nuclease-deficient mre11–H125N and mre11 variants with mutations at the putative DNA-binding interface (K62A, and R184A, JKM139 derivatives) were spotted onto YEPD plates with the indicated doses of genotoxic drugs and incubated for 2–3 days before being photographed. Drug sensitivity assays were also performed on strains deleted for SGS1, in which redundant resection pathways have been disabled. CPT, camptothecin; PHL, phleomycin. Shown is an example of spot assays performed three times independently.
E A graph showing the amount of un-resected DNA, measured by a Southern blot-based resection assay, in SLY1A (MRE11+, JKM139 derivatives) and mre11 mutant derivatives. Each point represents the average of at least three independent experiments ± SD.
F, G Graphs showing the amount of ssDNA at 0.7 or 5.7 kb distal to a DSB, measured by a PCR-based DNA resection assay. Each point represents the average of at least two independent experiments.
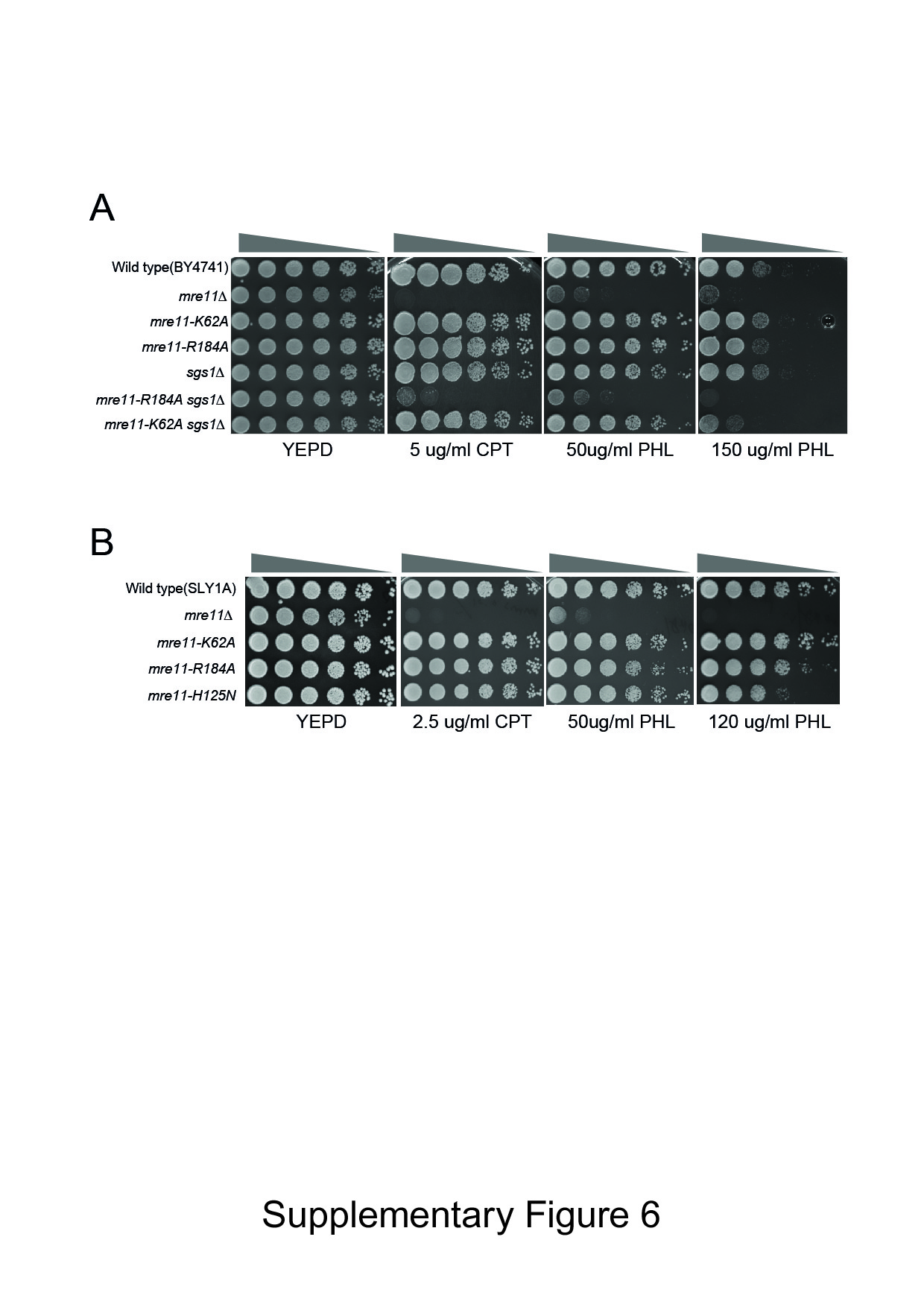
In budding yeast, the nuclease activity of Mre11 is dispensable for DSB resection and contributes little to cell survival under genotoxic stress due to the presence of other, functionally redundant resection pathways (Mimitou & Symington, 2010; Foster et al, 2011). To assess the effect of mre11 mutations on resection and genotoxic sensitivity, we deleted SGS1, which encodes a protein required for one of the two extensive resection pathways (Mimitou & Symington, 2008; Zhu et al, 2008), in mre11 mutant strains, and then examined the sensitivity of the resultant mutants to CPT or PHL treatment. As a control, we also included the mre11–H125N nuclease-deficient mutant in the analysis. We found that none of the mre11 mutants displayed apparent sensitivity to CPT or PHL treatment if SGS1 was intact (Supplementary Fig S6A and B). By contrast, in sgs1-deleted cells, mre11–R184A mutations caused a moderate but clear hypersensitivity to CPT and PHL; the level of sensitivity was comparable or slightly less than that in the mre11–H125N sgs1 mutant (Fig 5D, row 2 and 4, and Supplementary Fig S6A). mre11-K62A sgs1 mutation also showed a moderate sensitivity to PHL but not to CPT. The results support the importance of this residue in Mre11’s function in response to genotoxic stress.
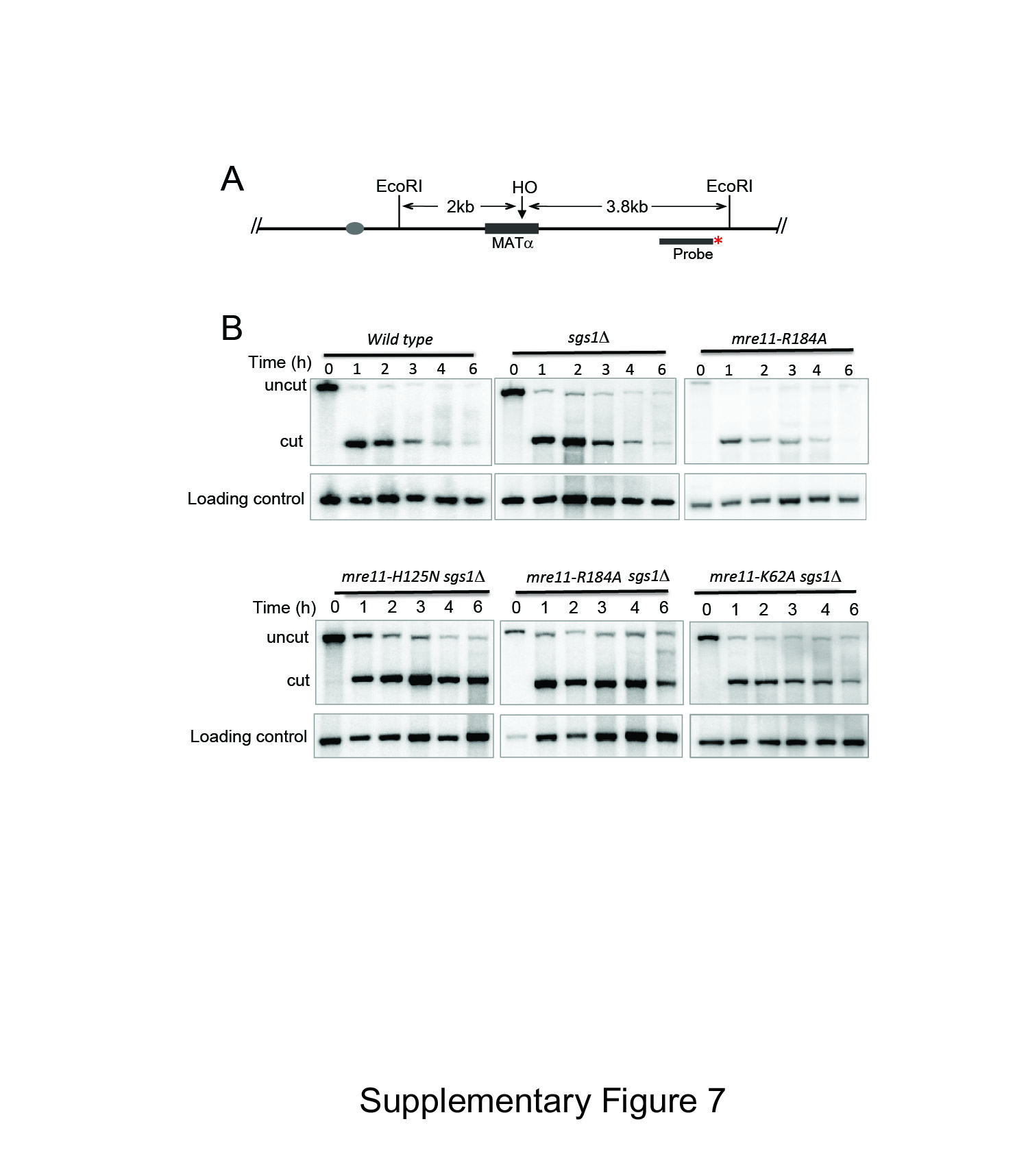
To further test the significance of Lys62 and Arg184 in Mre11 function, we also analyzed the effect of these mutations on resection of HO-induced DSB in donorless yeasts. Resection of HO-induced DSB rendered the DNA sequence flanking the HO break resistant to restriction enzyme digestion and triggered the disappearance of the HO-cleavage band in Southern blot-based assay using radiolabeled probe that annealed to the HO break site (Supplementary Fig S7A; Zhu et al, 2008). Due to the redundancy between Sgs1/Dna2 and Mre11 in regard to end resection (Mimitou & Symington, 2008; Zhu et al, 2008; Shim et al, 2010), we examined resection in cells deleted for SGS1 and expressing mutant mre11. We found that both K62A and R184A mutants exhibited moderate resection deficiency relative to the mre11 nuclease-deficient mutant H125N (Fig 5E, and Supplementary Fig S7B). Resection defects in yeasts expressing K62A and R184A mutant were further confirmed by a PCR-based assay (Zierhut & Diffley, 2008; Fig 5F and G, and Supplementary Fig S8). The results indicated that K62A and R184A are important for resection in vivo, validating the importance of these residues in Mre11 activity. These results are consistent with the premise that the newly identified Mre11–DNA interface represents a critical region for the repair of DNA breaks by HR and NHEJ.
Although the Lys62 (Arg55) and Arg184 (Lys132) of Mre11 are far from the sites for Rad50 or Xrs2 binding, the effects of Mre11 mutations on NHEJ, genotoxic sensitivity, and end resection could be caused by failure of MRX assembly, as observed in other Mre11 mutants (Limbo et al, 2012). Thus, we investigated in vivo assembly of the MRX complex using co-immunoprecipitation (co-IP) and yeast two-hybrid assays. Both co-IP and two-hybrid analyses revealed that neither K62A nor R184A mutation of Mre11 affected assembly of the MRX complex (Fig 6A and B).
Figure 6. Assembly of the MRX complex of the mre11 mutants.
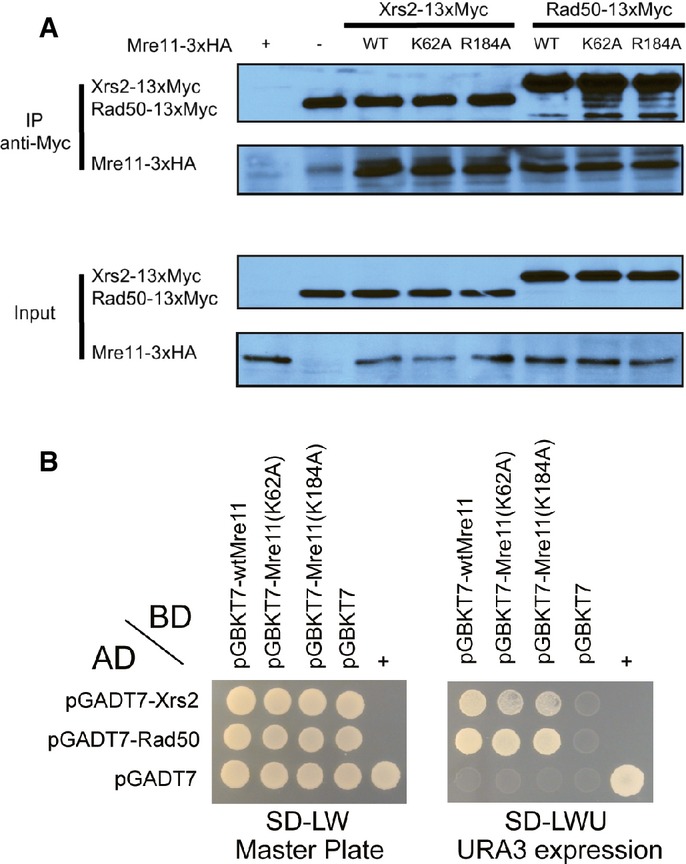
A Co-immunoprecipitation analysis of the interaction between Mre11 or its mutants (K62 and R184A), tagged with 3×HA, and Xrs2 and Rad50, tagged with 13×Myc. Mre11, Rad50, and Xrs2 were pulled down with anti-Myc antibody, and Mre11 proteins were detected using an anti-HA antibody.
B Yeast two-hybrid analysis of Mre11 proteins and other MRX components. Yeast strain (PBN204) was co-transformed with plasmids expressing various BD-Mre11 proteins and other AD-MRX subunits (Rad50 and Xrs2). Transformed yeast cells were spread on selective medium lacking leucine and tryptophan (SD-LW) to select for co-transformants (Master plate). Specific interactions between two proteins were monitored by growth on selective medium lacking leucine, tryptophan, and uracil (SD-LWU). The dimerization of polypyrimidine tract-binding protein served as the positive control (+), and the empty vector pGBKT7 and pGADT7 served as the negative control (−).
Source data are available online for this figure.
Discussion
The MR complex plays an important role in DSB repair by facilitating nucleolytic processing to form a recombinogenic ssDNA, as well as by catalyzing end synapsis for non-homologous and alternative end joining (Paull, 2010; Mimitou and Symington, 2011). In this study, we determined the structures of the Mre11 dimer bound to DNA molecules that more closely resemble a broken chromosomal end than DNA molecules used previously for structural studies. On the basis of our findings, we propose an alternative model for end recognition by Mre11.
Both the MjMre11–DNA1 and MjMre11–DNA2 structures showed that a whole Mre11 dimer binds one extended dsDNA, rather than two DNA molecules, and that the Mre11 dimer primarily recognizes the duplex DNA via its nuclease domain. This DNA recognition mode by Mre11 is markedly different from those described in previous studies showing that the PfMre11 dimer binds two DNA molecules for end tethering, or the capping domain of Mre11 interacts with the ssDNA overhang (Williams et al, 2008). The structure described here shows that a duplex with at least 14 bp is required for B-form DNA to cross the two nuclease domains of the Mre11 dimer and make full contact. No interactions between MjMre11 and ssDNA were observed. However, it is possible that ssDNA generated upon melting of DNA ends could interact with the capping domain, as observed in PfMre11-branched DNA. Furthermore, the DNA is tilted more toward one Mre11 molecule, contributing to a partially asymmetric interaction between the Mre11 dimer and DNA. Perturbing the Mre11 dimer resulted in the diminished nuclease activity, whereas increasing stability of the Mre11 dimer elevated the nuclease activity (Fig 4D and E). Our structure and biochemical analyses showing that the formation of a stable Mre11 dimer is important for DNA binding are consistent with previous analyses (Williams et al, 2008).
Arg55 and Arg89 in MjMre11 are important in binding and cleavage of DNA in vitro. Arg89 is not conserved in other species; the equivalent residue is serine or threonine in yeast or human Mre11, respectively (Fig 3A). Structural comparison reveals that this region in SpMre11 is disordered and Thr133 (HsMre11) is about 5 Å apart from the Arg89 (Supplementary Fig S3E). Although R89S mutant exhibited significantly diminished DNA-binding and nuclease activities, the activities we presented are relative to those of the wild-type MjMre11. An increased amount of the Mre11 mutants clearly cleaved the DNA substrate. Also, we do not exclude a possibility that the DNA-binding mode between archaeal Mre11 and eukaryotic Mre11 is somewhat different; for example, Ser138 (SpMre11) or Thr133 (HsMre11) may not be as critically involved in DNA binding as Arg89 in MjMre11.
One of the key features in our MjMre11–DNA structure is the recognition of DNA ends by the structurally conserved basic surface of Mre11, which is a result of the extended DNA across the whole Mre11 dimer. Mutation of two residues on this surface, Lys129 and Lys132, noticeably decreased DNA-binding and nuclease activity of MjMre11. Using two different resection assays, we found that the mutations of Lys184 (ScMre11) moderately reduced end resection. Cells expressing mre11–K184A were very sensitive to CPT or PHL treatment in the absence of the compensating nuclease/helicase (Sgs1/Dna2) complex. Both co-IP and two-hybrid analysis confirmed that the mutation did not impair MRX complex formation; therefore, the reduction in end resection is likely due to changes in the Mre11–DNA interaction. These results confirmed the importance of these residues in recognition of the DNA, and the validity of our structural model.
How might end recognition by the basic surface of Mre11 contribute to resection? In the MjMre11–DNA structure, the closest phosphate group of DNA is over 10 Å away from the active site and must be shifted substantially toward the active site for resection (Supplementary Fig S1D). Structural comparison of DNA-bound and DNA-unbound Mre11 dimers in the asymmetric unit revealed that each Mre11 undergoes rotation, such that the two capping domains move closer to each other (and toward the DNA) upon DNA binding. Because Lys129, Ser131, and Lys132 of each Mre11 bind or are closely located to DNA ends, rigid-body rotation of the Mre11 dimer could allow these residues to facilitate melting of the duplex end, which could then be subsequently placed in the active site (Supplementary Movies S1 and S2). In addition, rigid-body rotation is expected to push the two wedging residues, Asn17 and Asp19, into the central minor groove and to disrupt the base pairs in the middle region of the DNA. Based on these findings, we propose that the DNA end that is distant from the active site can be melted via interactions with the conserved basic surface, as well as subunit rotation. The melted DNA end could be guided to the active site, possibly via interaction with the capping domain as proposed by Williams et al (2008). Dynamic features of Mre11 dimer have been implicated in several previous studies. The structure of yeast Mre11–Nbs1 complex revealed that Nbs1 altered the arrangement of the Mre11 subunit such that it could interact more tightly with DNA (Schiller et al, 2012). Human Mre11 dimer also exhibited significant differences in the dimer arrangement (Park et al, 2011). The quaternary structure of Mre11 is altered by the binding of Rad50 in the MR complex (Lammens et al, 2011; Lim et al, 2011; Williams et al, 2011; Möckel et al, 2012). In addition, biochemical analysis showed that the phage Mre11 undergoes conformational changes during the exonuclease reaction, consistent with the results of structural studies of archaeal and eukaryotic Mre11 (Albrecht et al, 2012). Obtaining additional confirmation of these notions, as well as the molecular details of coordinated conformational changes of Mre11, DNA end melting, and placement in the active site, will require integrated approaches including single-molecule analysis, in addition to further structural studies.
The Mre11 dimer also contributes to end synapsis and serves as a platform for assembly and disassembly of core NHEJ proteins, such as Ku and DNA ligase IV, to DNA lesions in budding yeast (Zhang et al, 2007; Wu et al, 2008). In fission yeast, an Mre11 mutation that affects the DNA binding and positioning of DNA ends impairs NHEJ at telomeres (Reis et al, 2012) even if the role of MRN in NHEJ in vertebrate cells is not yet fully defined. Both R62A and K184A mutations exhibited moderately but clearly attenuated end joining following transient or persistent HO expression. The modest end-joining deficiency of these mutants and along with differences in the distributions of junction types among survivors between these mutants and the MRE11 gene deletion mutant suggest that additional Mre11–DNA contacts or contacts from Rad50/Xrs2 may confer residual DNA-binding capacity and/or end synapsis to sustain limited end joining.
An alternative model for the MjMre11–DNA complex presented here, together with the two previously published PfMre11–DNA structures, provides a better understanding of how an Mre11 dimer recognizes and repairs the DSB. The Mre11 dimer is required as an entity to bind extended B-form DNA near the end of broken chromosomes and to undergo DNA-induced changes in quaternary structure, explaining why dimerization is essential for both in vitro and in vivo function of Mre11. The Mre11–DNA structure likely reflects the DNA binding by ATP-unbound MR, in which disengagement of Rad50 allows the Mre11 dimer to be fully accessible for end resection in the HDR pathway (Lammens et al, 2011; Lim et al, 2011; Deshpande et al, 2014). In addition, the DNA-binding mode of the Mre11 dimer shown here may reflect the contribution of Mre11 to alternative end joining. In previous in vitro analysis, the mixture of wild-type Mre11 and the nuclease-deficient Mre11 mutant stimulated resection and DNA end bridging in the presence of DNA ligase (Paull & Gellert, 2000). Thus, the bridging of DNA through Mre11 is likely to be mediated by the oligomerization of the Mre11 dimer. This idea is supported by electron microscopy studies, in which the head domain of the MR complex oligomerizes on DNA in the presence or absence of ATP (de Jager et al, 2001). In the future, it will be intriguing to investigate whether the DNA-binding mode and the resulting quaternary structural changes are also observed in the mammalian MRN complex.
Materials and Methods
Protein expression and purification
A gene encoding MjMre11 core (residues 1–313) was inserted into pET28a and expressed in Escherichia coli Rosetta (DE3). Bacterial cells were grown to an OD600 of 0.7 and then induced using 0.5 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) for 18 h at 18°C. Cells were harvested by centrifugation and lysed in buffer (25 mM Tris pH 7.4, 300 mM NaCl, and 5 mM 2-mercaptoethanol) containing 1 mM phenylmethylsulfonyl fluoride (PMSF). Cells were lysed using a homogenizer (Thermo Fisher), sonicated, and insoluble material was removed by spinning at 100,000 × g for 1 h at 4°C. His-tagged MjMre11 core was purified by Ni2+-NTA affinity chromatography with a 0–400 mM imidazole gradient. Fractions containing MjMre11 core were further purified by ion-exchange (Mono-Q) chromatography with a 0–500 mM NaCl gradient and gel-filtration chromatography (Superdex 75) using a buffer containing 20 mM Bis-Tris-Propane-HCl (BTP-HCl) pH 7.0, 0.2 M NaCl, and 5 mM DTT. The MjMre11 core was concentrated to 5 and 0.5–2 mg/ml for crystallization and biochemical assays, respectively.
Crystallization and X-ray diffraction data collection
Crystals of MjMre11–DNA were grown at room temperature by the hanging-drop vapor-diffusion method. Crystals of the MjMre11–DNA1 or MjMre11–DNA2 were grown from the buffer containing 12% (w/v) 2-methyl-2,4-pentanediol (MPD), 0.1 M MgCl2, and 0.1 M sodium citrate, pH 5.6. Prior to flash freezing in liquid nitrogen, crystals were transferred to a reservoir buffer containing 30% (w/v) glycerol. Diffraction data for native crystals were collected at 0.9791 Å on the 5C beamline (Pohang Advanced Light Source) and processed using the HKL2000 package (Otwinowski & Minor, 1997). The DNA-bound MjMre11 crystals formed in the space group P21 with a = 91.5 Å, b = 185.6 Å, c = 106.2 Å, β = 99.9° (DNA1), and a = 90.5 Å, b = 184.0 Å, and c = 106.6 Å, and β = 99.5° (DNA2).
Structure determination and refinement
Both crystal forms contain one MjMre11–DNA and two MjMre11 dimer complexes in the asymmetric unit. Initial phases were obtained by the molecular replacement method using MjMre11 structure (3AUZ) as a search model and the Phaser program (McCoy et al, 2007; Lim et al, 2011). After density modification, an electron density map generated at a resolution of approximately 3.6 Å using the PHENIX program showed good quality, which allows to build both protein and DNA molecules (Adams et al, 2010). Successive rounds of model building using COOT (Emsley & Cowtan, 2004) and refinement with PHENIX using rigid-body, positional, overall and individual B-factors and TLS refinement were performed to build the complete model. A restrained non-crystallographic symmetry (NCS) was applied throughout the refinement process. Final refined models of the DNA1 at 3.55 Å (Rwork/Rfree = 18.4/23.6%) and DNA2 at 3.59 Å (Rwork/Rfree = 19.4/25.3%) exhibited good geometric parameters (Supplementary Table S1).
Nuclease assays
Reaction mixtures containing 20 nM 32P-labeled DNA substrate (DAR134, TP124/580) and MjMre11 (200 or 600 nM) proteins in reaction buffer (10 mM BTP-HCl, pH 7.5, 50 mM NaCl, 2 mM dithiothreitol [DTT] and 5% glycerol) were incubated at 55°C for 30 min. Nuclease reactions were stopped by addition of 0.1 volume of stop mixture (3% SDS, 50 mM EDTA, 0.5 mg/ml proteinase K), followed by incubation for 10 min at 37°C. Reaction products were boiled for 5 min and resolved on 20% denaturing polyacrylamide gels containing 7 M urea in TBE buffer. Gels were run for 400 min at 13 V/cm. The intensity of the uncleaved substrate bands was analyzed by ImageQuant TL (Amersham Biosciences). For nuclease assays of the cross-linking mutant under oxidized conditions, 4 mM H2O2 was added at the beginning of the reaction with no DTT present, and the reactions were incubated at 55°C for 30 min.
Cross-linking analysis of Mre11 dimeric interface mutants
V58C/E94 and V59C/L99C mutant proteins (5 μM) were incubated at 55°C for 10 min in buffer containing 25 mM MOPS pH 7.0, 10 mM BTP pH 7.4, 5 mM MgCl2, 5% glycerol, 50 mM NaCl, and 0.5 mM DTT in the presence or absence of 2 mM H2O2. Subsequently, the samples were resolved on native PAGE gels, followed by Coomassie blue staining.
End-joining assay
Logarithmically growing yeast cells were incubated in YEP-glycerol for 16 h, and then, serial dilutions were plated onto YEPD and YEP-galactose plates. To induce HO for shorter durations, 2% (w/v) galactose was added to logarithmically growing yeast cells in YEP-glycerol medium; after 1 h, aliquots of the culture were removed and plated onto YEPD to inhibit further HO endonuclease expression. Survival frequency was calculated by dividing the number of colonies surviving on YEP-galactose or on YEPD after galactose induction by the number of colonies growing on a YEPD plate without galactose induction. To analyze the repair-junction sequences, a 239-bp DNA fragment containing the HO cut site was amplified by PCR using the primers MATa-HOCS-F (TTGCAAAGAAATGTGGCATTACTCC) and MATa-HOCS-R (5′-GGCCAAATGTACAAACACA TCTTCC-3′) and then subjected to sequencing.
Co-immunoprecipitation
Yeast extracts expressing Mre11-3HA and Rad50-13Myc or Xrs2-13Myc were prepared by lysing cells with glass beads in 0.6 ml cold IP150 solution (20 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.5% NP-40) supplemented with protease inhibitor cocktail (Roche). Anti-Myc monoclonal antibody (4 μg; 9E10, Sigma) was added to pre-cleared cell lysate and incubated at 4°C for 90 min. Protein G Agarose slurry (40 μl) was then added to the lysate, and the mixture was incubated for an additional 30 min at 4°C. The beads were collected by centrifugation and washed extensively with IP150. Proteins released from the beads were separated by 7.5% SDS–PAGE and detected with anti-HA or anti-MYC antibodies.
Structure coordinate deposition
Coordinates and structure factors have been deposited to RCSB Protein Data Bank with the accession codes 4TUI for the MjMre11–DNA1 complex and 4TUG for the MjMre11–DNA2 complex.
Acknowledgments
We thank anonymous reviewers for helpful comments and suggestions. We also thank Kyuwon Baek for initial stage of structure determination, and Eun Yong Shim, Jung Soon Koh, and Kyong-Tai Kim for technical help and discussions. This work was supported by NIH research grant GM71011 (to SEL); grants from the National R&D Program for Cancer Control, Ministry for Health and Welfare (1020280), National Research Foundation of Korea (NRF) funded by the Korea government (MEST, No. 2012004028, No. 2012-054226, and No. 20120008833), a rising star program (POSTECH) and BK21 program (Ministry of Education) to YC.
Footnotes
#Correction added 28 August 2014, after first online publication. In Figure 5F and G, the y-axis labels “% unresected DSB end” was corrected to “% resected DSB end”.
Author contributions
SS carried out crystallization and structure determination; Y-B, SS, J-SK, JK and YC participated in biochemical experimental design and data analysis; FL, JC, and S-EL designed and performed yeast genetics experiment. A-KK and O-KS performed yeast two-hybrid analysis. YC and S-EL conceived of the project and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://emboj.embopress.org
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alani E, Padmore R, Kleckner N. Analysis of wild-type and rad50 mutants of yeast suggests an intimate relationship between meiotic chromosome synapsis and recombination. Cell. 1990;61:419–436. doi: 10.1016/0092-8674(90)90524-i. [DOI] [PubMed] [Google Scholar]
- Albrecht DW, Herdendorf TJ, Nelson SW. Disruption of the bacteriophage T4 Mre11 dimer interface reveals a two-state mechanism for exonuclease activity. J Biol Chem. 2012;287:31371–31381. doi: 10.1074/jbc.M112.392316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur LM, Gustausson K, Hopfner K, Carson CT, Stracker TH, Karcher A, Felton D, Weitzman MD, Tainer J, Carney JP. Structural and functional analysis of Mre11–3. Nucleic Acids Res. 2004;32:1886–1893. doi: 10.1093/nar/gkh343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buis J, Wu Y, Deng Y, Leddon J, Westfield G, Eckersdorff M, Sekiguchi JM, Chang S, Ferguson DO. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135:85–96. doi: 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, Kuhnlein J, Yang SH, Cheng A, Schindler D, Stark JM, Russell R, Paull TT. Visualization of local DNA unwinding by Mre11/Rad50/Nbs1 using single-molecule FRET. Proc Natl Acad Sci USA. 2013;110:18868–18873. doi: 10.1073/pnas.1309816110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Trujillo KM, Van Komen S, Roh DH, Krejci L, Lewis LK, Resnick MA, Sung P, Tomkinson AE. Effect of amino acid substitutions in the rad50 ATP binding domain on DNA double strand break repair in yeast. J Biol Chem. 2005;280:2620–2627. doi: 10.1074/jbc.M410192200. [DOI] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- Deshpande RA, Williams GJ, Limbo O, Williams RS, Kuhnlein J, Lee JH, Classen S, Guenther G, Russell P, Tainer JA, Paull TT. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J. 2014;33:482–500. doi: 10.1002/embj.201386100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Foster SS, Balestrini A, Petrini JH. Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol Cell Biol. 2011;31:4379–4389. doi: 10.1128/MCB.05854-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini G, Ristori E, Cerignoli F, Rinaldi C, Zani M, Viel A, Ottini L, Crescenzi M, Martinotti S, Bignami M, Frati L, Screpanti I, Gulino A. Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO Rep. 2002;3:248–254. doi: 10.1093/embo-reports/kvf044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner KP, Karcher A, Shin DS, Craig L, Arthur LM, Carney JP, Tainer JA. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 2000;101:789–800. doi: 10.1016/s0092-8674(00)80890-9. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell. 2001;105:473–485. doi: 10.1016/s0092-8674(01)00335-x. [DOI] [PubMed] [Google Scholar]
- Hopkins BB, Paull TT. The P. furious mre11/rad50 complex promotes 5′ strand resection at a DNA double-strand break. Cell. 2008;135:250–260. doi: 10.1016/j.cell.2008.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–1135. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- Lammens K, Bemeleit DJ, Möckel C, Clausing E, Schele A, Hartung S, Schiller CB, Lucas M, Angermüller C, Söding J, Sträßer K, Hopfner KP. The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell. 2011;145:54–66. doi: 10.1016/j.cell.2011.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Bressan DA, Petrini JH, Haber JE. Complementation between N-terminal Saccharomyces cerevisiae mre11 alleles in DNA repair and telomere length maintenance. DNA Repair. 2002;1:27–40. doi: 10.1016/s1568-7864(01)00003-9. [DOI] [PubMed] [Google Scholar]
- Lim HS, Kim JS, Park YB, Gwon GH, Cho Y. Crystal structure of the Mre11Rad50 ATPγS complex: understanding the interplay between Mre11 and Rad50. Genes Dev. 2011;25:1091–1104. doi: 10.1101/gad.2037811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbo O, Moiani D, Kertokalio A, Wyman C, Tainer JA, Russell P. Mre11 ATLD17/18 mutation retains Tel1/ATM activity but blocks DNA double-strand break repair. Nucleic Acids Res. 2012;40:11435–11449. doi: 10.1093/nar/gks954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 2010;29:3358–3369. doi: 10.1038/emboj.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. DNA end resection–unraveling the tail. DNA Repair (Amst) 2011;10:344–348. doi: 10.1016/j.dnarep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möckel C, Lammens K, Schele A, Hopfner KP. ATP driven structural changes of the bacterial Mre11:Rad50 catalytic head complex. Nucleic Acids Res. 2012;40:914–927. doi: 10.1093/nar/gkr749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncalian G, Lengsfeld B, Bhaskara V, Hopfner KP, Karcher A, Alden E, Tainer JA, Paull TT. The rad50 signature motif: essential to ATP binding and biological function. J Mol Biol. 2004;335:937–951. doi: 10.1016/j.jmb.2003.11.026. [DOI] [PubMed] [Google Scholar]
- Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Herrero F, de Jager M, Dekker NH, Kanaar R, Wyman C, Dekker C. Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature. 2005;437:440–443. doi: 10.1038/nature03927. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Park YB, Chae J, Kim YC, Cho Y. Crystal structure of human Mre11: understanding tumorigenic mutations. Structure. 2011;19:1591–1602. doi: 10.1016/j.str.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- Paull TT, Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999;13:1276–1288. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Gellert M. A mechanistic basis for Mre11-directed DNA joining at microhomologies. Proc Natl Acad Sci USA. 2000;97:6409–6414. doi: 10.1073/pnas.110144297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT. Making the best of the loose ends: Mre11/Rad50 complexes and Sae2 promote DNA double-strand break resection. DNA Repair. 2010;9:1283–1291. doi: 10.1016/j.dnarep.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis CC, Batista S, Ferreira MG. The fission yeast MRN complex tethers dysfunctional telomeres for NHEJ repair. EMBO J. 2012;31:4576–4586. doi: 10.1038/emboj.2012.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice PA, Correll CC. Protein-Nucleic Acid Interactions: Structural Biology. Cambridge: RSC Publisher; 2008. [Google Scholar]
- Schiller CB, Lammens K, Guerini I, Coordes B, Feldmann H, Schlauderer F, Möckel C, Hopfner KP. Structure of Mre11–Nbs1 complex yields insights into ataxia-telangiectasia–like disease mutations and DNA damage signaling. Nat Struct Mol Biol. 2012;19:693–700. doi: 10.1038/nsmb.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim EY, Chung WH, Nicolette ML, Zhang Y, Davis M, Zhu Z, Paull TT, Ira G, Lee SE. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 2010;29:3370–3380. doi: 10.1038/emboj.2010.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–587. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12:90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo KM, Sung P. DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50-Mre11 complex. J Biol Chem. 2001;276:35458–35464. doi: 10.1074/jbc.M105482200. [DOI] [PubMed] [Google Scholar]
- Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GJ, Williams RS, Williams JS, Moncalian G, Arvai AS, Limbo O, Guenther G, Sildas S, Hammel M, Russell P, Tainer JA. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat Struct Mol Biol. 2011;18:423–431. doi: 10.1038/nsmb.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Topper LM, Wilson TE. Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics. 2008;178:1237–1249. doi: 10.1534/genetics.107.083535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hefferin ML, Chen L, Shim EY, Tseng HM, Kwon Y, Sung P, Lee SE, Tomkinson AE. Role of Dnl4-Lif1 in nonhomologous end-joining repair complex assembly and suppression of homologous recombination. Nat Struct Mol Biol. 2007;14:639–646. doi: 10.1038/nsmb1261. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierhut C, Diffley JF. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 2008;27:1875–1885. doi: 10.1038/emboj.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.