

Abstract
We previously showed that peripheral noxious input after spinal cord injury (SCI) inhibits beneficial spinal plasticity and impairs recovery of locomotor and bladder functions. These observations suggest that noxious input may similarly affect the development and maintenance of chronic neuropathic pain, an important consequence of SCI. In adult rats with a moderate contusion SCI, we investigated the effect of noxious tail stimulation, administered one day after SCI, on mechanical withdrawal responses to von Frey stimuli from 1 to 28 days, post-treatment. In addition, because the pro-inflammatory cytokine tumor necrosis factor α (TNFα) is implicated in numerous injury-induced processes including pain hypersensitivity, we assessed the temporal and spatial expression of TNFα, TNF receptors, and several downstream signaling targets after stimulation. Our results showed that unlike sham surgery or SCI only, nociceptive stimulation following SCI induced mechanical sensitivity by 24 hours. These behavioral changes were accompanied by increased expression of TNFα. Cellular assessments of downstream targets of TNFα revealed that nociceptive stimulation increased the expression of caspase 8 and the active subunit (12 kDa) of caspase 3 at a time point consistent with the onset of mechanical allodynia, indicative of active apoptosis. In addition, immunohistochemical analysis revealed distinct morphological signs of apoptosis in neurons and microglia at 24 hours post-stimulation. Interestingly, expression of the inflammatory mediator NFκB was unaltered by nociceptive stimulation. These results suggest that noxious input caudal to the level of SCI can increase the onset and expression of behavioral responses indicative of pain, potentially involving TNFα signaling.
Introduction
Chronic neuropathic pain is a frequent consequence of spinal cord injury (SCI) [28,78]. Despite its prevalence, limited progress is made in the development of effective treatments, a reflection of how little is known about the underlying mechanisms. This lack of knowledge may result from the complex interactions between the primary insult and various secondary processes caused by the injury [4,71]. Additionally, noxious input arising from concomitant peripheral injuries may significantly contribute to maladaptive plasticity and chronic pain following SCI.
We previously investigated the effect of peripheral nociceptive stimulation on spinal plasticity and functional recovery following SCI [31,38]. In adult rats with a complete spinal transection, noxious input derived from electrical stimulation or peripheral inflammation, inhibited beneficial plasticity producing a spinal learning deficit that resembles learned helplessness [18,45]. Stimulation also increased withdrawal sensitivity to an innocuous stimulus [2,30], a behavioral response analogous to mechanical allodynia. As demonstrated by Baumbauer et al. [2], these negative effects are produced only when stimulation intensity reliably engages C-fibers, indicating that these behavioral effects reflect alterations in plasticity within pain pathways. When extended to a contusion SCI model, nociceptive stimulation decreased signaling in brain-derived neurotrophic factor pathways in the lumbar spinal cord [36], and undermined long-term locomotor recovery [39].
SCI produces several cellular and morphological changes, including proliferation and activation of microglia and astrocytes [22,42], excitotoxicity, and cell death resulting from necrotic and apoptotic processes [17]. One key mediator implicated in each of these processes is the cytokine tumor necrosis factor alpha (TNFα) [3,29,92]. TNFα has been shown to influence neural survival, exerting both neuroprotective and neurodegenerative actions [32,62,66]. The diverse actions of TNFα reflect the many signaling pathways it engages, among which are the pro-inflammatory, nuclear factor kappa B (NFκB) and the pro-apoptotic, caspase 8 pathways. TNFα also activates JNK and ERK1/2 pathways.
Although a large body of literature has detailed the expression and downstream effects of TNFα after SCI alone, little is known about the role peripheral input plays in altering the effects of TNFα in the injured spinal cord. Recent work suggested that peripheral injury can exacerbate nociceptive plasticity by overdriving TNFα expression [13]. Similarly, we have shown that the spinal learning deficit induced by peripheral nociceptive stimulation after complete SCI is mediated by TNFα [47]. Although apoptosis contributes to the loss of tissue after SCI [17,59], it is not known whether peripheral noxious input exacerbates apoptosis after SCI. In this study, we propose that the development and maintenance of chronic neuropathic pain following SCI is modulated by peripheral nociceptive input accompanying the injury and by increased TNFα expression. Using adult rats with a moderate thoracic contusion SCI, we examined the effect of nociceptive stimulation on hindlimb withdrawal threshold that may reflect mechanical allodynia. In addition, we investigated the temporal, spatial, and cell-specific expression of TNFα and its downstream targets. We show that peripheral nociceptive input after SCI induces a sustained mechanical sensitivity that is paralleled by increased TNFα expression and the engagement of the apoptotic caspase-signaling pathway in spinal neurons and glia.
Methods
Male Sprague-Dawley rats obtained from Harlan (Houston, TX) served as subjects. Rats were approximately 90–110 days old and weighed between 350 and 400 grams. They were housed individually and maintained on a 12-hour light/dark cycle, with all behavioral testing performed during the light cycle. Food and water were available ad libitum. All experiments were carried out in accordance with NIH standards for the care and use of laboratory animals (NIH publications No. 80–23), and were approved by the University Laboratory Animal Care Committee at Texas A&M University. Every effort was made to minimize suffering and limit the number of animals used.
Surgery and spinal contusion injury
Subjects were anesthetized with isoflurane (5%, gas). Once a stable level of anesthesia was achieved, the concentration of isoflurane was lowered to 2–3%. An area extending approximately 4.5 cm above and below the injury site was shaved and disinfected with iodine, and a 7.0 cm incision was made over the spinal cord. Next, two incisions were made on either side of the vertebral column, extending about 3 cm rostral and caudal to the T12 segment. The dorsal spinous processes at T12 were removed, and the spinal tissue exposed. The dura remained intact. For the contusion injury, administered to the lower thoracic spinal cord, the vertebral column was fixed within the MASCIS device [41] and a moderate injury was produced by allowing the 10g impactor (outfitted with a 2.5 mm tip) to drop 12.5 mm unto the dorsal surface of the spinal cord, with zero dwell time. The wound was then closed with Michel clips.
To help prevent infection, subjects were treated with 105 units/kg Pfizerpen (penicillin G potassium) immediately after surgery and again 2 days later. For the first 24 hours after surgery, rats were placed in a recovery room maintained at 26.6° C. To compensate for fluid loss, subjects were given a 2.5 ml intraperitoneal injection of 0.9% saline after surgery. Bladders were expressed twice daily (morning and evening) until the animals had empty bladders for three consecutive days at the times of expression.
Rats used as sham controls underwent the surgical procedure, but did not receive a contusion spinal cord injury.
Experimental Design
These experiments were designed to investigate the effect of peripheral nociceptive stimulation following SCI (SCI+STIM) on the temporal and spatial expression of the pro-inflammatory cytokine, TNFα, its receptors, and downstream signaling targets and on hindlimb mechanical withdrawal responses. The study consisted of 4 experiments, each involving a separate cohort of subjects. For all experiments, procedures for noxious tail shock or unshock (as described below) were conducted 24 hours following surgery. Each experiment consisted of three groups of subjects: contusion spinal cord injury alone (SCI), subjects administered noxious tail shock following injury (SCI+STIM), and sham operated controls (Sham), which did not receive any stimulation. The first experiment sought to establish the acute effect of contusion injury and the impact of nociceptive stimulation treatment following SCI on the expression of TNFα signaling genes. In this experiment, SCI+STIM subjects were sacrificed one hour following noxious stimulation (1 hour group). The subsequent experiments were conducted to extend the observations arising from experiment 1 and to determine the lasting behavioral and cellular effects of stimulation. In these experiments, SCI+STIM subjects were sacrificed at 24 hours (24 hour group), 7 days (7 day group) and 28 days (28 day group) following noxious stimulation. In all experiments, SCI and sham control subjects were sacrificed at the time equivalent to the SCI+STIM subjects within the individual group. In addition, to determine whether noxious stimulation had a differential effect across the dorsal/ventral regions of the spinal cord, the tissue was further divided to collect dorsal and ventral halves, which were subsequently processed for mRNA and protein, individually. This manipulation was introduced for subjects in the 24 hour, 7 day and 28 day groups (after the acute experiments were undertaken) because concurrent work had revealed that controllable stimulation (delivered when the leg is in an extended position) affected TrkB signaling in the dorsal, but not the ventral spinal cord [48]. Furthermore, work [36] showed that intermittent noxious stimulation following SCI differentially altered the expression of BDNF-TrkB signaling genes in the dorsal and ventral spinal cord. A total of 70 subjects were used as follows: Twenty-two sham controls [6 used for behavior and 16 (n=4, each time point) for cellular assays], 24 SCI (n=6, each time point) and 24 SCI+STIM (n=6, each time point).
Behavioral locomotor assessment
Twenty-four hours after surgery, before shock/unshock treatment, locomotor behavior was assessed using the Basso, Beattie, and Bresnahan (BBB) scale [1] in an open enclosure for all subjects to ensure the effectiveness of the contusion injury (for details, refer to Grau et al. [39]). Following baseline scores, the subjects were assigned to a treatment group (shock or unshock) in a manner that ensured that injury severity was balanced across groups before treatment. Additionally, subjects in the 28 day group were assessed at time points when the pain tests were conducted (7, 14 and 28 days after treatment). We previously reported BBB scores for subjects in the 7 day group over the entire post-surgical recovery period [36].
Peripheral Nociceptive Stimulation
Previously, we showed that just 6 minutes of intermittent nociceptive stimulation to the tail exerts the same detrimental effects on adaptive plasticity as uncontrollable leg shock [18]. In this study, we employed the tail shock paradigm to also assess the effect of nociceptive stimulation on mechanical withdrawal threshold, which was measured on the hind paws, bilaterally. Twenty-four hours after SCI, subjects were loosely restrained in Plexiglas tubes as previously described [18,36,39]. Electrical stimulation was applied to the tail using an electrode constructed from a modified fuse clip. The electrode was coated with electrode gel (Harvard Apparatus, Holliston, MA) and attached 2 cm from the tip of the tail with Orthaletic tape. Leads from the fuse clip were attached to a BRS/LVE shock generator (Model SG-903) and intermittent constant current 1.5-mA, AC (60 Hz) electrical stimulus was applied through the electrodes. Rats treated with intermittent nociceptive stimulation received 180, 80-ms tail shocks on a variable time schedule with a mean inter-stimulus interval of 2 s (range 0.2 –3.8 s). SCI only subjects were placed in the restraining tubes for equal amount of time as SCI+STIM subjects, had the electrodes attached, but did not receive the electrical stimuli.
Assessment of mechanical withdrawal responses
To assess the effect of noxious stimulation on mechanical sensitivity, we tested tactile thresholds using Semmes-Weinstein von Frey filaments (Stoelting, Wood Dale, IL, USA). Contusion subjects (SCI and SCI+STIM) used for mechanical assessment were also used for cellular assays at 7 and 28 days, post treatment (Fig. 2; 7 days group and 28 days group; respectively). The mechanical testing was done on subjects loosely restrained with the hind limbs hanging freely, in the Plexiglas tubes used for electrical tail stimulation. Each subject was first placed in the tube for a 15 minute acclimation period. Starting with the smallest von Frey filament (handle marking of 1.65), the filaments were pressed perpendicularly against the skin of the mid-plantar surface of both hind paws for 2–3 seconds, in an ascending order until a flexion response was elicited. All subjects were tested twice in a counterbalanced ABBA order (A left, B right), with tactile testing of the same leg separated by at least 2 minutes. The handle markings were used to calculate force (in grams) using the formula [Log10 (10 × Force in mg)]. A maximum force of 15 g was assigned for responses occurring at von Frey hairs greater than 5.18 and to subjects failing to respond to any hair. Typically, only sham controls failed to respond to stimulation at 15 g or less. The average force calculated from the 4 responses was determined for each subject.
Fig. 2.
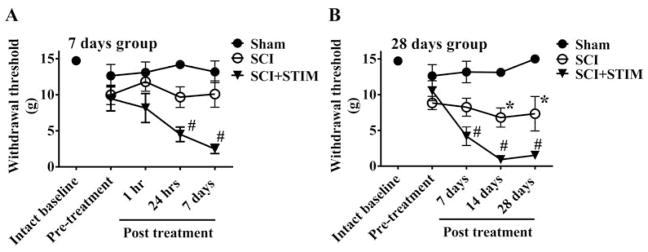
Nociceptive stimulation following SCI enhanced mechanical reactivity
Graphs show changes in mechanical reactivity to von Frey stimulation over time [In (A), 7 days group is assessed 1 hour, 24 hours and 7 days after treatment and in (B) 28 days group is assessed at 7, 14 and 28 days after treatment]. Subjects in the sham surgery group (black circles) did not demonstrate any changes in their withdrawal responses, expressed as force in grams, which were assessed at 1 hour, 24 hours, 7, 14 and 28 days. A) SCI only did not produce any changes in mechanical reactivity over the 7 day assessment period. In the SCI+STIM group, a significant decrease in tactile reactivity (mechanical allodynia) was first observed at 24 hrs and continued to 7 days after stimulation (#; p < .05). B) In SCI only subjects, there was a significant decrease in response threshold compared to sham controls at 14 days, which persisted to 28 days (*; p < .05). The SCI+STIM subjects demonstrated enhanced mechanical sensitivity at 7 days, 14 days and 28 days (#; p < .05). In addition to the early onset of mechanical sensitivity in SCI+STIM subjects, they were significantly more responsive (reduced withdrawal force) than SCI only subjects at 14 and 28 days (p < .001).
The following 3 tests of mechanical sensitivity were conducted: (i) Pre-surgery (intact baseline) response was determined for sham subjects only. (ii) Pre-treatment baseline was determined for all subjects 24 hours after surgery, but prior to treatment. (iii) Post-treatment assessments were conducted for contusion subjects and at the equivalent times for sham controls as follows: rats in the 7 days group were tested at 1 hour, 24 hours and 7 days after stimulation treatment, while those in the 28 days group were tested at 7 days, 14 days and 28 days after stimulation treatment. Care was taken to ensure that the investigator assessing mechanical threshold was blind to the subject’s treatment group. The subjects were sacrificed for cellular assays immediately after their last assessment of mechanical reactivity.
Sham controls
Twenty-two rats served as sham controls. Six subjects were used for assessment of their withdrawal responses to von Frey stimulation. These subjects were first tested prior to sham surgery to establish their baseline (Intact) responses. They were then tested 24 hours following surgery (pre-treatment) and at times that corresponded to the contusion groups behavioral time points of 1 hour, 24 hours, 7 days, 14 days and 28 days, post treatment. The remaining 16 subjects were used for cellular assays at each time point (n = 4).
RNA extraction and RT-PCR
We also investigated the effect noxious stimulation has on the expression of several members of the TNFα signaling pathway in lesioned epicenter. Animals were sacrificed at 1 hour, 24 hours, 7 days or 28 days following stimulation treatment or the equivalent times for unstimulated SCI and sham control subjects. All subjects were deeply anesthetized with pentobarbital (50mg/kg) and 1 centimeter of spinal cord around the lesioned epicenter was rapidly removed. For sham control subjects, one centimeter of spinal cord section equivalent to the lesioned area (approximately L1–L3) was removed. To determine the spatial (dorso-ventral) changes in the expression of genes of interest, the spinal cord tissue was further sectioned to yield dorsal and ventral portions in the 24 hours, 7 days and 28 days groups. The cord was processed for the extraction of both total RNA (RNeasy Mini Kit; Qiagen, Valencia, CA) and total protein (see below). Total RNA (100 ng) was converted into cDNA using TaqMan EZ RT-PCR Core reagents (Applied Biosystems, Carlsbad, CA) and the mRNA levels of all targets were measured by TaqMan quantitative real-time (RT)-PCR using a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Carlsbad, CA.). β-actin served as a control gene. The sequences of probes, forward and reverse primers for β-actin, c-jun, TNFα, TNF receptors 1 (TNFR1), TNFR2, nuclear factor kappa B (NFκB) and caspase 8 were obtained from Applied Biosystems, Carlsbad, CA. The mRNA expression for each gene of interest was normalized to β-actin expression, and presented as a fold change increase or decrease in experimental groups compared to the sham controls (normalized to 1).
Protein extraction and western blot
Subsequent to RNA extraction, total protein was extracted from the organic layer containing protein and DNA using the QIAzol™ lysis reagent protocol for isolation of genomic DNA and/or proteins from fatty tissue (Qiagen, Valencia, CA). Details on this procedure have been previously reported [49,79]. Following determination of the protein concentration using the Bradford Assay (BioRad, Hercules, CA), protein samples were diluted in Laemmli sample buffer and stored at −80° C at known concentrations of total protein (2–5μg/μl).
Western blotting was used for the protein quantification of c-Jun, TNFα, NFκB, caspase 8, caspase 3, ERK1/2 and pERK1/2. Equal amounts (30μg) of total protein were subjected to SDS-PAGE using 15% Tris-HCl precast gels (BioRad, Hercules, CA) gels for TNFα (estimated molecular weight of ~17 kDa), caspase 8 (~17–21 kDa), and caspase 3, (~12, 17 and 32 kDa) or 12% Tris-HEPES precast gels (Pierce, Rockford, IL) for c-Jun (~43 kDa), ERK1/2 and pERK1/2 (~42/44 kDa) and NFκB (~60 kDa). Following transfer onto PVDF membranes (Millipore, Bedford, MA), the blots for non-phosphorylated proteins were blocked for one hour in 5% blotting grade milk (BioRad, Hercules, CA) in Tris-Buffered Saline Tween-20 (TBST), whereas blots for pERK1/2 were blocked in 5% bovine serum albumin in TBST. After blocking, the blots were incubated overnight at 4° C in one of the following primary antibodies generated in rabbit: TNF alpha (1:500; #ARC3012 - Invitrogen, Camarillo, CA), caspase 8 (1:1000; #NB100-56116 - Novus Biological, Littleton, CO), caspase 3 (1:1500; #NB100-56113 - Novus Biological, Littleton, CO), c-Jun (1:2000; #NB110-55569 - Novus Biological, Littleton, CO), ERK1/2 (1:2000; #06-182 - Millipore, Temecula, CA), pERK1/2 (1:500; #07-467 - Millipore, Temecula, CA), NFκB p65 (1:500; NBP1-96139, a gift from Novus Biological Littleton, CO), or β-tubulin (~50 kDa, 1:1000; #05-661 - Upstate Cell Signaling, Lake Placid, NY), which was generated in mouse. All primary antibodies were diluted in blocking solution.
The following day, blots were washed in TBST (3 × 10 min) at room temperature then incubated in HRP-conjugated goat anti-rabbit or anti-mouse secondary antibodies (1:5,000; #31460 or 31430, respectively; Pierce, Rockford, IL) for 1 hour at room temperature. Following another 3 × 10 min series of washes, the blots were developed with ECL (Pierce, Rockford, IL) and imaged with Fluorchem HD2 (ProteinSimple, Santa Clara, CA). Ratios of the integrated densitometry of each protein of interest to the loading control (β-tubulin) were calculated, normalized to sham controls and averaged for animals within each group. The data is presented as a percentage change compared to sham control (100%).
Immunofluorescence
Fluorescent histochemistry was performed 24 hours after stimulation treatment to determine the effect of stimulation treatment on the expression of TNFα and caspase 3 proteins and to identify the cell type from which they are expressed. Thirteen subjects [Sham (n = 4), SCI (n = 4) and SCI+STIM (n = 5)] were anesthetized with pentobarbital (50mg/kg) and transcardially perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in PBS. The spinal cord was dissected and post-fixed in 4% PFA for 2 hours and then transferred to 30% sucrose for cryoprotection. Following at least 72 hours in sucrose, one centimeter of spinal cord around the lesioned epicenter was removed and cut into 3 sections (rostral, lesioned and caudal; approximately 3 mm each) and frozen into a mold with optimal cutting temperature (OCT) compound. Twenty micron (20μm) transverse sections were cut with a cryostat (Leica Microsystems, Buffalo Grove, IL) and mounted on superfrost plus slides. The slides were dried overnight and then stored at −80°C until the time of use. The sections were subsequently used for immunofluorescent labeling as described below.
The spinal cord sections were first washed (3 × 10 min) in Tris buffered saline (TBS), then incubated in blocking solution (3% normal goat serum, 0.1% Triton X-100 in TBS) for 1 hour at room temperature. For single fluorescent labeling, slides containing sections from Sham, SCI and SCI+STIM subjects were incubated overnight at room temperature in rabbit anti-TNF antibody (1:100; #NBP1-67821- Novus Biological, Littleton, CO) or anti-caspase 3 (1:2500; #NB100-56113 - Novus Biological, Littleton, CO) diluted in blocking solution. To determine the specific cell type expressing TNFα and caspase 3, double fluorescent labeling was conducted. Sections from SCI+STIM subjects were incubated in a combination of rabbit anti-TNFα (1:100) or anti-caspase 3 (1:2500) antibody with anti-mouse markers for neurons (NeuN, 1:400; #MAB377 - Millipore, Bedford, MA), astrocytes (Glial fibrillary acidic protein; GFAP, 1:500; #610565 - BD Biosciences, San Diego, CA) or microglia (OX42/CD11b/c, 1:1000; #550299 - BD Biosciences, San Diego, CA) in blocking solution overnight at room temperature. The following day, all slides were washed in cold TBS, following which they were incubated in the appropriate alexa fluor-conjugated secondary antibodies (goat anti-rabbit; #A11008 or goat anti-mouse; #A11030 - 1:300; Invitrogen, Eugene, OR) prepared in blocking solution for 1 hour at room temperature. Following another series of washes in TBS (3 × 15 min), the slides were mounted in Prolong Gold anti-fading mounting medium (Invitrogen, Eugene, OR) and coverslipped. Serial images focusing primarily on the epicenter of the lesioned spinal cord were taken with a confocal microscope (Olympus BX61 with a Plan Apo N 60x; N.A. = 1.42 oil immersion objective and a Plan Apo N 40x; N.A. = 1.30 oil immersion objective) using Olympus Fluoview version 5. Control slides were exposed to blocking solution instead of the primary antibody.
Estimations of caspase 3 and its co-expression with NeuN in the gray matter of the lesioned epicenter were made using the optical fractionator method [88] (Stereo Investigator, version 9, MBF Biosciences, Williston, VA, USA). Four animals in each group were used for these estimations. The entire gray matter (average area of 6.0 × 106 μm2 microns) of one spinal section of the lesioned epicenter (or equivalent for sham) was scanned. An average number of 72 ± 3 sites per section were counted. The following parameters were used: height of optical dissector, 15 μm; base of optical dissector, 60 × 60 μm2, average grid size, 300 × 300 μm2, and average section thickness, 20 μm.
Statistical Analyses
All data were analyzed using repeated measures analysis of variance (ANOVA) with an a priori alpha value of 0.05 or below considered significant [* indicates p < .05, ** indicates p < .01 relative to the sham control group, and # indicates p < .05 of the SCI+STIM group relative to the SCI group]. Mean group differences were evaluated using Duncan’s New Multiple Range post hoc test. In both text and figures, all data are presented as Mean ± SEM. The statistical analyses were conducted with SuperANOVA (Abacus Concepts).
Results
This study assessed the effect of noxious stimulation on withdrawal responses to mechanical stimulation, and on the temporal and spatial expression of the cytokine TNFα, its receptors, TNFR1, TNFR2; and subsequent downstream targets (NFκB, caspase 8, caspase 3, c-jun and ERK1/2) during the acute and early chronic periods following SCI.
BBB Locomotor Scores
All subjects were assessed for baseline BBB scores 1 day after surgery (D1) (Fig. 1). Only subjects with a D1 BBB score less than 8 were used in this study. The average baseline BBB score for contused animals was 3.4 ± 0.4 (SCI: 3.0 ± 0.6, SCI and STIM: 3.9 ± 0.4). There were no differences in D1 scores in SCI and SCI+STIM subjects across the different groups [1 hour group (3.8 ± 0.7 vs. 3.8 ± 1), 24 hour group (3.3 ± 0.9 vs 3.7 ± 1.0), 7 day group (4.2 ± 0.6 vs 3.2 ± 0.8) and 28 day group (3.6 ± 0.4 vs 2.8 ± 1.0)]. Previously, we reported that the shock paradigm used here significantly impaired the recovery of locomotor function (lower BBB scores) over the 7 day recovery period [36], an observation that was consistent with our prior report [39]. In this study, we also evaluated BBB locomotor scores in the 28 day group subjects on days when von Frey assessments were undertaken (days 7, 14 and 28 days after shock/unshock treatment). In these subjects, shock treatment impaired locomotor performance at 7 days [28 day group: SCI, 11.3 ± 0.5 and SCI+STIM, 8.5 ± 1). Thus, shock treatment delayed locomotor recovery but did not affect terminal performance, in the 28 day group. All sham subjects had a day 1 BBB score of 21 (not shown).
Fig. 1.
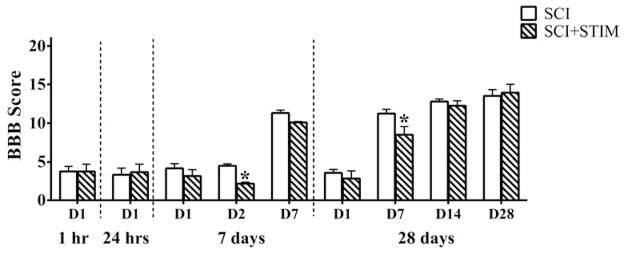
BBB scores
Histogram shows the average baseline BBB locomotor scores for SCI and SCI+STIM rats at post surgery day 1 (D1) and when pain tests were undertaken. There were no differences in D1 BBB scores for SCI and SCI+STIM rats in the 1 hr, 24 hr, 7 day and 28 day groups. In the 7 day group, noxious stimulation significantly decreased BBB scores at D2, whereas in the 28 day group a significant effect of noxious shock was seen at D7. There were no differences in BBB scores in SCI and SCI+STIM groups at D14 or D28 in the 28 day group. [*, p < .05].
Noxious stimulation fosters the development of enhanced mechanical sensitivity after SCI
The effects of SCI and noxious stimulation on mechanical withdrawal responses are illustrated in Fig. 2. We first assessed the effect of surgery on mechanical reactivity for the sham operated controls. While subjects were slightly more reactive to mechanical stimulation a day after surgery (Pre-treatment; 12.6 ± 1.6 g) relative to their pre-surgery scores (Intact baseline; 14.7 ± 0.3 g), this difference was not statistically significant, F(1, 10)=2.09, p > .05. We then looked at whether sham operated rats exhibited a shift in mechanical reactivity 1–28 days after injury, using an analysis of covariance (ANCOVA) to control for variation in Pre-treatment scores. Again, no statistically significant differences were found, F(3,12)=1.83, p > .05. We also examined whether the contusion injury had an acute effect on mechanical reactivity. At 24 hours after surgery, the contused groups did not differ from the sham operated controls, F(4, 25) < 1.0, p > .05. The finding that a moderate contusion injury fails to induce changes in mechanical sensitivity shortly after injury was previously reported by [58].
Next, we examined the effect of contusion injury (SCI) or injury plus noxious stimulation (SCI+STIM) on withdrawal responses to von Frey stimuli. Rats in the 7 day group were tested at 1 hour, 24 hours and 7 days after treatment (Fig. 2A). Noxious stimulation (SCI+STIM) induced a mechanical allodynia that emerged within 24 hours. An analysis of variance (ANOVA) confirmed that there was a main effect of group treatment, F(2, 15)=11.05, p < .005, and that the impact of shock depended upon time of testing, F(4, 28)=3.32, p < .05. Post hoc comparisons showed that the SCI+STIM treated group differed from the other two (p < .05). No other comparisons were statistically significant. To further explore the nature of the group by time interactions, additional analyses were conducted at each time point. These revealed that noxious stimulation had a significant impact at both 24 hours and 7 days (p < .05).
Rats in the 28 day group were tested at 7, 14 and 28 days after treatment (Fig. 2B). In these subjects, an effect of both SCI (SCI versus Sham) and noxious stimulation (SCI+STIM versus SCI) were observed. Relative to the sham controls, SCI alone enhanced mechanical sensitivity, and this effect was further enhanced in subjects that received nociceptive stimulation. An ANOVA confirmed that there was a significant effect of stimulation treatment condition, F(2, 14)=45.24, p < .0001. Post hoc comparisons confirmed that each group differed from the other two (p < .05). Again, additional post hoc analyses were conducted at each time point. Overall, when compared to sham control, the effect of noxious stimulation treatment emerged relatively early; at 24 hours. However, SCI only rats were not significantly different to sham controls until day 14.
Both noxious stimulation and spinal cord injury increase TNFα mRNA/protein expression
We then examined the impact of noxious stimulation on TNFα mRNA and protein expression after SCI. As illustrated in Fig. 3A, noxious stimulation following SCI (SCI+STIM) produced an increase in TNFα mRNA levels, that began to emerge at 1 hour, reached significance at 24 hours, and remained elevated (compared to both the SCI and sham controls) for 7 days (p < .05). SCI alone also had an effect that emerged more slowly. A significant difference between shams and the SCI group was only observed in the dorsal spinal cord at 7 days (p < .05). At 28 days, injured rats (both SCI and SCI+STIM groups) exhibited lower levels of TNFα mRNA expression (p < .001).
Fig. 3.

TNFα mRNA and protein were increased by SCI and SCI+STIM
A) qRT-PCR analyses showed that neither SCI nor SCI+STIM treatments altered TNFα mRNA levels at 1 hour group after treatment. TNFα mRNA levels in both dorsal and ventral spinal cords were significantly elevated by SCI plus noxious stimulation at 24 hours, post treatment compared to sham surgery. In the dorsal spinal cord the mRNA levels were also significantly increased when compared to SCI alone. Elevated expression of TNFα mRNA in the SCI+STIM was maintained for 7 days, with levels been significantly higher than both sham and SCI group in both dorsal and ventral spinal cord. In the dorsal spinal cord, TNFα mRNA levels in SCI only subjects was significantly increased compared to shams, although levels were still lower than in SCI+STIM subjects. By 28 days, TNFα was decreased in both treatment groups compared to shams. B) At 1 hour and in the dorsal spinal cord at 7 days after treatment, SCI significantly increased TNFα protein levels. Though levels were significantly increased compared to sham, noxious stimulation did not produce any greater effect than SCI only. A significant effect of noxious stimulation was observed in the dorsal and ventral spinal cord at 24 hour. At 28 days, although TNFα levels in the dorsal spinal cord of SCI and SCI+STIM subjects had returned to sham control levels, TNFα levels in the ventral spinal cord were significantly reduced in both treatment groups. [* indicates p < .05 relative to the sham control group, and # indicates p < .05 of the SCI+STIM group relative to the SCI group]. C) Examples of representative western blot images for TNFα. β-tubulin served as a loading control protein.
The effect of noxious stimulation on TNFα protein expression, illustrated in Fig. 3B & C, was also assessed in this study. At 1 hour, western blot analyses revealed a significant increase in TNFα protein levels in both SCI and SCI+STIM groups (208 ± 38% and 305 ± 32%, respectively; p < .05) relative to sham controls. At 24 hours, SCI+STIM subjects had significantly greater TNFα levels in both dorsal (312 ± 53%) and ventral (284 ± 39%) spinal cord (p < .05). At 7 days, injury per se increased TNFα protein expression in the dorsal horn in both the SCI+STIM and SCI groups, relative to the sham controls (p < .05). At 28 days, protein expression in injured rats was reduced in the ventral spinal cord (p < .05), but not the dorsal (p > .05) spinal cord.
Noxious stimulation increases TNFα immunofluorescence in microglia at 24 hours
As our western blot data showed that noxious stimulation produced a significant increase in TNFα protein expression 24 hours after stimulation, we sought to further investigate the spatial and cellular localization of TNFα protein expression at this time point. Using fluorescent immunohistochemistry, we found that TNFα expression was increased in both the superficial dorsal and ventral lesioned spinal cord of SCI+STIM subjects compared to SCI and sham controls (Fig. 4A, green). In the SCI+STIM subjects, dense TNFα labeling is observed in the superficial dorsal horn, and extending into the deeper laminae, compared to SCI only subjects. No TNFα labeling was present in control tissue (no primary antibody).
Fig. 4.

TNFα immunofluorescent labeling
A) At 24 hours after treatment, noxious stimulation increased TNFα expression in the lesioned spinal cord (dorsal; top panel and ventral; bottom panel) compared to sham and SCI subjects. The dorsal and ventral spinal cords are devoid of TNFα labeling in tissue from sham controls. In SCI+STIM subjects, TNFα is strongly expressed in the superficial dorsal horn which extends into the deeper laminae of the dorsal horn. TNFα is also strongly expressed in the mid-ventral spinal cord. No labeling was observed on control slides. [Sections depict the dorsal and ventral gray matter. The dotted white lines indicate the gray/white matter border]. B) Further histological characterization of TNFα labeling in the dorsal spinal cord of the lesioned area at 24 hours after stimulation shows that TNFα (green; a, e and i) was co-expressed with the microglial marker, OX-42 (red; f–h). There was no co-labeling of TNFα with NeuN (red; b–d) or GFAP in the gray matter (red; j–l). In the adjacent lateral white matter, TNFα (green; m) is co-localized with GFAP (red; n–p). To further illustrate TNFα co-labeling with NeuN, OX-42 and GFAP, the areas indicated by the white rectangles are enlarged in (d), (h) and (l & p), respectively. C) TNFα expression (green; a, e and i) is negligible in sham control tissue. There is no co-localization of TNFα with NeuN (red; b–d), OX-42 (red; f–h) and GFAP (red; j–l) in sham subjects. Sections in B and C are from the dorsal spinal cord of the lesioned epicenter.
We examined the lesioned tissue for co-localization of TNFα with cellular markers for neurons, astrocytes and microglia. Examination of the dorsal gray matter revealed TNFα co-localization with the microglial marker, OX-42 (Fig. 4B; e–h). There was no co-labeling with NeuN (Fig. 4B; a–d) suggesting that TNFα is not expressed in neurons. There was minimal GFAP labeling in the lesioned epicenter (Figs. 4B and 11A). TNFα was not co-expressed with GFAP in the gray matter of the dorsal spinal cord of the lesioned area (Fig. 4B; i–l), although there was sparse TNFα/GFAP co-labeling in the surrounding white matter (Fig. 4B; m–p). As a positive control, we examined GFAP expression rostral and caudal to the injury and observed high levels of GFAP (data not shown). This confirms the loss of astrocytes in the lesion epicenter. These findings suggest that TNFα is expressed primarily in microglia in the acute stages of injury. Compared to SCI and SCI+STIM subjects (Fig. 4A), tissue from sham subjects had very little TNFα labeling in the dorsal spinal cord (Fig. 4C; a, e, i), although there was strong labeling of the cellular markers NeuN (Fig. 4C; b–d), OX-42 (Fig. 4C; f–h) and GFAP (Fig. 4C; j–l). Similar patterns of TNFα, NeuN, OX-42 and GFAP labeling were observed in the ventral spinal cord of sham control (not shown).
Fig. 11.

Caspase 3 immunofluorescent labeling in the lesioned spinal cord
A) Noxious stimulation increased caspase 3 (green) expression in the lesioned spinal cord. Caspase 3 is co-labeled with OX-42, (red, top panel), but not with GFAP (red, bottom panel). The areas in the white square boxes are enlarged in the panels to the right to further illustrate co-labeling with OX-42 (top) and GFAP (bottom). B) (i) Caspase 3 (green) is also co-labeled with NeuN (red). Extensive caspase 3 labeling is also observed in processes and in areas bordering cavities. (ii) Varying stages of apoptosis (white arrows in Bi, merged image) are demonstrated in neurons. Cells labeled 1, 2, and 3 are enlarged to illustrate (1) a neuron during the early onset of apoptosis, containing a large condensed nucleus, (2) a neuron farther along in the process, containing a shriveled nucleus, and (3) a neuron undergoing late apoptosis, containing a fragmented NeuN-immunopositive nucleus. (iii) Unbiased stereology was used to estimate the average caspase 3 expression in the gray matter of the lesioned epicenter, as well as caspase 3 co-expressed with NeuN depicting the different morphologies (undergoing the varying stages of apoptosis). Caspase 3 expression was greatest in SCI+STIM subjects (p < .01). Caspase was also significantly elevated in SCI subjects (p < .05) compared to sham controls (also see C and D). Caspase 3 was mostly co-expressed with NeuN in the late stage of apoptosis (NeuN-Fragment). Although SCI subjects showed significant caspase 3/NeuN-Fragment co-labeling compared to sham (p < .01), levels in SCI+STIM subjects were significantly greater compared to both SCI (p < .05) and sham groups (p < .001). Caspase 3 expression was less in cells with the NeuN-Bulge and NeuN-Condense morphologies. Yet, caspase 3/NeuN-Bulge and caspase 3-NeuN-Condense co-expressions were significantly greater in the SCI only (p < .01) and SCI+STIM & SCI groups (p < .01), respectively compared to sham. Overall NeuN labeling was comparable across all groups; and neurons with normal morphology, including sham subjects had minimal caspase 3 expression (inset; also see D). [*, indicates significance compared to sham controls and #, indicates significance compared to SCI]. C and D illustrate caspase 3 (green) and NeuN (red) labeling in SCI only and sham subject, respectively. All spinal cord sections are from the mid dorsal spinal cord of the lesioned epicenter.
Spinal injury increases the expression of the TNFα receptor mRNA (TNFR1 and TNFR2) up to day 7
Next, we assessed the effect of nociceptive stimulation and injury on the downstream signal pathway, beginning with the mRNA expression of TNFα receptors, TNFR1 and TNFR2 (Fig. 5). Both receptor subtypes were robustly increased by injury, with contused rats (SCI+STIM and SCI) differing from the sham operated controls at 1 hour post treatment (p < .05). Similarly, there were robust increases in the dorsal spinal cord expression of both TNFR1 and TNFR2 at 24 hours and 7 days (p < .05). The ventral spinal cord expression of TNFR1 and TNFR2 were unchanged at 24 hours, but were increased in both SCI and SCI+STIM groups at 7 days (p < .05). At no time point was there a difference in SCI+STIM and SCI mRNA levels for either receptor. By 28 days post treatment, TNFR1 and TNFR2 levels were significantly decreased compared to shams in the dorsal spinal cord (p < .01), although ventral spinal cord levels were unchanged.
Fig. 5.

SCI increased TNF receptors mRNA expression
SCI and SCI+STIM increased (A) TNFR1 and (B) TNF2 mRNA levels at 1 hour, dorsal spinal cord at 24 hours and 7 days, compared to sham control. Noxious stimulation had no additional effect on TNFRs mRNA levels. By 28 days, TNFR1 and TNFR2 mRNA levels were decreased in the dorsal spinal cord compared to shams. [* indicates p < .05 relative to the sham control group].
Both noxious stimulation treatment and SCI increase c-jun mRNA and protein expression
We examined mRNA and protein expression levels of c-jun, an immediate early gene that can be induced by TNFα signaling and is associated with apoptotic pathways in neurons [26]. Elevation in c-Jun is seen in other forms of injury, including peripheral nerve injury [50,51] and brain injury [23]. As shown in Fig. 6A, in the 1 hour group, c-jun mRNA was significantly increased in SCI (2.2 ± 0.2) and SCI+STIM (2.3 ± 0.3) subjects compared to sham controls (p < .05). C-jun mRNA levels in both groups were significantly reduced in the dorsal spinal cord at 24 hours (p < .001). Though no changes were observed in the dorsal spinal cord at 7 days, noxious stimulation produced a significant increase in c-jun expression in the ventral spinal cord compared to SCI and sham-operated controls (p < .05). At 28 days, c-jun mRNA expression was reduced in contused rats (SCI+STIM and SCI) relative to the sham operated controls in both the dorsal and ventral horn (p < .001).
Fig. 6.

Noxious stimulation increased c-jun expression
A) In both SCI only and SCI+STIM groups, c-jun mRNA levels were increased in the 1 hour group, but decreased in the dorsal spinal cord at 24 hours and at 28 days. SCI+STIM subjects had increased c-jun levels only in the ventral spinal cord at 7 days. B) At 24 hours post treatment, c-Jun protein in the dorsal spinal cord was significantly increased in SCI+STIM subjects. At 7 days (dorsal and ventral spinal cord) and in the dorsal spinal cord only at 28 days, c-Jun levels were increased in both SCI and SCI+STIM groups compared to sham controls. There were no differences between the two treatment groups. [* indicates p < .05 relative to the sham control group, and # indicates p < .05 of the SCI+STIM group relative to the SCI group]. C) Examples of representative western blot images for c-jun protein. β-tubulin served as a loading control protein.
The effect of noxious stimulation on c-Jun protein levels is illustrated in Fig. 6B and C. Noxious stimulation induced a significant increase in c-Jun expression in the dorsal spinal cord at 24 hours (142 ± 11%; p < .05). Injury, per se, increased c-Jun protein levels in both SCI+STIM and SCI groups at 7 days (dorsal and ventral spinal cord expression) and in the dorsal cord at 28 days (p < .05). There were no differences in the expression of c-Jun protein levels in the ventral spinal cord at 24 hours and 28 days among the three groups. Interestingly, the emergence of these effects over time paralleled the behavioral results, with the effect of SCI plus noxious stimulation emerging sooner than that of just SCI. The overall pattern of results generally paralleled the changes observed in TNFα mRNA and protein. Generally, nociceptive stimulation induced a significant effect at 24 hours, followed by an effect of SCI at 7 days.
Injury increases pERK expression at 28 days
In our prior report, we assessed the expression of ERK1/2 and pERK1/2 protein at 1 hour, 24 hours and 7 days after SCI, and the effect of SCI plus nociceptive stimulation [36]. We found that spinal injury reduced ERK1/2 expression at 24 hours. Likewise, pERK was generally down-regulated by injury. Noxious stimulation further reduced the expression of ERK2 (at 24 hours and 7 days) and pERK1 (at 7 days) within the dorsal horn (see Table 1). Here we extend these observations by assessing ERK1/2 and pERK1/2 at 28 days post treatment. As observed at earlier time points, ERK1 level was significantly decreased (Fig. 7) in the dorsal spinal cord of SCI+STIM subjects (76 ± 6%) compared to sham controls (100 ± 10%; p < .05). The expressions of ERK2 in the dorsal and ventral spinal cord and ERK1 in the ventral cord were unchanged. The activated forms, pERK1 and pERK2, were significantly up-regulated in both SCI and SCI+STIM groups (p < .05) in the ventral spinal cord although their levels in the dorsal cord were unchanged. We also assessed the ratios of phosphorylated ERKs to total ERKs (pERK1/ERK1 and pERK2/ERK2). There were no differences in these ratios, consistent with results observed at earlier time points [36].
Table 1.
| Target | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mRNA/protein | Time after Treatment | |||||||||||||
| 1 hour | 24 hours | 7 days | 28 days | |||||||||||
| Dorsal | Ventral | Dorsal | Ventral | Dorsal | Ventral | |||||||||
| SCI | SCI+ STIM | SCI | SCI+ STIM | SCI | SCI+ STIM | SCI | SCI+ STIM | SCI | SCI+ STIM | SCI | SCI+ STIM | SCI | SCI+ STIM | |
| mRNA | ||||||||||||||
| c-jun | + | + | − | − | o | o | o | o | o | ++ | − | − | − | − |
| TNFα | o | o | o | ++ | o | + | + | ++ | o | + | − | − | − | − |
| TNFR1 | + | + | + | + | o | o | + | + | + | + | − | − | o | o |
| TNFR2 | + | + | + | + | o | o | + | + | + | + | − | − | o | o |
| NFκB | − | − | − | − | o | o | o | o | o | o | − | − | − | − |
| Casp 8 | o | o | o | o | o | o | + | + | + | + | o | ++ | + | + |
| Protein | ||||||||||||||
| c-Jun | o | o | o | + | o | o | + | + | + | + | + | + | o | o |
| TNFα | + | + | o | ++ | o | ++ | + | + | o | o | o | o | − | − |
| NFκB | − | − | − | − | o | o | o | o | o | o | o | o | o | − |
| Casp 8 | o | o | o | ++ | o | ++ | + | + | o | o | + | + | o | o |
| Casp 3 – 32 kDa | o | o | o | + | o | + | + | o | + | o | o | o | o | o |
| Casp 3 – 17 kDa | o | o | o | + | o | o | + | + | + | + | o | o | o | o |
| Casp 3 – 12 kDa | o | + | o | + | o | ++ | o | + | o | + | + | o | o | o |
| ERK1~ | o | o | − | − | − | − | o | − | o | o | o | − | o | o |
| ERK2~ | o | o | − | # | o | o | − | # | o | − | o | o | o | o |
| pERK1~ | o | o | o | o | o | o | + | # | o | o | o | o | + | + |
| pERK2~ | o | o | o | o | o | o | o | − | o | o | o | o | + | + |
o, no change
+ significant increase compared to sham
++ significant increase compared to SCI
− significant decrease compared to sham
# significant decrease compared to SCI
~ some parts reported previously, (Garraway et al., 2011)
Fig. 7.

ERK and pERK expression at 28 days post treatment
A) Noxious stimulation decreased ERK1 levels in the dorsal spinal cord compared to sham. B) Both pERK1 and pERK2 were increased in the ventral spinal cord of SCI and SCI+STIM subjects, although there were no additional effects of noxious stimulation. [* indicates p < .05 relative to the sham control group]. (C) Examples of representative western blot images for ERK1/2 and pERK1/2. β-tubulin served as a loading control protein.
Spinal injury reduces mRNA and protein expression of NFκB
NFκB is a transcription factor activated by a number of proteins including TNFα. It is engaged by both TNFα receptors and is thought to promote the inflammatory actions of TNFα. Like TNFα, NFκB has previously been implicated in the development of inflammatory and neuropathic pain [56,83]. We anticipated that NFκB would be up-regulated by SCI and noxious stimulation, but found the opposite. At 1 hour, SCI reduced NFκB mRNA (Fig. 8A) and protein (Fig. 8B and C) expression, yielding a significant difference between the two injured groups (SCI+STIM and SCI) and the sham operated controls (p < .05). A similar pattern was observed in the dorsal horn at 24 hours (p < .05). While no differences were observed at 7 days, NFκB mRNA expression was significantly reduced by injury in both the dorsal and ventral horn at 28 days. Noxious stimulation did not have an additional effect, except at 28 days, where it produced a significant decrease in protein expression within the ventral horn (77 ± 8%; p < .05). The observation that changes in NFκB mRNA and protein expression were opposite to that of TNFα suggests that the cellular nociceptive actions of TNFα after SCI and noxious stimulation may involve an alternative signaling pathway.
Fig. 8.

SCI decreased NFκB mRNA and protein expression
A) qRT-PCR analyses showed that NFκB mRNA levels were decreased at 1 hour, in the dorsal spinal cord at 24 hours, and in both dorsal and ventral spinal cord at 28 days in the SCI and SCI+STIM groups, relative to sham controls. There were no differences between the two treatment groups. B) NFκB protein levels were decreased in SCI and SCI+STIM subjects compared to shams at 1 hour and in the dorsal spinal cord at 24 hours. By 28 days, only in the ventral spinal cord of SCI+STIM subjects was NFκB protein level decreased. [* indicates p < .05 relative to the sham control group]. (C) Examples of representative western blot images for NFκB p65. β-tubulin served as a loading control protein.
Both injury and noxious stimulation enhance caspase 8 mRNA and protein expression
Caspase 8, an important initiator of the pro-apoptotic cell death pathway, is also a major downstream target of TNFα. Unlike NFκB, caspase 8 is engaged primarily by TNFα signaling through TNFR1, and does not appear to exhibit an inflammatory role. Changes in the expression of caspase 8 followed the general pattern of change seen with TNFα. qRT-PCR analyses showed that injury increased caspase 8 mRNA expression at day 7, yielding a significant difference between the injured (SCI+STIM and SCI) and sham operated controls (p < .05). At 28 days, a similar pattern was observed in the ventral horn (p < .05). However, in the dorsal horn, only the SCI+STIM group exhibited a significant increase in caspase 8 (1.5 ± 0.1) relative to SCI (1.1 ± 0.1) and sham controls (1.0 ± 0.0; p < .05).
Noxious stimulation also produced a robust increase in caspase 8 protein at 24 hours in both the dorsal (212 ± 35%) and ventral (186 ± 30%) horn (p < .05) (Fig. 9B and C). SCI alone did not change caspase 8 protein levels until 7 days, at which point it was significantly increased in the dorsal spinal cord of SCI+STIM and SCI. This increase in the dorsal spinal cord was maintained up to 28 days (p < .05). These results suggest that the activation of TNFα by injury and noxious stimulation engages the caspase 8 signaling pathway.
Fig. 9.

Nociceptive stimulation increased caspase 8 mRNA and protein expression
A) Caspase 8 mRNA was unaltered by SCI or SCI+STIM in the acute time points. At 7 days and in the ventral spinal cord at 28 days, caspase 8 mRNA was increased in both SCI and SCI+STIM groups. At 28 days, in the dorsal spinal cord of SCI+STIM subjects, caspase 8 mRNA levels was significantly increased relative to sham controls and SCI. B) Western blot analyses showed that noxious stimulation significantly increased caspase 8 protein levels at 24 hours. At the later time points, dorsal spinal cord levels of caspase 8 were significantly up-regulated in SCI and SCI+STIM subjects compared to shams, while ventral levels were unchanged. [* indicates p < .05 relative to the sham control group, and # indicates p < .05 of the SCI+STIM group relative to the SCI group]. (C) Examples of representative western blot images for caspase 8. β-tubulin served as a loading control protein.
Noxious stimulation and SCI increase the expression of activated caspase 3
To further investigate whether nociceptive stimulation after SCI engages an apoptotic signaling cascade, we next assessed caspase 3, a major executor of apoptosis that is a downstream target of caspase 8. We measured the protein expression of the processed forms of caspase 3, consisting of large (17 kDa) and small (12 kDa) subunits, and the unprocessed (inactive) precursor (32 kDa). As depicted in Fig. 10A, there were no differences in the expression pattern of the inactive caspase 3-32 kDa at 1 hour. However at 24 hours, there was a significant increase in its expression in SCI+STIM subjects in both dorsal and ventral spinal cord compared to sham operated control (p < .05). Interestingly, at 7 days, SCI subjects had elevated levels of caspase 3-32 kDa compared to sham control (p < .05). The 12 kDa subunit of caspase 3 was increased only in SCI+STIM subjects at 1 hour and in both dorsal and ventral spinal cord at 24 hours and 7 days (p < .05) (Fig. 10C). Levels for SCI alone were not different to sham control, except at 28 days in the dorsal spinal cord (163 ± 19; p < .05). There were no differences in the expression of caspase 3-17 kDa at 1 hour. At 24 hours in the dorsal spinal cord only, noxious stimulation increased its expression levels (SCI+STIM: 345 ± 97%) compared to sham controls (100 ± 11%) (p < .05). At 7 days, both SCI+STIM and SCI levels were enhanced in the dorsal and ventral spinal cord compared to sham controls (p < .05), although no differences existed between the two treatment groups (Fig. 10B). For the active subunits, their expression mirrored the results observed for both TNFα and caspase 8, with noxious stimulation treatment up-regulating expression within the first 24 hours and SCI alone exerting an effect at 7 days. For all three ligands, this overall pattern resembles the shift observed in mechanical reactivity (Fig. 2).
Fig. 10.

Noxious stimulation increased caspase 3 protein expression
A) Uncleaved caspase 3-32 kDa protein was increased in SCI+STIM subjects at 24 hours post treatment compared only to sham control, while at 7 days, only in SCI subjects was caspase 3-32 kDa significantly increased. B) Like the uncleaved caspase 3, the active caspase 3-17 kDa was significantly increased in the dorsal spinal cord of SCI+STIM subjects at 24 hours. At 7 days, its levels were increased in the dorsal and ventral spinal cords of both SCI and SCI+STIM groups. No other changes in caspase 3-17 kDa levels were observed. C) The active caspase 3-12 kDa was significantly increased only in the SCI+STIM group at 1 hour, 24 hours and 7 days, post treatment. By 28 days, only in the dorsal spinal cord of SCI subjects, was caspase 3-12 kDa protein levels increased. [*, indicates significance relative to sham controls and #, indicates significance relative to SCI]. (C) Representative western blot images for the uncleaved and cleaved products of caspase 3 are shown (note: only the 24 hour time point is shown).
Caspase 3 expression co-localizes with morphological signs of apoptosis 24 hours after noxious stimulation
In order to seek converging evidence of apoptotic activity after nociceptive stimulation, we next investigated caspase 3 expression by immunofluorescence. Consistent with the western blot results, at 24 hours post treatment, there was stronger immunolabeling of caspase 3 (green) in SCI+STIM subjects (Fig. 11A and B) compared to SCI only (Fig. 11C) and sham controls (Fig. 11D). This was supported by unbiased stereological estimations (Fig. 11B: iii) which showed that caspase 3 was significantly elevated in the SCI+STIM group (p < .01), compared to the sham controls. Caspase 3 expression was also significantly elevated in SCI only subjects (p < .05) compared to sham controls. Detailed analyses showed that caspase 3 was intensely expressed in the penumbra of the injury-induced cavities. This observation is consistent with previous work by Crowe et al. [17] suggesting that following SCI, necrotic cell death that produces large cavities may precede apoptosis. Caspase 3 was co-labeled mostly with NeuN (Fig. 11B, C), but also with OX-42 (Fig. 11A; top panel), primarily in the gray matter, indicating that caspase 3 is expressed in spinal neurons and microglia. There was no co-localization of caspase 3 with GFAP in the gray matter around the lesion (Fig. 11A; bottom panel) although minimal co-localization was observed in the ventral white matter (not shown). As depicted in Fig. 11B: ii, NeuN-immunopositive neurons were observed exhibiting morphological characteristics of three distinct stages of apoptosis. (1) Neurons exhibited nuclear and cytoplasmic blebbing as well as a large, bulging nuclei characteristic of early onset of apoptosis. (2) Other neurons appeared further along in the apoptotic process exhibiting nuclear condensation and lacking a distinctive cytoplasmic structure. (3) Some neurons contained fragmented NeuN-immunopositive nuclei, indicative of late-stage apoptosis. We used unbiased stereology to estimate the number of neurons undergoing these distinct stages of apoptosis and expressing caspase 3. As illustrated in Fig. 11B: iii, neurons in the late stage of apoptosis (NeuN-Fragment) were significantly more abundant in SCI+STIM subjects, compared to SCI only (p < .05) and sham control animals (p < .01). Furthermore, cells in this category represented the greatest proportion of caspase 3 expression in neurons. The number of cells in the early onset (NeuN-Bulge) and intermediate (NeuN-Condense) stages of apoptosis was smaller than those undergoing late stage apoptosis. Despite the small number of neurons in these categories (focused on in inset), there was significantly more NeuN/caspase 3 co-expression in SCI and/or SCI+STIM groups compared to sham. Specifically, caspase 3 co-expressed with NeuN-bulge was significantly greater in SCI only subjects compared to shams (p < .01) and SCI+STIM (p < .05); and caspase 3 expression in neurons in the mid-apoptosis stage (NeuN-Condense), was greater in both SCI and SCI+STIM groups than in sham (p < .01). The expression of caspase 3 in NeuN positive cells with healthy normal morphology (including shams) was negligible (Fig. 11B: iii and D) and total NeuN expression was comparable in all three groups. These findings suggest that the deleterious effects observed following nociceptive input to the injured spinal cord may be driven in part by apoptosis.
Dorsal versus ventral spinal cord
We sought to determine the spatial regulation of TNFα targets, by assessing dorsal versus ventral expressions at 24 hours, 7 and 28 days. Although, similar patterns of changes in gene expression were observed dorsally and ventrally, in general increases in mRNA and protein expression in the dorsal spinal cord preceded those ventrally. This was true for TNFα mRNA in SCI+STIM, where levels were significantly elevated compared to both shams and SCI at 24 hours and for TNFRs mRNA in SCI and SCI+STIM subjects. Also at 24 hours, c-jun and caspase 3-17 kDa protein levels in SCI+STIM subjects were significantly increased in the dorsal spinal cord, but not ventrally. Similar changes were observed for TNFα protein level at 7 days and caspase 8 at 7 and 28. Only pERK1/2 at 28 days had elevated levels in the ventral spinal cord, but not dorsally in SCI and SCI+STIM subjects.
Discussion
This study investigated the effect of peripheral noxious input after SCI on enhanced mechanical reactivity, a potential behavioral correlate of central sensitization [72,89]. Our results concur with previous reports demonstrating threshold alterations following a moderate SCI. Sham subjects responded minimally to von Frey filaments [14]. Compared to shams, SCI rats showed increased sensitivity to von Frey hair. This effect began to emerge 7 days after injury and was statistically significant at 14 days [19,58,70]. A low thoracic contusion injury has been shown to elicit mechanical allodynia caudal to the injury [43]. Because the damage may have extended to the lumbar region, the present results could reflect converging at-level and below-level mechanical hypersensitivity.
We investigated a potential interaction between the behavioral changes induced by stimulation following SCI and expression profiles of key components of the TNFα signaling pathway at the lesion site (summarized in Table 1). Overall, the results suggested that tactile sensitivity is associated with increased TNFα signaling. This finding agrees with the presumed role TNFα plays in a variety of pathologies ranging from inflammatory and peripheral injury-induced pain [74,94] to CNS neurological disorders, such as Alzheimer’s disease [64,85] and Parkinson’s disease [65]. The involvement of TNFα in inflammatory pain is particularly relevant to a role in SCI-induced pain. Both processes require central-sensitization-like mechanisms.
Like TNFα, TNFR mRNAs were robustly increased by SCI. This is consistent with trauma-induced increases observed elsewhere in the nervous system, ranging from the dorsal root ganglion to the brain [7,73]. TNFα activates NFκB, ERK, JNK and caspase 8 pathways thereby initiating inflammatory and apoptotic processes. NFκB and ERK were decreased, at least acutely, whereas caspase 8, caspase 3 and c-jun were increased. These divergent results may depend on which TNFR is engaged, even though both subtypes are increased by injury. While there are some conflicting reports, most studies agree with proposals made by Chen and Goeddel [10], that soluble TNFα stimulates TNFR1 and by Grell et al. [40] that TNFR2 is activated primarily by membrane-bound TNFα. How each receptor affects downstream signal pathways also remains controversial. The association of both receptors with TNFR-associated protein (TRAF)-2 molecule renders them capable of activating JNK, ERK and NFκB [9,60,80]. However, the clearest distinction in the actions of TNFR1 and TNFR2 is in relation to the apoptotic pathway, which is activated by TNFR1 interacting with TRADD (tumor necrosis factor receptor type 1-associated death domain), an association not found with TNFR2 [16,46,77].
The development of mechanical allodynia was not associated with increased expression of NFκB, and in this way our results differ from those obtained in other pain models. NFκB is implicated in the development of inflammatory [56] and neuropathic pain resulting from nerve injury [83], and although there is currently no report directly linking NFκB to pain after SCI its involvement has been inferred. Increased levels of NFκB [6,90] and a potential role in SCI-induced inflammatory processes [8] have been reported. It is unclear why the present results failed to provide a link between NFκB and SCI pain. Because cleaved (active) NFκB translocates from the cytoplasm to the nucleus, the possibility remains that our protein assessment methods were insufficiently sensitive to detect nuclear expression of NFκB. However, these methods have been successfully tested for preservation of nuclear protein [49,79] and similar changes were observed in both mRNA and protein levels.
NFκB inhibits caspase activity and offers protection from apoptotic cell death [5,86,87]. The overwhelming increase in caspase 8 and 3 by SCI and/or noxious stimulation suggests a diminution of NFκB-mediated suppression of caspase, which is supported by the concurrent decrease in NFκB expression. It is possible that distal to the lesioned epicenter, areas with minimal caspase activity and apoptosis, NFκB levels are not decreased. However, the current observation infers that caspase and NFκB mediate opposite and potentially competing actions; so that an increase in one is accompanied by a decrease in the other. These findings, coupled with previous studies, make clear the complex nature of TNFα signaling and the engagement of caspase versus NFκB pathways after SCI, and suggest the need for further exploration.
Caspase 8, an initiator of TNFα-induced apoptosis and the active component of caspase 3, a downstream executor of apoptosis, most closely mirrored TNFα levels. These changes were consistent with previous demonstrations of increased caspase expression or activity after SCI [15,55,81]. That caspase and not NFκB is increased after SCI and noxious stimulation suggests that mechanical allodynia after SCI associates with apoptosis. Work by Joseph and Levine [53] suggests that apoptotic genes might contribute to acute pain hypersensitivity. While the role of apoptosis in the development of chronic neuropathic pain has not received much attention, there are several interesting commonalities between both processes. Neuropathic pain and apoptotic death of glia and neurons are critical consequences of SCI [4,17,59]. Our data suggest both processes are co-occurring, and may depend on TNFα signaling mechanism. What is not clear is whether apoptosis plays a causal role in SCI- and noxious stimulation-induced mechanical sensitivity. SCI pain and apoptosis could reflect separate mechanisms that share TNFα as a root cause. For example, TNFα can directly drive neuronal or glial activity through an unexpected mechanism: rapid trafficking of glutamate receptors into the plasma membrane [4,29,82].
This non-canonical mechanism of TNFα signaling provides mechanistic links between apoptosis and pain and may account for the current discovery of an NFκB-independent mechanism for SCI pain. TNFα directly enhances neuronal excitability [11] and increases trafficking of Ca2+-permeable AMPA receptors (CP-AMPARs) to the plasma membrane, an effect that can ultimately lead to cell death [29,92]. CP-AMPARs and intracellular calcium are also implicated in a variety of processes associated with spinal nociception [33,35,52]. After SCI, persistent pain behavior [27] and TNFα-induced apoptosis [57] are linked to nitric oxide mechanisms. Treatments that produce neuropathic pain enhance the expression of apoptotic genes, specifically in pain processing regions such as the dorsal root ganglia [76] and superficial dorsal horn [61]. Finally, apoptosis can induce neuronal sensitization, while promoting a loss of inhibitory neurons [69,75]. A selective loss of GABAergic neurons following SCI has been shown to promote pain hypersensitivity [24,93], presumably by producing disinhibition of excitatory transmission. Consequently, the resulting glutamate excitotoxicity can lead to apoptotic or necrotic cell death [54,63]. These observations imply neuropathic pain and apoptosis share overlapping cellular mechanisms, a scenario that is likely to exist after SCI.
ERK [21,91,96] and JNK/c-Jun [34,96] pathways are implicated in spinal pain processing. The decrease in ERK expression at earlier time points, an effect that could result from neuronal death [91], suggests ERK had a limited role in the present paradigm. In contrast, the increase in c-jun levels and decreased NFκB levels are consistent with apoptotic mechanisms. JNK directly induces apoptosis [12,21], while NFκB suppresses TNFα-induced apoptosis [5,86,87] and inhibits JNK activation [20,84]. A reduction in NFκB will likely unleash the apoptotic process mediated by the caspases and JNK. The interactions involving the caspases, NFκB, JNK and ERK suggest that multiple TNFα signaling pathways contribute to enhanced mechanical reactivity following SCI. The current results suggest that sustained expression of mechanical allodynia induced by peripheral nociceptive input after SCI is associated with caspase-mediated apoptotic pathway. Further work is needed to unravel the nature of this interaction.
Generally, gene changes in the dorsal cord preceded or were more robustly elevated than in the ventral cord. Because the contusion injury is administered dorsally, these changes may reflect a spatial relation to the injury site. Nonetheless, studies have identified plasticity in the dorsal spinal cord as critical to the development of inflammatory or neuropathic pain (e.g. [24,37,61,91]). The current observations are supported by these prior reports. An important observation is the demonstration of converging plasticity across multiple spinal networks. Nociceptive plasticity in the lumbar spinal cord is modified by events occurring at rostral and caudal sites. Spinal neurons are susceptible to various manipulations such as injury and noxious activity (reviewed by [25]). It has been established that SCI profoundly modifies spinal circuitry below the level of injury, primarily due to disruption of brain-mediated descending control. However, for the first time we show that peripheral noxious input caudal to the level of SCI modifies nociceptive plasticity and pain behavior. Stimulation that induces wind-up [68,67] of spinal neurons, when given at or adjacent to the segmental level has been associated with several behavioral pain states (see [44]). In concordance with those reports, we show that noxious stimulation (0.5 Hz) that can induce wind-up exacerbates nociceptive plasticity, even when the stimulation is administered to distal dermatomes.
Conclusion
The highly plastic nature of the spinal cord creates an environment that can support beneficial as well as maladaptive outcomes. Especially after injury, spinal plasticity is often steered towards maladaptive consequences which include chronic pain. This study contributes to this line of work by identifying peripheral noxious stimulation as an important contributor to the development of mechanical hypersensitivity following SCI. It also suggests that the cytokine, TNFα can impact nociceptive processes in an unexpected way; by engaging an alternative TNF-caspase pathway that leads to apoptosis.
Summary statement.
Concomitant peripheral nociceptive stimulation after SCI produces maintained mechanical sensitivity, possibly indicative of persistent pain. Behavioral changes are in parallel with increased TNFα-induced apoptosis.
Acknowledgments
This work was funded by National Institute of Neurological Disorders and Stroke (NS41548, NS069537, NS081606 and DA031197) and National Institute of Child Health and Human Development (HD058412). The authors wish to thank Derek Irish and Michael Preston for assistance with western blot. The authors would like to thank Jacqueline C. Bresnahan and Michael S. Beattie for comments on a prior version of this manuscript.
Footnotes
Conflict of interest statement
The authors report no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. Journal of neurotrauma. 1995;12(1):1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- 2.Baumbauer KM, Hoy KC, Jr, Huie JR, Hughes AJ, Woller SA, Puga DA, Setlow B, Grau JW. Timing in the absence of supraspinal input I: variable, but not fixed, spaced stimulation of the sciatic nerve undermines spinally-mediated instrumental learning. Neuroscience. 2008;155(4):1030–1047. doi: 10.1016/j.neuroscience.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beattie MS, Ferguson AR, Bresnahan JC. AMPA-receptor trafficking and injury-induced cell death. The European journal of neuroscience. 2010;32(2):290–297. doi: 10.1111/j.1460-9568.2010.07343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beattie MS, Hermann GE, Rogers RC, Bresnahan JC. Cell death in models of spinal cord injury. Progress in brain research. 2002;137:37–47. doi: 10.1016/s0079-6123(02)37006-7. [DOI] [PubMed] [Google Scholar]
- 5.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274(5288):782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 6.Bethea JR, Castro M, Keane RW, Lee TT, Dietrich WD, Yezierski RP. Traumatic spinal cord injury induces nuclear factor-kappaB activation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1998;18(9):3251–3260. doi: 10.1523/JNEUROSCI.18-09-03251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Botchkina GI, Meistrell ME, 3rd, Botchkina IL, Tracey KJ. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Molecular medicine. 1997;3(11):765–781. [PMC free article] [PubMed] [Google Scholar]
- 8.Brambilla R, Bracchi-Ricard V, Hu W-H, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor B reduces inflammation and improves functional recovery after spinal cord injury. The Journal of Experimental Medicine. 2005;202(1):145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carpentier I, Declercq W, Malinin NL, Wallach D, Fiers W, Beyaert R. TRAF2 plays a dual role in NF-kappaB-dependent gene activation by mediating the TNF-induced activation of p38 MAPK and IkappaB kinase pathways. FEBS letters. 1998;425(2):195–198. doi: 10.1016/s0014-5793(98)00226-9. [DOI] [PubMed] [Google Scholar]
- 10.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296(5573):1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Pang RP, Shen KF, Zimmermann M, Xin WJ, Li YY, Liu XG. TNF-alpha enhances the currents of voltage gated sodium channels in uninjured dorsal root ganglion neurons following motor nerve injury. Experimental neurology. 2011;227(2):279–286. doi: 10.1016/j.expneurol.2010.11.017. [DOI] [PubMed] [Google Scholar]
- 12.Chen YR, Wang X, Templeton D, Davis RJ, Tan TH. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. Duration of JNK activation may determine cell death and proliferation. The Journal of biological chemistry. 1996;271(50):31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- 13.Choi JI, Svensson CI, Koehrn FJ, Bhuskute A, Sorkin LS. Peripheral inflammation induces tumor necrosis factor dependent AMPA receptor trafficking and Akt phosphorylation in spinal cord in addition to pain behavior. Pain. 2010;149(2):243–253. doi: 10.1016/j.pain.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christensen MD, Everhart AW, Pickelman JT, Hulsebosch CE. Mechanical and thermal allodynia in chronic central pain following spinal cord injury. Pain. 1996;68(1):97–107. doi: 10.1016/S0304-3959(96)03224-1. [DOI] [PubMed] [Google Scholar]
- 15.Citron BA, Arnold PM, Sebastian C, Qin F, Malladi S, Ameenuddin S, Landis ME, Festoff BW. Rapid upregulation of caspase-3 in rat spinal cord after injury: mRNA, protein, and cellular localization correlates with apoptotic cell death. Experimental neurology. 2000;166(2):213–226. doi: 10.1006/exnr.2000.7523. [DOI] [PubMed] [Google Scholar]
- 16.Cleveland JL, Ihle JN. Contenders in FasL/TNF death signaling. Cell. 1995;81(4):479–482. doi: 10.1016/0092-8674(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 17.Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nature medicine. 1997;3(1):73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- 18.Crown ED, Ferguson AR, Joynes RL, Grau JW. Instrumental learning within the spinal cord: IV. Induction and retention of the behavioral deficit observed after noncontingent shock. Behav Neurosci. 2002;116(6):1032–1051. doi: 10.1037//0735-7044.116.6.1032. [DOI] [PubMed] [Google Scholar]
- 19.Crown ED, Ye Z, Johnson KM, Xu GY, McAdoo DJ, Hulsebosch CE. Increases in the activated forms of ERK 1/2, p38 MAPK, and CREB are correlated with the expression of at-level mechanical allodynia following spinal cord injury. Experimental neurology. 2006;199(2):397–407. doi: 10.1016/j.expneurol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 20.De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature. 2001;414(6861):308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 21.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 2003;115(1):61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 22.Detloff MR, Fisher LC, McGaughy V, Longbrake EE, Popovich PG, Basso DM. Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Experimental neurology. 2008;212(2):337–347. doi: 10.1016/j.expneurol.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dragunow M, Young D, Hughes P, MacGibbon G, Lawlor P, Singleton K, Sirimanne E, Beilharz E, Gluckman P. Is c-Jun involved in nerve cell death following status epilepticus and hypoxic-ischaemic brain injury? Brain research Molecular brain research. 1993;18(4):347–352. doi: 10.1016/0169-328x(93)90101-t. [DOI] [PubMed] [Google Scholar]
- 24.Drew GM, Siddall PJ, Duggan AW. Mechanical allodynia following contusion injury of the rat spinal cord is associated with loss of GABAergic inhibition in the dorsal horn. Pain. 2004;109(3):379–388. doi: 10.1016/j.pain.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 25.Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends in neurosciences. 1992;15(3):96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- 26.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. The Journal of cell biology. 1994;127(6 Pt 1):1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fairbanks CA, Schreiber KL, Brewer KL, Yu CG, Stone LS, Kitto KF, Nguyen HO, Grocholski BM, Shoeman DW, Kehl LJ, Regunathan S, Reis DJ, Yezierski RP, Wilcox GL. Agmatine reverses pain induced by inflammation, neuropathy, and spinal cord injury. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(19):10584–10589. doi: 10.1073/pnas.97.19.10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Felix ER, Cruz-Almeida Y, Widerstrom-Noga EG. Chronic pain after spinal cord injury: what characteristics make some pains more disturbing than others? Journal of rehabilitation research and development. 2007;44(5):703–715. doi: 10.1682/jrrd.2006.12.0162. [DOI] [PubMed] [Google Scholar]
- 29.Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, Bresnahan JC, Beattie MS. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28(44):11391–11400. doi: 10.1523/JNEUROSCI.3708-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferguson AR, Crown ED, Grau JW. Nociceptive plasticity inhibits adaptive learning in the spinal cord. Neuroscience. 2006;141(1):421–431. doi: 10.1016/j.neuroscience.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 31.Ferguson AR, Huie JR, Crown ED, Baumbauer KM, Hook MA, Garraway SM, Lee KH, Hoy KC, Grau JW. Maladaptive spinal plasticity opposes spinal learning and recovery in spinal cord injury. Frontiers in physiology. 2012;3:399. doi: 10.3389/fphys.2012.00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22(7):RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gangadharan V, Wang R, Ulzhofer B, Luo C, Bardoni R, Bali KK, Agarwal N, Tegeder I, Hildebrandt U, Nagy GG, Todd AJ, Ghirri A, Haussler A, Sprengel R, Seeburg PH, MacDermott AB, Lewin GR, Kuner R. Peripheral calcium-permeable AMPA receptors regulate chronic inflammatory pain in mice. The Journal of clinical investigation. 2011;121(4):1608–1623. doi: 10.1172/JCI44911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao YJ, Xu ZZ, Liu YC, Wen YR, Decosterd I, Ji RR. The c-Jun N-terminal kinase 1 (JNK1) in spinal astrocytes is required for the maintenance of bilateral mechanical allodynia under a persistent inflammatory pain condition. Pain. 2010;148(2):309–319. doi: 10.1016/j.pain.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garraway SM, Petruska JC, Mendell LM. BDNF sensitizes the response of lamina II neurons to high threshold primary afferent inputs. The European journal of neuroscience. 2003;18(9):2467–2476. doi: 10.1046/j.1460-9568.2003.02982.x. [DOI] [PubMed] [Google Scholar]
- 36.Garraway SM, Turtle JD, Huie JR, Lee KH, Hook MA, Woller SA, Grau JW. Intermittent noxious stimulation following spinal cord contusion injury impairs locomotor recovery and reduces spinal brain-derived neurotrophic factor-tropomyosin-receptor kinase signaling in adult rats. Neuroscience. 2011;199:86–102. doi: 10.1016/j.neuroscience.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garraway SM, Xu Q, Inturrisi CE. siRNA-mediated knockdown of the NR1 subunit gene of the NMDA receptor attenuates formalin-induced pain behaviors in adult rats. The journal of pain : official journal of the American Pain Society. 2009;10(4):380–390. doi: 10.1016/j.jpain.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grau JW, Huie JR, Garraway SM, Hook MA, Crown ED, Baumbauer KM, Lee KH, Hoy KC, Ferguson AR. Impact of behavioral control on the processing of nociceptive stimulation. Frontiers in physiology. 2012;3:262. doi: 10.3389/fphys.2012.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grau JW, Washburn SN, Hook MA, Ferguson AR, Crown ED, Garcia G, Bolding KA, Miranda RC. Uncontrollable stimulation undermines recovery after spinal cord injury. Journal of neurotrauma. 2004;21(12):1795–1817. doi: 10.1089/neu.2004.21.1795. [DOI] [PubMed] [Google Scholar]
- 40.Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83(5):793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 41.Gruner JA. A monitored contusion model of spinal cord injury in the rat. Journal of neurotrauma. 1992;9(2):123–126. doi: 10.1089/neu.1992.9.123. discussion 126–128. [DOI] [PubMed] [Google Scholar]
- 42.Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26(16):4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hains BC, Yucra JA, Hulsebosch CE. Reduction of pathological and behavioral deficits following spinal cord contusion injury with the selective cyclooxygenase-2 inhibitor NS-398. Journal of neurotrauma. 2001;18(4):409–423. doi: 10.1089/089771501750170994. [DOI] [PubMed] [Google Scholar]
- 44.Herrero JF, Laird JM, Lopez-Garcia JA. Wind-up of spinal cord neurones and pain sensation: much ado about something? Progress in neurobiology. 2000;61(2):169–203. doi: 10.1016/s0301-0082(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 45.Hook MA, Huie JR, Grau JW. Peripheral inflammation undermines the plasticity of the isolated spinal cord. Behav Neurosci. 2008;122(1):233–249. doi: 10.1037/0735-7044.122.1.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84(2):299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 47.Huie JR, Baumbauer KM, Lee KH, Bresnahan JC, Beattie MS, Ferguson AR, Grau JW. Glial tumor necrosis factor alpha (TNFalpha) generates metaplastic inhibition of spinal learning. PloS one. 2012;7(6):e39751. doi: 10.1371/journal.pone.0039751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huie JR, Garraway SM, Baumbauer KM, Hoy KC, Jr, Beas BS, Montgomery KS, Bizon JL, Grau JW. Brain-derived neurotrophic factor promotes adaptive plasticity within the spinal cord and mediates the beneficial effects of controllable stimulation. Neuroscience. 2012;200:74–90. doi: 10.1016/j.neuroscience.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hummon AB, Lim SR, Difilippantonio MJ, Ried T. Isolation and solubilization of proteins after TRIzol extraction of RNA and DNA from patient material following prolonged storage. BioTechniques. 2007;42(4):467–470. 472. doi: 10.2144/000112401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jenkins R, Hunt SP. Long-term increase in the levels of c-jun mRNA and jun protein-like immunoreactivity in motor and sensory neurons following axon damage. Neuroscience letters. 1991;129(1):107–110. doi: 10.1016/0304-3940(91)90731-8. [DOI] [PubMed] [Google Scholar]
- 51.Jenkins R, McMahon SB, Bond AB, Hunt SP. Expression of c-Jun as a response to dorsal root and peripheral nerve section in damaged and adjacent intact primary sensory neurons in the rat. The European journal of neuroscience. 1993;5(6):751–759. doi: 10.1111/j.1460-9568.1993.tb00539.x. [DOI] [PubMed] [Google Scholar]
- 52.Jones TL, Sorkin LS. Calcium-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/kainate receptors mediate development, but not maintenance, of secondary allodynia evoked by first-degree burn in the rat. The Journal of pharmacology and experimental therapeutics. 2004;310(1):223–229. doi: 10.1124/jpet.103.064741. [DOI] [PubMed] [Google Scholar]
- 53.Joseph EK, Levine JD. Caspase signalling in neuropathic and inflammatory pain in the rat. The European journal of neuroscience. 2004;20(11):2896–2902. doi: 10.1111/j.1460-9568.2004.03750.x. [DOI] [PubMed] [Google Scholar]
- 54.Kim GW, Copin JC, Kawase M, Chen SF, Sato S, Gobbel GT, Chan PH. Excitotoxicity is required for induction of oxidative stress and apoptosis in mouse striatum by the mitochondrial toxin, 3-nitropropionic acid. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2000;20(1):119–129. doi: 10.1097/00004647-200001000-00016. [DOI] [PubMed] [Google Scholar]
- 55.Knoblach SM, Huang X, VanGelderen J, Calva-Cerqueira D, Faden AI. Selective caspase activation may contribute to neurological dysfunction after experimental spinal cord trauma. J Neurosci Res. 2005;80(3):369–380. doi: 10.1002/jnr.20465. [DOI] [PubMed] [Google Scholar]
- 56.Ledeboer A, Gamanos M, Lai W, Martin D, Maier SF, Watkins LR, Quan N. Involvement of spinal cord nuclear factor kappaB activation in rat models of proinflammatory cytokine-mediated pain facilitation. The European journal of neuroscience. 2005;22(8):1977–1986. doi: 10.1111/j.1460-9568.2005.04379.x. [DOI] [PubMed] [Google Scholar]
- 57.Lee YB, Yune TY, Baik SY, Shin YH, Du S, Rhim H, Lee EB, Kim YC, Shin ML, Markelonis GJ, Oh TH. Role of tumor necrosis factor-alpha in neuronal and glial apoptosis after spinal cord injury. Experimental neurology. 2000;166(1):190–195. doi: 10.1006/exnr.2000.7494. [DOI] [PubMed] [Google Scholar]
- 58.Lindsey AE, LoVerso RL, Tovar CA, Hill CE, Beattie MS, Bresnahan JC. An analysis of changes in sensory thresholds to mild tactile and cold stimuli after experimental spinal cord injury in the rat. Neurorehabilitation and neural repair. 2000;14(4):287–300. doi: 10.1177/154596830001400405. [DOI] [PubMed] [Google Scholar]
- 59.Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW. Neuronal and glial apoptosis after traumatic spinal cord injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17(14):5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996;87(3):565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 61.Maione S, Siniscalco D, Galderisi U, de Novellis V, Uliano R, Di Bernardo G, Berrino L, Cascino A, Rossi F. Apoptotic genes expression in the lumbar dorsal horn in a model neuropathic pain in rat. Neuroreport. 2002;13(1):101–106. doi: 10.1097/00001756-200201210-00024. [DOI] [PubMed] [Google Scholar]
- 62.Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel UL. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. The Journal of biological chemistry. 2004;279(31):32869–32881. doi: 10.1074/jbc.M311766200. [DOI] [PubMed] [Google Scholar]
- 63.Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis. Brain research bulletin. 1998;46(4):281–309. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- 64.McAlpine FE, Lee JK, Harms AS, Ruhn KA, Blurton-Jones M, Hong J, Das P, Golde TE, LaFerla FM, Oddo S, Blesch A, Tansey MG. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009;34(1):163–177. doi: 10.1016/j.nbd.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCoy MK, Martinez TN, Ruhn KA, Szymkowski DE, Smith CG, Botterman BR, Tansey KE, Tansey MG. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26(37):9365–9375. doi: 10.1523/JNEUROSCI.1504-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. Journal of neuroinflammation. 2008;5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mendell LM. Physiological properties of unmyelinated fiber projection to the spinal cord. Experimental neurology. 1966;16(3):316–332. doi: 10.1016/0014-4886(66)90068-9. [DOI] [PubMed] [Google Scholar]
- 68.Mendell LM, Wall PD. Responses of Single Dorsal Cord Cells to Peripheral Cutaneous Unmyelinated Fibres. Nature. 1965;206:97–99. doi: 10.1038/206097a0. [DOI] [PubMed] [Google Scholar]
- 69.Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22(15):6724–6731. doi: 10.1523/JNEUROSCI.22-15-06724.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nesic O, Lee J, Johnson KM, Ye Z, Xu GY, Unabia GC, Wood TG, McAdoo DJ, Westlund KN, Hulsebosch CE, Regino Perez-Polo J. Transcriptional profiling of spinal cord injury-induced central neuropathic pain. Journal of neurochemistry. 2005;95(4):998–1014. doi: 10.1111/j.1471-4159.2005.03462.x. [DOI] [PubMed] [Google Scholar]
- 71.Rossignol S, Schwab M, Schwartz M, Fehlings MG. Spinal cord injury: time to move? The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27(44):11782–11792. doi: 10.1523/JNEUROSCI.3444-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sang CN, Gracely RH, Max MB, Bennett GJ. Capsaicin-evoked mechanical allodynia and hyperalgesia cross nerve territories. Evidence for a central mechanism Anesthesiology. 1996;85(3):491–496. doi: 10.1097/00000542-199609000-00007. [DOI] [PubMed] [Google Scholar]
- 73.Schafers M, Sorkin LS, Geis C, Shubayev VI. Spinal nerve ligation induces transient upregulation of tumor necrosis factor receptors 1 and 2 in injured and adjacent uninjured dorsal root ganglia in the rat. Neuroscience letters. 2003;347(3):179–182. doi: 10.1016/s0304-3940(03)00695-5. [DOI] [PubMed] [Google Scholar]
- 74.Schafers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23(7):2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scholz J, Broom DC, Youn DH, Mills CD, Kohno T, Suter MR, Moore KA, Decosterd I, Coggeshall RE, Woolf CJ. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25(32):7317–7323. doi: 10.1523/JNEUROSCI.1526-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sekiguchi M, Sekiguchi Y, Konno S, Kobayashi H, Homma Y, Kikuchi S. Comparison of neuropathic pain and neuronal apoptosis following nerve root or spinal nerve compression. European spine journal : official publication of the European Spine Society, the European Spinal Deformity Society, and the European Section of the Cervical Spine Research Society. 2009;18(12):1978–1985. doi: 10.1007/s00586-009-1064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shu HB, Takeuchi M, Goeddel DV. The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(24):13973–13978. doi: 10.1073/pnas.93.24.13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Siddall PJ, Loeser JD. Pain following spinal cord injury. Spinal cord. 2001;39(2):63–73. doi: 10.1038/sj.sc.3101116. [DOI] [PubMed] [Google Scholar]
- 79.Simoes AE, Pereira DM, Amaral JD, Nunes AF, Gomes SE, Rodrigues PM, Lo AC, D’Hooge R, Steer CJ, Thibodeau SN, Borralho PM, Rodrigues CM. Efficient recovery of proteins from multiple source samples after TRIzol((R)) or TRIzol((R))LS RNA extraction and long-term storage. BMC genomics. 2013;14:181. doi: 10.1186/1471-2164-14-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Song HY, Regnier CH, Kirschning CJ, Goeddel DV, Rothe M. Tumor necrosis factor (TNF)-mediated kinase cascades: bifurcation of nuclear factor-kappaB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(18):9792–9796. doi: 10.1073/pnas.94.18.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Springer JE, Azbill RD, Knapp PE. Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nature medicine. 1999;5(8):943–946. doi: 10.1038/11387. [DOI] [PubMed] [Google Scholar]
- 82.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440(7087):1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 83.Sun T, Song WG, Fu ZJ, Liu ZH, Liu YM, Yao SL. Alleviation of neuropathic pain by intrathecal injection of antisense oligonucleotides to p65 subunit of NF-kappaB. British journal of anaesthesia. 2006;97(4):553–558. doi: 10.1093/bja/ael209. [DOI] [PubMed] [Google Scholar]
- 84.Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, Lin A. Inhibition of JNK activation through NF-kappaB target genes. Nature. 2001;414(6861):313–317. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 85.Tweedie D, Ferguson RA, Fishman K, Frankola KA, Van Praag H, Holloway HW, Luo W, Li Y, Caracciolo L, Russo I, Barlati S, Ray B, Lahiri DK, Bosetti F, Greig NH, Rosi S. Tumor necrosis factor-alpha synthesis inhibitor 3,6′-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. Journal of neuroinflammation. 2012;9:106. doi: 10.1186/1742-2094-9-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274(5288):787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 87.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS. NF-kappa B antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281(5383):1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 88.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. The Anatomical record. 1991;231(4):482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 89.Willis WD. Long-term potentiation in spinothalamic neurons. Brain research reviews. 2002;40(1–3):202–214. doi: 10.1016/s0165-0173(02)00202-3. [DOI] [PubMed] [Google Scholar]
- 90.Xu J, Fan G, Chen S, Wu Y, Xu XM, Hsu CY. Methylprednisolone inhibition of TNF-α expression and NF-kB activation after spinal cord injury in rats. Molecular Brain Research. 1998;59(2):135–142. doi: 10.1016/s0169-328x(98)00142-9. [DOI] [PubMed] [Google Scholar]
- 91.Xu QH, Garraway SM, Weyerbacher AR, Shin SJ, Inturrisi CE. Activation of the Neuronal Extracellular Signal-Regulated Kinase 2 in the Spinal Cord Dorsal Horn Is Required for Complete Freund’s Adjuvant-Induced Pain Hypersensitivity. Journal of Neuroscience. 2008;28(52):14087–14096. doi: 10.1523/JNEUROSCI.2406-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yin HZ, Hsu CI, Yu S, Rao SD, Sorkin LS, Weiss JH. TNF-α triggers rapid membrane insertion of Ca(2+) permeable AMPA receptors into adult motor neurons and enhances their susceptibility to slow excitotoxic injury. Experimental neurology. 2012;238(2):93–102. doi: 10.1016/j.expneurol.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang AL, Hao JX, Seiger A, Xu XJ, Wiesenfeldhallin Z, Grant G, Aldskogius H. Decreased Gaba Immunoreactivity in Spinal-Cord Dorsal Horn Neurons after Transient Spinal-Cord Ischemia in the Rat. Brain research. 1994;656(1):187–190. doi: 10.1016/0006-8993(94)91383-8. [DOI] [PubMed] [Google Scholar]
- 94.Zhang L, Berta T, Xu ZZ, Liu T, Park JY, Ji RR. TNF-alpha contributes to spinal cord synaptic plasticity and inflammatory pain: Distinct role of TNF receptor subtypes 1 and 2. Pain. 2011;152(2):419–427. doi: 10.1016/j.pain.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114(1–2):149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 96.Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: Respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. Journal of Neuroscience. 2006;26(13):3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]