

Abstract
15(S)-Hydroxyeicosatetraenoic acid (15(S)-HETE), the major 15-lipoxygenase 1/2 (15-LO1/2) metabolite of arachidonic acid (AA), induces CD36 expression through xanthine oxidase and NADPH oxidase-dependent ROS production and Syk and Pyk2-dependent STAT1 activation. In line with these observations, 15(S)-HETE also induced foam cell formation involving ROS, Syk, Pyk2 and STAT1-mediated CD36 expression. In addition, peritoneal macrophages from Western diet-fed ApoE−/− mice exhibited elevated levels of xanthine oxidase and NADPH oxidase activities, ROS production, Syk, Pyk2, and STAT1 phosphorylation and CD36 expression compared to those from ApoE−/−:12/15-LO−/− mice and these events correlated with increased lipid deposits, macrophage content and lesion progression in the aortic roots. Human atherosclerotic arteries also showed increased 15-LO1 expression, STAT1 phosphorylation and CD36 levels as compared to normal arteries. Together, these findings suggest that 12/15-LO metabolites of AA, particularly 12/15(S)-HETE might play a crucial role in atherogenesis by enhancing foam cell formation.
INTRODUCTION
Atherosclerosis is a chronic inflammatory disease of blood vessels and is one of the major causes of death and disability in the world (1, 2). It is characterized by lipid-laden foam cell accumulation in the sub-endothelial space as well as calcification (2, 3). Since its discovery that epitopes of oxidized low-density lipoprotein (OxLDL) co-localized with 15-lipoxygenase mRNA and protein and that the atherosclerotic arteries exhibited increased 15-lipoxygenase activity (4–7), a number of studies using genetic and pharmacological approaches have demonstrated that lipoxygeases (LOs), mainly 5- and 15-LOs, play a role in atherogenesis (8–11). In addition, the most appreciated mechanism of the role of LOs in atherogenesis was their apparent involvement in the oxidation of LDL (12, 13). LOs catalyze stereospecific insertion of molecular oxygen into cis-polyunsaturated fatty acids including arachidonic acid (AA) and linoleic acid (LA) resulting in the formation of hydroperoxyeicosatetraenoic acids (HpETEs) and hydroperoxyoctadecadienoic acids (HpODEs) (14–16). HpETEs and HpODEs are nonenzymatically converted to hydroxyeicosatetraenoic acids (HETEs) and hydroxyoctadecadienoic acids (HODEs), respectively. 15-LO1 and 15-LO2 metabolize AA mainly to 15(S)-HpETE (14–17), while the murine ortholog of 15-LO1, 12/15-LO, converts AA to 12/15(S)-HpETE (17) and atherosclerotic arteries upon incubation with AA produced more of 15-HETE compared to normal arteries (18, 19). Furthermore, many cardiovascular disease risk factors such as hypercholesterolemia, diabetes, obesity and smoking all have been shown to be associated with increased expression and/or activity of 12/15-LO (20–23). In addition, HETEs are prooxidants (24) and a convincing body of evidence links oxidant stress to the pathogenesis of a variety of diseases, including cardiovascular diseases, cancer and rheumatoid arthritis (25–28). Thus, while a mounting amount of data point out a pro-atherogenic role for 12/15-LO (8–10), one study showed that this lipoxygenase exerts anti-atherogenic effects (29). Despite its controversial role in atherogenesis, no mechanisms supporting its pro or anti-atherogenic effects were explored.
Since many studies have shown that cardiovascular risk factors such as hypercholesterolemia, diabetes, obesity and smoking increase 15-LO activity (20–23) and atherosclerotic arteries produce 15-HETE (18, 19), a major 15-LO1/2 metabolite of AA, we asked the question whether this eicosanoid has any influence in the pathogenesis of atherogenesis. Towards this end, we have previously reported that 15(S)-HETE by inducing IL-17A expression triggers inflammation (30). In this study, we report that 15(S)-HETE also enhances CD36 expression and foam cell formation. In addition, we show that 15(S)-HETE-induced CD36 expression and foam cell formation require xanthine oxidase and NADPH oxidase-dependent ROS production and Syk and Pyk2-mediated STAT1 activation. Furthermore, peritoneal macrophages from Western diet (WD)-fed ApoE−/− mice exhibited substantially higher levels of xanthine oxidase and NADPH oxidase activities, ROS production, Syk, Pyk2, and STAT1 phosphorylation as well as CD36 expression as compared to those from ApoE−/−:12/15-LO−/− mice. These observations correlated with increased lipid deposits and plaque progression in aortic roots of ApoE−/− mice compared to ApoE−/−:12/15-LO−/− mice in response to WD feeding. Interestingly, human atherosclerotic arteries showed increased 15-LO1 levels, STAT1 phosphorylation and CD36 expression as compared to normal arteries. Thus, these findings provide additional evidence for the role of 12/15-LO in foam cell formation and hence in the pathogenesis of atherogenesis.
MATERIALS AND METHODS
Reagents
5(S)-HETE (34230), 12(S)-HETE (34570), 15(R)-HETE (34710), 15(S)-HETE (34720), anti-CD36 (100011), total cholesterol assay kit (10007640), and triglycerides assay kit (1001030) were obtained from Cayman Chemicals (Ann Arbor, MI). Anti-pJak2 (3771), anti-pPyk2 (3291), anti-pSrc (2101), anti-pSTAT1 (7649), anti-pSTAT3 (9133), anti-pSTAT5 (9351), anti-pSTAT6 (9364) and anti-pSyk (2715) antibodies were purchased from Cell Signaling Technology (Beverly, MA). Anti-CD68 (SC-9139 & SC9154), anti-CREB (SC-58), anti-Mac3 (SC-19991), anti-p38MAPK (SC-538), anti-p47Phox (SC-14015), anti-SR-A1 (SC-20660), anti-SR-B1 (SC-67098), anti-STAT1 (SC464), anti-STAT3 (SC-482), anti-STAT5B (SC-1656), anti-STAT6 (SC981), anti-Syk (SC-573), anti-β-tubulin (SC-9104), and anti-xanthine oxidase (SC-20991) antibodies and normal mouse serum (SC-45051) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Pyk2 antibodies (ab32571), HDL assay kit, and LDL/VLDL cholesterol assay kit (ab65390) were purchased from Abcam (Cambridge, MA). Anti-hSTAT-5A (MAB2174) antibody was obtained from R&D Systems (Minneapolis, MN). Brewer-modified thioglycolate medium (21176) was purchased from BD Biosciences (San Jose, CA). Apocynin (A10809), allopurinol (A8003) and Oil-red-O (234117) were obtained from Sigma-Aldrich Chemicals (St. Louis, MO). Diphenyleneiodonium chloride (BML-CN240) was from Enzo Life Sciences (Farmingdale, NY). AG-490 (658401) was purchased from Calbiochem (Billerica, MA). Alexa Fluor 488-conjugated goat anti-rabbit secondary antibodies (A11034), CM-H2DCFDA [5-(6)-chloromethyl-2',7'-dichlorodihydrofluorescein diacetate] (C6827), Lipofectin transfection reagent (15596018) and TRIzol reagent were obtained from Invitrogen (Grand Island, NY). pGL3 basic vector and Luciferase assay system (E4530) were purchased from Promega (Madison, WI). Apolipoproteins (BT927), Dil-OxLDL (BT-920), and OxLDL (BT-910) were obtained from Biomedical Technologies (Stoughton, MA). Quick-change site-directed mutagenesis kit was purchased from Stratagene (La Jolla, CA). T4 polynucleotide kinase was obtained from New England Biolabs (Ipswich, MA). [γ32P]-ATP (S.A. 3000 Ci/mmol) was from MP Biomedicals (Irvine, CA). [3H]-Cholesterol (S.A. 53 Ci/mmol) was bought from PerkinElmer (Waltham, MA). The enhanced chemiluminescence (ECL) Western blotting detection reagents (RPN2106) were obtained from GE Healthcare. All the phosphorothioate-modified antisense oligonucleotides (ASOs) and primers were synthesized by IDT (Coralville, IA). The phosphorothioate-modified ASOs used in this study are as follows: hControl ASO, 5'-GGGGGUTCTCTGCGTACGGTGCUAGU-3'; hCD36 (NM_000072) ASO, 5'-CCACAGTTCCGGTCACAGCC-3'; hCREB (NM_004379) ASO, 5'-GCUGCTTCCCTGTTCUUCAU-3'; hp47Phox (NM_000265) ASO, 5'-GUUGGGCTCAGGGTCTTCCGUCUC-3'; hPyk2 (NM_173175) ASO, 5'-CCUGUGTCCATAGCCCAGAGUACC-3'; hSTAT1 (NM_007315) ASO, 5'-GGUCUCGTGTTCTCTGUUCU-3'; hSTAT5B (NM_012448) ASO, 5'-GGUGCTCTGCCTTCTUCUGC-3'; hSyk (NM_001174168) ASO, 5'-UUCCCTGTCTTGTCTUUGUC-3'; and hXO (NM_000379) ASO, 5'-GCCUCCTCCCATTCTCTTCACUCG-3'.
Cell culture
THP1 cell cultures were maintained in a humidified 95% air and 5% CO2 atmosphere at 37°C as described previously (30).
ROS detection
Intracellular ROS generation was measured using membrane permeable 5-(6)-chloromethyl-2',7'-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) dye as described previously (30) and expressed as relative fluorescence units (RFU).
RT-PCR
Total cellular RNA was extracted from THP1 cells using TRIzol reagent according to the manufacturer's protocol. Reverse transcription was performed with a high capacity cDNA reverse transcription kit (Applied Biosystems). Complementary DNA (cDNA) was then used as a template for amplification using the following primers: human SR-A1 (NM_002445), forward, 5'-CCTCGTGTTTGCAGTTCTCA-3' and reverse, 5'-CCATGTTGCTCATGTGTTCC-3'; human SR-B1 (NM_001082959), forward, 5'-CTGTGGGTGAGATCATGTGG-3' and reverse, 5'- GCCCTTCCTTTGGAGTAACC-3'; human CD36 (NM_000072), forward, 5'- ACAGATGCAGCCTCATTTCC-3' and reverse, 5'- GCCTTGGATGGAAGAACAAA-3'; human β-actin (NM_001101), forward, 5'-AGCCATGTACGTTGCTAT-3' and reverse, 5'-GATGTCCACGTCACACTTCA-3'. The amplification was performed using Gene AMP PCR system 2400 (Applied Biosystems). The amplified PCR products were separated on 1.5% agarose gels, stained with ethidium bromide and the images were captured using Kodak In Vivo Imaging System.
Western blotting
Western blotting was performed as described previously (30).
NADPH oxidase and XO activities
NADPH oxidase and XO activities were measured as described previously (30).
Transfections
Transfections using the indicated ASO was performed as described previously (30).
CD36 promoter cloning
Using HRMVEC genomic DNA as a template, CD36 promoter fragment from −724 nucleotides (nt) to +83 nt was amplified by polymerase chain reaction using a forward primer, 5'-GGTACCTTTTGGTTGAAGAAATTTAAAGAGTT-3' incorporating a KpnI restriction enzyme site at the 5'-end and a reverse primer, 5'-AGATCTTTCAATCAAATGCTCCAACA-3' incorporating BglII restriction site at the 5'-end. The resulting 0.807 kb PCR product was digested with KpnI and BglII and cloned into KpnI and BglII sites of the pGL3 basic vector (Promega) to yield pGL3-hCD36. The underlined regions are KpnI and BglII sites in both the forward and reverse primers, respectively. Site-directed mutations within the STAT binding element at −107 nt were introduced by using the Quick-Change site-directed mutagenesis kit according to manufacturer's instructions and using the following primers: forward, 5'-CTTTCAATGCATCTAGGAAACAAACCA-3' and reverse, 5'-TGGTTTGTTTCCTAGATGCATTGAAAG-3' to yield pGL3-hCD36m. The boldface letters indicate the mutated bases. The clones were verified by DNA sequencing using pGL3 vector-specific primers.
Luciferase assay
THP1 cells were transfected with pGL3 empty vector or pGL3-hCD36 promoter with and without the indicated mutations using Lipofectamine transfection reagent. After growth arresting in serum-free medium for 12 hrs, cells were treated with and without 0.1 μM 15(S)-HETE for 6 hrs, washed with cold PBS and lysed in 200 μl of lysis buffer. The cell extracts were cleared by centrifugation at 12,000 rpm for 2 min at 4⍛C. The supernatants were assayed for luciferase activity using luciferase assay system (Promega) and a single tube luminometer (TD20/20; Turner Designs, Sunnyvale, CA) and expressed as relative luciferase units (RLU).
Electrophoretic mobility shift assay
Nuclear extracts of THP1 cells with and without appropriate treatments were prepared as described previously (31). The protein content of the nuclear extracts was determined using micro BCA method. [32P]-labeled double-stranded oligonucleotides encompassing STAT-binding element at −107 nt (5'-ATTTTTTTTTTCTTTCAATTTCTCTAGGAAACAAACCACACACTG-3') were used as a probe. Protein-DNA complexes were formed by incubating 5 μg of nuclear extract in a total volume of 20 μl consisting 15 mM HEPES, pH 7.9, 3 mM Tris-HCl, pH 7.9, 60 mM KCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, 4.5 μg of bovine serum albumin, 2 μg of poly (dI-dC), 15% glycerol and 100,000 cpm of [32P]-labeled oligonucleotide probe for 30 min on ice. The protein-DNA complexes were resolved by electrophoresis on a 6% polyacrylamide gel using 1X Tris-glycine-EDTA buffer (25 mM Tris-HCl, pH 8.5, 200 mM glycine and 0.1 mM EDTA). Double-stranded oligonucleotides were labeled with [γ-32P]-ATP using T4 polynucleotide kinase following the supplier's instructions. To perform supershift electrophoretic mobility shift assay, the complete reaction mix was incubated with the indicated antibodies for 1 hr on ice before separating it by electrophoresis. Normal serum was used as a negative control.
Chromatin immunoprecepitation (ChIP) assay
ChIP assay was performed on THP1 cells with and without the indicated treatments or the primary mouse peritoneal macrophages isolated from Apo E−/− and ApoE−/−:12/15-LO−/− mice fed with WD using a kit and following the supplier's protocol (Upstate Biotechnology Inc., Lake Placid, NY). STAT1-DNA complexes were immunoprecipitated using anti-STAT1 antibodies. Preimmune mouse or rabbit serum was used as a negative control. The immunoprecipitated DNA was uncross-linked, subjected to Proteinase K digestion, purified using QIAquick columns (Cat. No. 28104, Qiagen, Valenica, CA), and used as a template for PCR amplification with primers, forward: 5'-GGGGAAACTCAGCAAGTCAG-3' and reverse: 5'-AGTGTCAGATCCCAGTGG-3' that would amplify 228 bp fragment encompassing the STAT-binding site at −107 nt. The resulting PCR products were resolved on 1.8% agarose gels, stained with ethidium bromide and the images were captured using Kodak In Vivo Imaging System.
Isolation of peritoneal macrophages and collection of plasma and aorta
ApoE−/− and ApoE−/−:12/15-LO−/− mice were bred and maintained according to the Institutional Animal Care and Use facility guidelines. All the experiments involving the use of animals were approved by the Animal Care and Use Committee of the University of Tennessee Health Science Center, Memphis, TN. Six weeks old male ApoE−/− and ApoE−/−:12/15-LO−/− mice were fed with WD (protein, 15.2%; carbohydrate, 42.7% and fat, 42%) (Harlan, Madison, WI, Cat. No. TD.88137) for 3 months and the peritoneal macrophages, plasma and aortas were collected as described previously (30).
Oil Red O staining
After fixing with 4% (v/v) paraformaldehyde, the aortic root sections were stained with Oil Red O and counter stained with hematoxylin. The sections were observed under Nikon Eclipse 50i microscope with 4X/0.10 or 10X/0.25 magnification and the images were captured with a Nikon Digital Sight DS-L1 camera.
Dil-OxLDL uptake assay
THP1 cells that were treated with and without the indicated doses of 15(S)-HETE or the indicated HETE (0.1 μM) for 6 hrs or the peritoneal macrophages isolated from ApoE−/− and ApoE−/−:12/15-LO−/− mice that were fed with WD for 12 weeks were incubated with Dil-OxLDL (10 μg/ml) for 6 hrs at 37°C. After incubation with Dil-OxLDL, cells were washed with PBS, re-suspended in PBS and analyzed by FACS calibur flow cytometer (BD Biosciences, San Jose, CA) with an acquired capacity of 10,000 cells. The data were analyzed using cellquest software and Dil-OxLDL uptake is presented as a percentage of the total cells.
Cholesterol efflux assay
THP1 cells and peritoneal macrophages were seeded into 12-well plates at a density of 6 × 105 cells/well. Cells were labeled with [3H]-cholesterol (1 μCi/ml) for 24 hrs followed by extensive washings with PBS. Cells were then equilibrated in serum-free DMEM containing 0.2% fatty acid free-bovine serum albumin (FAF-BSA) for 2 hrs. After equilibration, medium was replaced with fresh DMEM containing 0.2% FAF-BSA and 10 μg/ml of Apolipoprotein A-I and incubation was continued for 4 hrs at 37⍛C. An aliquot of the efflux medium (100 μl) was collected for radioactivity determination. Cells were then rinsed with PBS, dried and isopropanol was added for overnight extraction of cholesterol at room temperature. An aliquot of the extract (100 μl) was collected for radioactivity determination. Cholesterol efflux was determined as % of total cellular radioactivity released into the medium.
Cell surface CD36 expression assay
THP1 cells that were treated with and without 0.1 μM 15(S)-HETE for 6 hrs or the peritoneal macrophages isolated from ApoE−/− and ApoE−/−:12/15-LO−/− mice that were fed with WD for 12 weeks were washed with cold FACS buffer (2% BSA, 0.1% sodium azide in PBS) and incubated with rabbit anti-human CD36 antibody or isotype control serum for 30 min on ice. After repeated washings with FACS buffer, cells were re-suspended in 100 μl FACS buffer containing Alexa Fluor 488-conjugated goat anti-rabbit secondary antibodies for 30 min on ice, washed again with FACS buffer, re-suspended in 200 μl of fixation buffer (1% paraformaldehyde in FACS buffer) and analyzed by FACS.
Foam cell assay
THP1 cells that were treated with and without the indicated doses of 15(S)-HETE or the indicated HETE (0.1 μM) for 6 hrs or the peritoneal macrophages isolated from ApoE−/− and ApoE−/−:12/15-LO−/− mice that were fed with WD for 12 weeks were incubated with OxLDL (10 μg/ml) for 6 hrs at 37⍛C. Cells were then fixed with 4% paraformaldehyde for 30 min, stained with Oil red O for 10 min and counterstained with hematoxylin. Cell staining was observed under a Nikon Eclipse 50i microscope with 40X/0.65 magnification and the images were captured with a Nikon Digital Slight DS-L1 camera. After capturing the images, the Oil red O stain was eluted by incubating the slides with isopropanol for 15 min at room temperature and the optical density was measured at 500 nm in a SpectraMax 190 spectrophotometer (Molecular Devices).
Plasma lipid profile
The concentration of total cholesterol, HDL, LDL and triglycerides in the plasma were measured using kits following the manufacturers' instructions.
Human normal and atherosclerotic artery specimens
Human normal and atherosclerotic artery samples were collected as described previously following the approved IRB protocols (32). The sections were deparaffinixed with xylene and the antigen was unmasked by treating the sections with antigen unmasking solution for 30 min at 99⍛C. The sections after permeabilization in 0.5% Triton-X100 for 15 min and after blocking in normal goat serum were probed with mouse anti-human Mac3 antibodies (1:100) in combination with rabbit anti-human 15-LO1 antibodies (1:100), rabbit anti-human pSTAT1 or rabbit anti-human CD36 antibodies followed by incubation with AlexaFluor 488-conjugated goat anti-mouse and AlexaFluor 568-conjugated goat anti-rabbit secondary antibodies. The sections were observed under Zeiss inverted microscope (Zeiss AxioVision Observer Z1; original magnification X40/NA 0.6) and the fluorescence images were captured by Zeiss AxioCam MRm camera using the microscope operating and image analysis software AxioVision 4.7. 2 (Carl Zeiss Imaging solutions GmbH).
Statistics
All the experiments were repeated three times and the data are presented as Mean ± SD. The treatment effects were analyzed by Student t test, and the p values <0.05 were considered statistically significant. In the case of RT-PCR and Western blotting, histochemistry, immunohistochemistry and immunofluorescence staining, one representative set of data is shown. In the case of animals, each group consists of 6 animals.
RESULTS
15(S)-HETE induces foam cell formation via CD36 expression
Foam cell formation is a crucial event in the pathogenesis of atherogenesis (2, 33). To understand the role of 15-LO1 in atherogenesis, we studied the effects of 15(S)-HETE, a major AA metabolite of 15-LO1/2 and whose production has been reported to be increased in atherosclerotic arteries (17–19), on foam cell formation. 15(S)-HETE stimulated OxLDL uptake and foam cell formation in a dose-dependent manner with maximum effects at 0.1 μM (Figure 1A & B). To determine the specificity, we compared its effects with those of 5(S)-HETE and 12(S)-HETE, the 5-LO and 12-LO metabolites of AA, respectively. While 5(S)-HETE has relatively little effect, 12(S)-HETE induced both OxLDL uptake and foam cell formation to ~70% of the capacity of 15(S)-HETE (Figure 1C & D). 15(R)-HETE, the enantiomer of 15(S)-HETE, although induced OxLDL uptake and foam cell formation, its effects were found to be much lower than those of 15(S)-HETE (Figure 1C & D). To understand the mechanisms of 15(S)-HETE-induced OxLDL uptake and foam cell formation, we examined its effects on scavenger receptor expression. 15(S)-HETE while having no effect on SR-A1 and SR-B1 expression induced the mRNA and protein levels of CD36 in a time-dependent manner with near maximum effects at 4 hrs (Figure 1E & F). In regard to specificity, 15(S)-HETE exhibited more potent effects on CD36 expression than 5(S)-HETE, 12(S)-HETE or 15(R)-HETE (Figure 1G). Furthermore, FACS analysis showed that 15(S)-HETE-induced CD36 expression also led to an increase in its cell surface levels (Figure 1H). Since 15(S)-HETE induced CD36 expression, we examined its role in foam cell formation. ASO-mediated downregulation of CD36 levels attenuated 15(S)-HETE-induced OxLDL uptake and foam cell formation (Figure 1I & J). These results suggest that 15(S)-HETE induces OxLDL uptake and foam cell formation via enhancing CD36 expression.
Figure 1.

15(S)-HETE induces OxLDL uptake and foam cell formation via CD36 expression. A–D. Quiescent THP1 cells were treated with vehicle or the indicated doses of 15(S)-HETE or 0.1 μM of the indicated HETE for 4 hrs and subjected to Dil-OxLDL uptake (panels A & C) and foam cell formation (panels B & D) as described in “Materials and Methods.” E–G. Quiescent THP1 cells were treated with vehicle or 0.1 μM 15(S)-HETE for the indicated time periods or 0.1 μM of the indicated HETE for 4 hrs and either RNA was isolated or protein extracts were prepared. The RNA and protein extracts were analyzed by RT-PCR and Western blotting for the indicated scavenger receptor mRNA (panel E) and protein (panels F & G) levels using their specific primers and antibodies, respectively, and normalized to β-actin mRNA and β-tubulin protein levels, respectively. H. After treatment of quiescent THP1 cells with vehicle or 15(S)-HETE (0.1 μM) for 4 hrs, the cell surface CD36 expression was measured by FACS analysis. A representative histogram is shown in the upper panel and the quantitative analysis is shown as a bar graph in the lower panel. I & J. THP1 cells were transfected with the indicated ASO, quiesced, treated with vehicle or 0.1 μM 15(S)-HETE for 4 hrs and subjected to Dil-OxLDL uptake (panel I) and foam cell formation (panel J) as described in “Materials and Methods”. The bar graphs represent Mean ± SD values of three experiments. *, p<0.01 versus vehicle control or control ASO; **, p<0.01 versus 15(S)-HETE or control ASO + 15(S)-HETE.
15(S)-HETE-induced CD36 expression and foam cell formation require ROS production and Syk & Pyk2 activation
Previously, we have shown that 15(S)-HETE induces ROS production in XO-dependent NADPH oxidase activation in monocytes (30). To explore the mechanisms of CD36 expression, we examined the role of ROS. Pharmacological inhibitors of XO or NADPH oxidase (26–28) or ASO-mediated downregulation of XO or a NADPH oxidase component, p47Phox, levels suppressed 15(S)-HETE-induced CD36 expression (Figure 2A & B). ASO-mediated downregulation of XO or p47Phox levels also blocked 15(S)-HETE-induced OxLDL uptake and foam cell formation (Figure 2C & D). These findings reveal that XO and NADPH oxidase-dependent ROS production is required for 15(S)-HETE-induced CD36 expression, OxLDL uptake and foam cell formation. Our previous results showed that ROS production leads to activation of non-receptor tyrosine kinases (NRTKs) such as Syk and Pyk2 in monocytes in response to 15(S)-HETE (30). In order to test the role of NRTKs, we first studied the time course effects of 15(S)-HETE on tyrosine phosphorylation of various NRTKs. 15(S)-HETE stimulated the tyrosine phosphorylation of Jak2, Pyk2, Src and Syk in a time-dependent manner (Figure 3A). Since there is a lack of correlation between the time courses of Src activation and CD36 expression, we only examined the role of Jak2, Pyk2 and Syk in 15(S)-HETE-induced CD36 expression. ASO-mediated downregulation of XO or p47Phox while having little effect on Jak2 phosphorylation blocked 15(S)-HETE-induced Pyk2 and Syk phosphorylation (Figure 3B). Similarly, pharmacological inhibition Syk (34) or Pyk2 (35) but not Jak2 (36) attenuated 15(S)-HETE-induced CD36 expression (Figure 3C). ASO-mediated downregulation of Syk or Pyk2 levels also inhibited 15(S)-HETE-induced CD36 expression (Figure 3D). In line with these observations, while ASO-mediated downregulation of Syk or Pyk2 levels attenuated 15(S)-HETE-induced OxLDL uptake and foam cell formation, the inhibition of Jak2 had no effect (Figure 3E & F).
Figure 2.

15(S)-HETE-induced CD36 expression and foam cell formation require XO and NADPH oxidase activities. A. Quiescent THP1 cells were treated with vehicle or 0.1 μM 15(S)-HETE in the presence and absence of apocynin (100 μM), DPI (10 μM) or allopurinol (100 μM) for 4 hrs and either RNA was isolated or protein extracts were prepared. RNA and protein extracts were analyzed by RT-PCR and Western blotting for CD36 mRNA (upper panel) and protein (lower panel) levels using its specific primers and antibodies, respectively, and normalized to β-actin mRNA and β-tubulin protein levels, respectively. B. THP1 cells were transfected with the indicated ASO and quiesced before subjecting to the indicated treatments and analyzing for CD36 mRNA (upper panel) and protein (lower panel) levels as described in panel A. The RNA was also analyzed by RT-PCR for β-actin mRNA levels for normalization. The CD36 Western blot was reprobed for p47Phox, XO or β-tubulin levels to show the effects of ASOs on their target and off-target molecules. C & D. THP1 cells were transfected with the indicated ASO, quiesced, treated with vehicle or 0.1 μM 15(S)-HETE for 4 hrs, and subjected to Dil-OxLDL uptake (panel C) and foam cell formation (panel D) as described in “Materials and Methods.” The bar graphs represent Mean ± SD values of three experiments. *, p<0.01 versus vehicle control or control ASO; **, p<0.01 versus 15(S)-HETE or control ASO + 15(S)-HETE.
Figure 3.

Syk and Pyk2 mediate 15(S)-HETE-induced CD36 expression and foam cell formation. A. Quiescent THP1 cells were treated with vehicle or 0.1 μM 15(S)-HETE for the indicated time periods and cell extracts were prepared. Cell extracts consisting of equal amounts of protein from control and each treatment were analyzed by Western blotting for pJak2, pPyk2, pSrc and pSyk levels using their phospho-specific antibodies and normalized to their total levels. B. After transfection with the indicated ASO and quiescence, THP1 cells were treated with vehicle or 0.1 μM 15(S)-HETE for 2 hrs, cell extracts were prepared and analyzed by Western blotting for phospho and total levels of Jak2, Pyk2, and Syk as described in panel A. Cell extracts were also analyzed by Western blotting for p47Phox and XO levels using their specific antibodies to show the effects of the ASOs on their target molecules. C. Quiescent THP1 cells were treated with vehicle or 0.1 μM 15(S)-HETE in the presence and absence of BAY61-3606 (10 μM), PF431396 (5 μM) or AG490 (25 μM), the Syk, Pyk2 and Jak2 inhibitors, respectively, for 4 hrs and either RNA was isolated or protein extracts were prepared. The RNA and protein extracts were analyzed by RT-PCR and Western blotting for CD36 mRNA (upper panels) and protein (lower panels) levels, respectively, followed by normalization to β-actin mRNA and β-tubulin protein levels, respectively. D. THP1 cells were transfected with the indicated ASO and quiesced before subjecting to the indicated treatments and analyzing for CD36 mRNA (upper panel) and protein (lower panel) levels as described in panel C. Cell extracts were also analyzed by Western blotting for Syk, Pyk2 and β-tubulin levels to show the effects of the ASOs on their target and off-target molecules. E. THP1 cells were transfected with the indicated ASO, quiesced, treated with vehicle or 0.1 μM 15(S)-HETE for 4 hrs and subjected to Dil-OxLDL uptake and foam cell formation as described in “Materials and Methods.” F. Quiescent THP1 cells were treated with vehicle or 0.1 μM 15(S)-HETE in the presence and absence of AG490 (25 μM) for 4 hrs and Dil-OxLDL uptake and foam cell formation were measured as described in panel E. The bar graphs represent Mean ± SD values of three experiments. *, p<0.01 versus vehicle control or control ASO; †, p<0.01 versus 15(S)-HETE or control ASO + 15(S)-HETE.
15(S)-HETE-induced CD36 expression and foam cell formation require STAT1 activation
Previously we have shown that Syk and Pyk2 mediate 15(S)-HETE-induced CREB activation in the induction of IL-17A expression (30). In order to identify the transcriptional factors involved in 15(S)-HETE-induced CD36 expression and thereby in OxLDL uptake and foam cell formation, we first tested the role of CREB. ASO-mediated downregulation of CREB levels had no effect on 15(S)-HETE-induced CD36 expression, OxLDL uptake or foam cell formation (Figure 4A & B). Since CREB had no role in 15(S)-HETE-induced CD36 expression, OxLDL uptake and foam cell formation, we next tested the time course effects of 15(S)-HETE on activation of STATs. 15(S)-HETE stimulated tyrosine phosphorylation of STAT1, STAT5 and STAT6 (Figure 4C, upper panel). To identify the subtype of STAT5 activated by 15(S)-HETE, we next performed an immunoprecipitation assay using PY20 antibodies. Immunoprecipitation of proteins from control and various time periods of 15(S)-HETE-treated THP1 cells with PY20 antibodies followed by immunoblotting for STAT5A and STAT5B showed that 15(S)-HETE activates predominantly STAT5B over STAT5A (Figure 4C, lower panel). The effects of 15(S)-HETE on STAT5A and STAT6 phosphorylation are much lower as compared to STAT1 and STAT5B phosphorylation and their time courses of activation do not correlate with the sustained time course of CD36 expression. Therefore, we only examined the role of STAT1 and STAT5B in 15(S)-HETE-induced foam cell formation. Despite the robust activation of both STAT1 and STAT5B by 15(S)-HETE, downregulation of STAT1 but not STAT5B blunted 15(S)-HETE-induced CD36 expression, OxLDL uptake and foam cell formation (Figure 4D & E). To find the upstream signaling of STAT1 activation, we examined the role of ROS and NRTKs. Pharmacological inhibition of XO or NADPH oxidase (26–28) or ASO-mediated downregulation of XO or p47Phox levels suppressed 15(S)-HETE-induced STAT1 phosphorylation (Figure 4F). Similarly, ASO-mediated downregulation of Syk or Pyk2 levels also blocked 15(S)-HETE-induced STAT1 phosphorylation (Figure 4G). To gain more insights into the mechanisms of STAT1 role in 15(S)-HETE-induced CD36 expression, we cloned ~800 bp of its promoter and characterized it for TF binding sites by TRANSFAC analysis. One putative STAT-binding site was identified at −107 nt relative to the transcription start site (Figure 5A). EMSA using STAT-binding element at −107 nt as a [32P]-labeled probe showed that 15(S)-HETE induces nuclear protein binding to STAT-binding site in the CD36 promoter (Figure 5B). Supershift EMSA showed the presence of STAT1 in 15(S)-HETE-induced protein-CD36 promoter DNA complexes (Figure 5B). In addition, ChIP assays showed that STAT1 binds to CD36 promoter in a time-dependent manner in response to 15(S)-HETE (Figure 5C, upper panel). Furthermore, downregulation of XO, p47Phox, Syk or Pyk2 levels attenuated 15(S)-HETE-induced STAT1 binding to CD36 promoter (Figure 5C, middle and bottom panels). Cloning the ~800 bp CD36 promoter region into pGL3 vector and transfection into THP1 cells resulted in a 3-fold increase in its activity upon treatment with 15(S)-HETE and this response was found to be sensitive to inhibition of XO, NADPH oxidase, Syk or Pyk2 (Figure 5D, upper and middle panels). To confirm the role of STAT-binding site in 15(S)-HETE-induced CD36 promoter activity, we next performed site-directed mutagenesis. Site-directed mutagenesis of STAT-binding site at −107 nt attenuated 15(S)-HETE-induced CD36 promoter activity (Figure 5D, bottom panel). To validate the role of 15(S)-HETE in foam cell formation, we also studied its effects in peritoneal macrophages of wild type (WT) mice. 15(S)-HETE stimulated Pyk2, Syk and STAT1 tyrosine phosphorylation and CD36 expression in a time-dependent manner in these cells as well (Figure 6A). In addition, 15(S)-HETE enhanced the capacity of these cells to uptake OxLDL and become foam cells (Figure 6B).
Figure 4.


STAT1 mediates 15(S)-HETE-induced CD36 expression and foam cell formation. A & B. Cells were transfected with control or CREB ASO, quiesced, treated with vehicle or 0.1 μM 15(S)-HETE for 4 hrs and either cell extracts were prepared and analyzed for CD36 expression by Western blotting (panel A) or subjected to Dil-OxLDL uptake (upper panel B) and foam cell formation (lower panel B) as described in “Materials and Methods.” C. Equal amounts of protein from vehicle or various time periods of 0.1 μM 15(S)-HETE-treated THP1 cells were analyzed by Western blotting for phospho and total STAT1, STAT3, STAT5 and STAT6 levels using their phospho-specific and normal antibodies. Cell extracts were also immunoprecipitated with PY20 antibodies and the immunocomplexes were analyzed by Western blotting for STAT5A and STAT5B levels using their specific antibodies. D. THP1 cells were transfected with the indicated ASO, quiesced, treated with vehicle or 0.1 μM 15(S)-HETE for 4 hrs and either RNA was isolated or protein extracts were prepared. The RNA and protein extracts were analyzed by RT-PCR and Western blotting for CD36 mRNA (upper panel) and protein (lower panel) levels using its specific primers and antibodies, respectively, and normalized to β-actin mRNA and β-tubulin protein levels, respectively. The protein extracts were also analyzed by Western blotting for STAT1 and STAT5B levels using their specific antibodies to show the effects of the ASOs on their target molecules. E. All the conditions were the same as in panel D except that after quiescence cells were subjected to OxLDL uptake (upper panel) and foam cell formation (lower panel) as described in “Materials and Methods.” F. Left panel: Quiescent THP1 cells were treated with vehicle or 0.1 μM 15(S)-HETE in the presence and absence of apocynin (100 μM), DPI (10 μM) or allopurinol (100 μM) for 2 hrs and cell extracts were prepared. An equal amount of protein from control and each treatment was analyzed by Western blotting for pSTAT1 using its phospho-specific antibodies and normalized to its total levels. Right panel: All the conditions were the same as in the left panel except that cells were transfected with the indicated ASOs and quiesced before subjecting to treatment with vehicle or 0.1 μM 15(S)-HETE and analyzing for phospho and total STAT1 levels. The protein extracts were also analyzed for p47Phox and XO levels to show the effects of the ASOs on their target molecules. G. All the conditions were the same as in panel F except that cells were transfected with the indicated ASOs and quiesced before subjecting to treatment with vehicle or 0.1 μM 15(S)-HETE and analyzing for phospho and total STAT1 levels. The protein extracts were analyzed for Syk and Pyk2 levels to show the effects of the ASOs on their target molecules. The bar graphs represent Mean ± SD values of three experiments. *, p<0.01 versus vehicle control or control ASO; **, p<0.01 versus 15(S)-HETE or control ASO + 15(S)-HETE.
Figure 5.

STAT1 mediates 15(S)-HETE-induced CD36 promoter activity. A. CD36 promoter encompassing from −724 nt to +83 nt was cloned and sequenced. B. Left panel: Nuclear extracts of control and various time periods of 0.1 μM 15(S)-HETE-treated cells were analyzed by EMSA for STAT binding using STAT binding site at −107 nt as a [32P]-labeled probe. Right panel: Nuclear extracts of control and 2 hrs of 15(S)-HETE (0.1 μM)-treated cells were analyzed for the presence of STAT1 in the STAT-DNA complex by supershift EMSA. C. Upper panel: Control and various time periods of 0.1 μM 15(S)-HETE-treated cells were analyzed for STAT1 binding to CD36 promoter by ChIP assay. Middle & bottom panels: The levels of p47Phox, XO, Syk and Pyk2 were downregulated using their respective ASOs, quiesced, treated with vehicle or 0.1 μM 15(S)-HETE for 2 hrs and analyzed for STAT1 binding to CD36 promoter by ChIP assay. D. Upper panel: The CD36 promoter encompassing from −724 nt to +83 nt was cloned into pGL3 vector, transfected THP1 cells with the empty vector or pGL3-hCD36 promoter plasmid DNA, growth-arrested, treated with vehicle or 0.1 μM 15(S)-HETE for 6 hrs and the luciferase activities were measured. Middle panel: All the conditions were the same as in the upper panel except that after transfection and quiescence, cells were pretreated with Apocyanin (100 μM), DPI (10 μM), Allopurinol (100 μM), PF431396 (5 μM) or Bay61-3606 (10 μM) for 30 min before subjecting to treatment with vehicle or 0.1 μM 15(S)-HETE for 6 hrs and analyzing for luciferase activity. Bottom panel: THP1 cells were transfected with empty vector or pGL3-hCD36 promoter plasmid DNA with and without the mutated STAT-binding site (TTC was mutated to GCA), quiesced, treated with vehicle or 0.1 μM 15(S)-HETE for 6 hrs and luciferase activity was measured. *, p<0.01 versus vehicle control; **, p<0.01 versus 15(S)-HETE.
Figure 6.

15(S)-HETE stimulates Pyk2, Syk and STAT1 activation, CD36 expression, OxLDL uptake and foam cell formation in the peritoneal macrophages of WT mice. A. Mouse peritoneal macrophages were treated with vehicle or 0.1 μM 15(S)-HETE for the indicated time periods and cell extracts were prepared. An equal amount of protein from control and each treatment was analyzed by Western blotting for pPyk2, pSyk, and pSTAT1 levels using their phospho-specific antibodies and normalized to their total levels. The same extracts were also analyzed by Western blotting for CD36 levels and normalized to β-tubulin levels. B. Mouse peritoneal macrophages were treated with vehicle or 0.1 μM 15(S)-HETE for 4 hrs and subjected to OxLDL uptake and foam cell formation as described in Materials and Methods. The bar graphs represent Mean ± SD values of three experiments. *, p<0.01 versus vehicle control.
Western Diet triggers STAT1 activation and its binding to CD36 promoter enhancing CD36 expression, OxLDL uptake and foam cell formation in ApoE−/− but not in ApoE−/−:12/15-LO−/− mice
To validate the in vitro findings in vivo, we used ApoE−/− mice, an experimental model of atherosclerosis, along with ApoE−/−:12/15-LO−/− mice. Peritoneal macrophages isolated from these mice fed with WD for 3 months were examined for ROS production, XO and NADPH oxidase activities, Syk, Pyk2 and STAT1 phosphorylation and CD36 expression. The ROS production, XO and NADPH oxidase activities and Syk, Pyk2 and STAT1 phosphorylation were all found to be substantially higher in macrophages of ApoE−/− mice as compared to those of ApoE−/−:12/15-LO−/− mice (Figure 7A & B). CD36 expression as measured by its mRNA and protein levels was also found to be significantly higher in the peritoneal macrophages of WD-fed ApoE−/− mice as compared to those of ApoE−/−:12/15-LO−/− mice (Figure 7C). Similarly, FACS analysis showed increased cell surface CD36 expression in the peritoneal macrophages of WD-fed ApoE−/− mice as compared to those of ApoE−/−:12/15-LO−/− mice (Figure 7D). No differences were observed in the peritoneal macrophage cell surface CD36 expression between ApoE−/− and ApoE−/−:12/15-LO−/− mice that were fed with CD (Figure 7D). ChIP assay showed increased STAT1 binding to CD36 promoter in the peritoneal macrophages of WD-fed ApoE−/− mice as compared to those of ApoE−/−:12/15-LO−/− mice (Figure 7E). In line with these observations, the peritoneal macrophages from WD-fed ApoE−/− mice exhibited increased capacity of OxLDL uptake and foam cell formation as compared to those of ApoE−/−:12/15-LO−/− mice (Figure 7F & G). Levels of the total cholesterol and cholesterol esters were also found to be 3-fold higher in the peritoneal macrophages of WD-fed ApoE−/− mice as compared to those of ApoE−/−:12/15-LO−/− mice (Figure 7H). The plasma total cholesterol, LDL cholesterol and triglycerides levels were found to be higher in WD-fed ApoE−/− mice as compared to ApoE−/−:12/15-LO−/− mice; whereas the HDL cholesterol levels were observed to be more in ApoE−/−:12/15-LO−/− mice as compared to ApoE−/− mice (Figure 8A). Since the levels of total cholesterol and its esters were higher in the peritoneal macrophages of ApoE−/− mice as compared to those of ApoE−/−:12/15-LO−/− mice, we wanted to find whether 15(S)-HETE also affects cholesterol efflux. It is interesting to note that while 5(S)-HETE had little effect, both 12(S)-HETE and 15(S)-HETE decreased cholesterol efflux substantially (Figure 8B). 15(R)-HETE, the enantiomer of 15(S)-HETE, had no effect on cholesterol efflux (Figure 8B). In agreement with these findings, the peritoneal macrophages from ApoE−/−:12/15-LO−/− mice exhibited 2-fold higher cholesterol efflux as compared to those of ApoE−/− mice (Figure 8C). ATP-binding cassette transporters A1/G1 (ABCA1/G1) have been shown to play an important role in cholesterol efflux (37). Therefore, to find the cause for the decreased cholesterol efflux by 15(S)-HETE, we tested its effects on ABCG1 expression. Surprisingly, 15(S)-HETE decreased ABCG1 levels in a time-dependent manner in THP1 cells and 12/15-LO deficiency led to its enhanced levels in peritoneal macrophages as compared to ApoE−/− mice (Figure 8D & E). In accordance with these findings, the lipid deposits in the aortic roots of WD-fed ApoE−/− mice were found to be substantially higher as compared to those of ApoE−/−:12/15-LO−/− mice (Figure 8F). To extrapolate and validate these findings in humans, we obtained normal and atherosclerotic human artery sections and performed immunofluorescence staining for 15-LO1, STAT1 phosphorylation and CD36 expression. It is interesting to note that 15-LO1 levels, STAT1 phosphorylaion and CD36 expression were all increased in human atherosclerotic arteries as compared to normal arteries (Figure 8G).
Figure 7.

Deletion of 12/15-LO gene blunts OxLDL uptake and foam cell formation capacity of macrophages. A. Peritoneal macrophages from ApoE−/− and ApoE−/−:12/15-LO−/− mice that were fed with WD for 3 months were analyzed for ROS production, XO and NADPH oxidase activities. B. Peritoneal macrophages from ApoE−/− and ApoE−/−:12/15-LO−/− mice that were fed with WD for 3 months were analyzed by Western blotting for pPyk2, pSyk, and pSTAT1 levels using their phospho-specific antibodies and normalized to their total levels. C. CD36 mRNA and protein levels were measured by RT-PCR and Western blotting, respectively, in the peritoneal macrophages of ApoE−/− and ApoE−/−:12/15-LO−/− mice that were fed with WD for 3 months. The RNA was normalized for β-actin transcripts and the CD36 Western blot was reprobed for β-tubulin levels for lane loading control. D. Peritoneal macrophages isolated from CD and WD-fed ApoE−/− and ApoE−/−:12/15-LO−/− mice were analyzed for cell surface CD36 expression by FACS. Representative histograms are shown in the left panels and the quantitative analysis is shown as a bar graph in the right panel. E. Peritoneal macrophages from ApoE−/− and ApoE−/−:12/15-LO−/− mice that were fed with WD for 3 months were analyzed for STAT1 binding to CD36 promoter by ChIP assay. F & G. Peritoneal macrophages from ApoE−/− and ApoE−/−:12/15-LO−/− mice that were fed with WD for 3 months were analyzed for OxLDL uptake and foam cell formation as described in Materials and Methods. H. Peritoneal macrophages from ApoE−/− and ApoE−/−:12/15-LO−/− mice that were fed with WD for 3 months were analyzed for total cholesterol and its esters. *, p<0.01 versus ApoE−/− mice (n=6).
Figure 8.

Deletion of 12/15-LO gene decreases macrophage cholesterol efflux. A. Plasma lipid profiles were measured in ApoE−/− and ApoE−/−:12/15-LO−/− mice fed with WD for 3 months. B. THP1 cells that were incubated with [3H]-cholesterol were treated with vehicle or 0.1 μM of the indicated HETE for 2 hrs and cholesterol efflux was measured. C. Peritoneal macrophages were isolated from ApoE−/− and ApoE−/−:12/15-LO−/− mice fed with WD for 3 months and subjected to cholesterol efflux assay. D. THP1 cells were treated with vehicle or 0.1 μM 15(S)-HETE for the indicated time periods and analyzed by Western blotting for ABCG1 levels followed by normalization to β-tubulin. E. ABCG1 levels were measured in peritoneal macrophages of ApoE−/− and ApoE−/−:12/15-LO−/− mice that were fed with WD for 3 months. F. Atherosclerotic lesion formation was assessed by Oil red O-staining of the cryosections of the aortic roots from 12 wks of WD fed ApoE−/− and ApoE−/−:12/15-LO−/− mice. Left: Images of the representative sections; right: pooled data based on the staining intensities. G. Human normal and atherosclerotic artery sections were analyzed by double immunofluorescence staining for 15-LO1, pSTAT1 or CD36 along with Mac3. *, p<0.01 versus ApoE−/− mice (n=6) or vehicle control versus the indicated HETE.
DISCUSSION
While a substantial body of literature suggests that 15-LO1 and its murine ortholog 12/15-LO play a role in atherogenesis (8–10), a few reports indicated a protective role of this lipoxygenase against atherogenesis (29). However, besides its role in LDL oxidation (12, 13), the molecular mechanisms supporting the pro or anti-atherogenic role of 15-LO1 are lacking. Since, some reports showed that atherosclerotic arteries generate 15-HETE, a major 15-LO1 metabolite of AA, predominantly (18, 19), we asked if this molecule might be involved in the pro or anti-atherogenic role of 15-LO1. To address the role of 15(S)-HETE in atherogenesis, we have previously reported that it enhances neointima formation in response to injury (31, 38) and angiogenesis in response to ischemia (39, 40). Having observed that 15(S)-HETE possesses the capacity to stimulate smooth muscle cell and endothelial cell migration, we further reasoned that it might also promote monocyte/macrophage migration, and thereby, atherogenesis. Towards this end, in our previous study we showed that 15(S)-HETE via activating CREB and enhancing IL-17A expression stimulates monocyte migration (30). In exploring additional mechanisms of its possible role in atherogenesis, in the present study, we show that 15(S)-HETE stimulates OxLDL uptake and foam cell formation. Among the many scavenger receptors, CD36 has been shown to be a major mediator of OxLDL uptake (41, 42). In line with this view, our findings show that 15(S)-HETE induces CD36 expression in monocytes. Furthermore, since downregulation of CD36 expression substantially blocked 15(S)-HETE-induced OxLDL uptake, it is conceivable that this scavenger receptor plays a determinant role in 15(S)-HETE-induced foam cell formation. A substantial body of literature suggests that oxidant stress plays a crucial role in vascular diseases (25–28). Because some reports showed that 15-HpETE possess the pro-oxidant capacity and able to enhance LDL oxidation in the presence of copper (24), we asked the question whether 15(S)-HETE has any role in oxidant production. In this aspect, we have previously shown that 15(S)-HETE induces ROS production via xanthine oxidase-mediated NADPH oxidase activation (30). In this study, we found that xanthine oxidase and NADPH oxidase-dependent ROS production is also required for 15(S)-HETE-induced CD36 expression. In addition, 15(S)-HETE by stimulating ROS production might also enhance LDL oxidation, facilitating its uptake by CD36. Because the previous studies have shown that 15-HETE mediates LDL oxidation and our present results show that it induces CD36 expression, it might be conceivable that 15(S)-HETE by both mediating LDL oxidation and enhancing CD36 expression could exacerbate OxLDL uptake and foam cell formation. Foam cell formation may take place due to increased OxLDL uptake or decreased cholesterol efflux (43). In this aspect, other studies have reported that 12/15-LO exerts an inhibitory effect on the expression of ABCG1 that plays a role in cholesterol efflux (44). Based on this information, it may be speculated that in addition to oxidation of LDL and its uptake, 15(S)-HETE might also prevent cholesterol and phospholipid efflux leading to their retention in the cell. In this context, it was interesting to note that 15(S)-HETE in addition to inducing the expression of CD36 inhibits ABCG1 levels in macrophages. As ABCG1 by binding to cholesterol and phospholipids facilitates their efflux (43), its decreased expression by 15(S)-HETE might also enhance cholesterol retention in macrophages and facilitate foam cell formation.
Since ROS via inhibition of PTPs can influence the tyrosine phosphorylation of proteins, including receptor and non-receptor tyrosine kinases (45, 46), we examined and found that 15(S)-HETE activates Jak2, Pyk2 and Syk. It is interesting to note that inhibition of XO or NADPH oxidase while blocking Syk and Pyk2 phosphorylation had no effect on Jak2 phosphorylation, indicating a lack of a role for Jak2 in 15(S)-HETE-induced CD36 expression, OxLDL uptake and foam cell formation. Indeed, inhibition of Jak2 did not affect CD36 expression, OxLDL uptake or foam cell formation. In contrast, inhibition of either Syk or Pyk2 reduced 15(S)-HETE-induced CD36 expression, OxLDL uptake and foam cell formation. These findings suggest that both Syk and Pyk2 are involved in 15(S)-HETE-induced CD36 expression and foam cell formation. Syk and Pyk2 have been shown to play a role in atherogenesis (47, 48). Recently, we have shown that ROS production and Syk and Pyk2 stimulation are required for 15(S)-HETE-induced CREB activation in the regulation of IL-17A expression (30). Because Syk and Pyk2 are mediating 15(S)-HETE-induced CREB activation in the induction of IL-17A expression in macrophages, we suspected that CREB plays a role in 15(S)-HETE-induced CD36 expression and foam cell formation. However, downregulation of CREB had no effect on 15(S)-HETE-induced CD36 expression and foam cell formation, pointing to a role for other transcriptional factors in these effects. In this context, 15(S)-HETE activated STAT1, STAT5A, STAT5B and STAT6 in THP1 cells. However, its effects on STAT5A and STAT6 are marginal and their time course of stimulation do not correlate with the sustained time course of CD36 expression. Therefore, it may be unlikely that STAT5A or STAT6 play a role in 15(S)-HETE-induced CD36 expression and foam cell formation. Similarly, STAT5B depletion had no effect on 15(S)-HETE-induced CD36 expression, indicating a lack of a role for STAT5B in 15(S)-HETE-induced foam cell formation. Instead, the findings that 15(S)-HETE activated STAT1 robustly and its depletion attenuated 15(S)-HETE-induced CD36 expression suggest that STAT1 mediates 15(S)-HETE-induced foam cell formation. The promoter analysis and ChIP assay further confirm the role of STAT1 in 15(S)-HETE-induced CD36 expression. Since downregulation of STAT1 also blocked foam cell formation, it may be suggested that CD36 might be the major mechanism of foam cell formation by 15(S)-HETE. A recent study showed that deletion of STAT1 leads to reduced CD36 expression and protects mice against atherogenesis in response to high-fat diet feeding; thus suggesting a possible link between STAT1 and CD36 expression (49). In this context, our results for the first time reveal a direct binding of STAT1 to CD36 promoter mediating its activity in monocytes in response to 15(S)-HETE in vitro. Previous studies have also shown that 15-HETE induces CD36 expression via activation of PPARγ (50). Based on these observations as well as the present findings, it may be inferred that 15(S)-HETE can induce CD36 expression and foam cell formation via different mechanisms, including STAT1 and PPARγ, alone or in combination and contribute in atherogenesis.
In evaluating the role of 15(S)-HETE in atherogenesis, we used ApoE−/− mice as an experimental model of atherogenesis (9). We found that deletion of 12/15-LO gene on ApoE−/− background diminishes ROS production, XO and NADPH oxidase activities, Syk, Pyk2 and STAT1 phosphorylation and CD36 expression as compared to their levels in ApoE−/− mice in response to WD feeding. In addition, the CD36 promoter in peritoneal macrophages of ApoE−/−:12/15-LO−/− mice exhibited less binding of STAT1 as compared to those of ApoE−/− mice in response to WD feeding. This observation further confirms the role of STAT1 in 12/15-LO-12/15(S)-HETE-induced CD36 expression in macrophages in response to WD feeding in vivo as well. In this context it should also be noted that WD induces 12/15-LO expression in the endothelium disrupting its permeability (51). In corroboration with these findings, peritoneal macrophages from ApoE−/−:12/15-LO−/− mice took up reduced amounts of OxLDL and exhibited attenuated foam cell formation as compared to those from ApoE−/− mice upon WD feeding. In accordance with these observations, the lipid deposits and the plaque sizes in aortic roots were more in ApoE−/− mice as compared to ApoE−/−:12/15-LO−/− mice and resulted in enhanced lesion formation. Since deletion of 12/15-LO gene diminished these effects leading to attenuation of lesions, it may be viewed that 15-LO via production of 15(S)-HETE contributes to the pathogenesis of atherogenesis. In addition, the findings that human atherosclerotic arteries show more 15-LO1 expression, STAT1 phosphorylation and CD36 expression compared to age matched control subjects further exemplifies the role of 15(S)-HETE in atherogenesis. Together, these results for the first time show that 15(S)-HETE by both its ability to enhance OxLDL uptake via increased expression of CD36 and its capacity to decrease cholesterol efflux via inhibition of ABCG1 could play a major role in atherogenesis. As 15(R)-HETE, the enantiomer of 15(S)-HETE that can be produced by cytochrome P450 monooxygenase or aspirin-modified COX2-dependent conversion of AA (52), also induced CD36 expression, OxLDL uptake and foam cell formation, at least to some extent, besides 12/15-LO-12/15(S)-HETE axis a role for other HETEs such as 5(S)-HETE and 15(R)-HETE in atherogenesis can not be excluded. Indeed, deletion of both 12/15-LO and 5-LO conferred enhanced protection against diet-induced atherogenesis (53).
In summary, the present observations as depicted in Figure 9 show that 12/15-LO-12/15(S)-HETE enhances atherogenesis via XO-dependent ROS production, leading to Syk and Pyk2-mediated STAT1 activation and CD36 expression.
Figure 9.
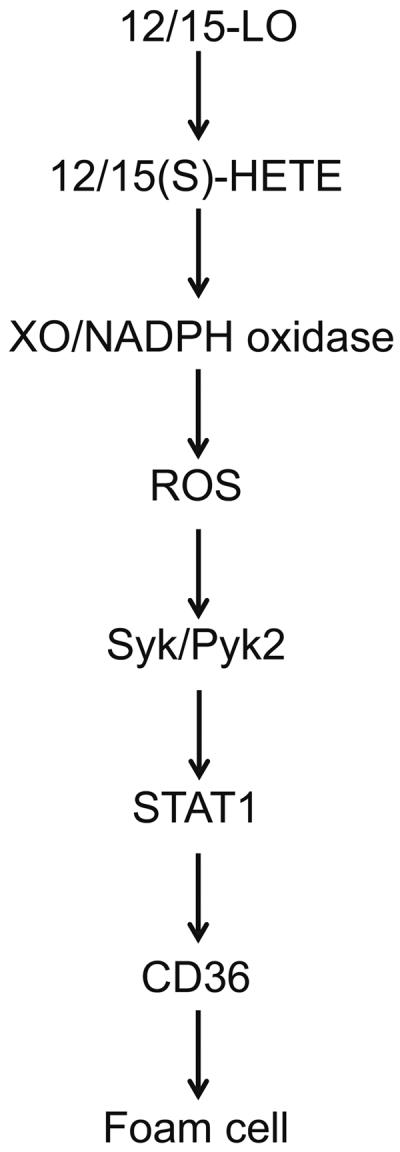
Schematic diagram that shows the explored signaling of 12/15-LO-12/15(S)-HETE on CD36 expression and foam cell formation.
HIGHLIGHTS.
15(S)-HETE induces CD36 expression and foam cell formation.
15(S)-HETE-induced CD36 expression and foam cell formation require xanthine oxidase and NADPH oxidase-dependent reactive oxygen species production leading to Syk and Pyk2-mediated STAT1 activation.
12/15-LO deletion attenuates xanthine oxidase and NADPH oxidase activities, reactive oxygen species production and Syk, Pyk2 and STAT1 phosphorylation in the peritoneal macrophages of ApoE−/− mice fed with Western diet.
12/15-LO deletion diminishes CD36 expression in the peritoneal macrophages of ApoE−/− mice fed with Western diet and lowers their capacity to uptake OxLDL and become foam cell.
ACKNOWLEDGEMENTS
This work was supported by grants HL064165 and HL074860 to GNR and HL098435 to AWO from the National Heart, Lung and Blood Institute of National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat. Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- [2].Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- [3].Demer L, Tintut Y. The roles of lipid oxidation products and receptor activator of nuclear factor-κB signaling in atherosclerotic calcification. Circ. Res. 2011;108:1482–1493. doi: 10.1161/CIRCRESAHA.110.234245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ylä-Herttuala S, Palinski W, Rosenfeld ME, Parthasarathy S, Carew TE, Butler S, Witztum JL, Steinberg D. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J. Clin. Invest. 1989;84:1086–1095. doi: 10.1172/JCI114271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ylä-Herttuala S, Rosenfeld ME, Parthasarathy S, Glass CK, Sigal E, Witztum JL, Steinberg D. Colocalization of 15-lipoxygenase mRNA and protein with epitopes of oxidized low-density lipoprotein in macrophage-rich areas of atherosclerotic lesions. Proc. Natl. Acad. Sci. USA. 1990;87:6959–6963. doi: 10.1073/pnas.87.18.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Simon TC, Makheja AN, Bailey JM. The induced lipoxygenase in atherosclerotic aorta converts linoleic acid to the platelet chemorepellant factor 13-HODE. Thromb. Res. 1989;55:171–178. doi: 10.1016/0049-3848(89)90433-7. [DOI] [PubMed] [Google Scholar]
- [7].Hiltunen T, Luoma J, Nikkari T, Ylä-Herttuala S. Induction of 15-lipoxygenase mRNA and protein in early atherosclerotic lesions. Circulation. 1995;92:3297–3303. doi: 10.1161/01.cir.92.11.3297. [DOI] [PubMed] [Google Scholar]
- [8].Sendobry SM, Cornicelli JA, Welch K, Bocan T, Tait B, Trivedi BK, Colbry N, Dyer RD, Feinmark SJ, Daugherty A. Attenuation of diet-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase inhibitor lacking significant antioxidant properties. Br. J. Pharmacol. 1997;120:1199–1206. doi: 10.1038/sj.bjp.0701007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF, Funk CD. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J. Clin. Invest. 1999;103:1597–1604. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Harats D, Shaish A, George J, Mulkins M, Kurihara H, Levkovitz H, Sigal E. Overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2000;20:2100–2105. doi: 10.1161/01.atv.20.9.2100. [DOI] [PubMed] [Google Scholar]
- [11].Mehrabian M, Allayee H, Wong J, Shi W, Wang XP, Shaposhnik Z, Funk CD, Lusis AJ. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ. Res. 2002;91:120–126. doi: 10.1161/01.res.0000028008.99774.7f. [DOI] [PubMed] [Google Scholar]
- [12].Ylä-Herttuala S, Luoma J, Viita, Hiltunen T, Sisto T, Nikkari T. Transfer of 15-lipoxygenase gene into rabbit iliac arteries results in the appearance of oxidation-specific lipid-protein adducts characteristic of oxidized low density lipoprotein. J. Clin. Invest. 1995;95:2692–2698. doi: 10.1172/JCI117971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cyrus T, Praticò D, Zhao L, Witztum JL, Rader DJ, Rokach J, FitzGerald GA, Funk CD. Absence of 12/15-lipoxygenase expression decreases lipid peroxidation and atherogenesis in apolipoprotein e-deficient mice. Circulation. 2001;103:2277–2282. doi: 10.1161/01.cir.103.18.2277. [DOI] [PubMed] [Google Scholar]
- [14].Brash AR. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J. Biol. Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- [15].Kühn H, Barnett J, Grunberger D, Baecker P, Chow J, Nguyen B, Bursztyn-Pettegrew H, Chan H, Sigal E. Overexpression, purification andcharacterization of human recombinant 15-lipoxygenase. Biochim. Biophys. Acta. 1993;1169:80–89. doi: 10.1016/0005-2760(93)90085-n. [DOI] [PubMed] [Google Scholar]
- [16].Brash AR, Boeglin WE, Chang MS. Discovery of a second 15(S)-lipoxygenase in humans. Proc. Natl. Acad. Sci. USA. 1997;94:6148–6152. doi: 10.1073/pnas.94.12.6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Harmon GS, Lam MT, Glass CK. PPARs and lipid ligands in inflammation and metabolism. Chem. Rev. 2011;111:6321–6340. doi: 10.1021/cr2001355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Henriksson P, Hamberg M, Diczfalusy U. Formation of 15-HETE as a major hydroxy-eicosatetraenoic acid in the atherosclerotic vessel wall. Biochim. Biophys. Acta. 1985;834:272–274. doi: 10.1016/0005-2760(85)90166-3. [DOI] [PubMed] [Google Scholar]
- [19].Simon TC, Makheja AN, Bailey JM. Formation of 15-hydroxyeicosatetraenoic acid (15-HETE) as the predominant eicosanoid in aortas from Watanabe Heritable Hyperlipidemic and cholesterol-fed rabbits. Atherosclerosis. 1989;75:31–38. doi: 10.1016/0021-9150(89)90204-9. [DOI] [PubMed] [Google Scholar]
- [20].Bailey JM, Makheja AN, Lee R, Simon TH. Systemic activation of 15-lipoxygenase in heart, lung, and vascular tissues by hypercholesterolemia: relationship to lipoprotein oxidation and atherogenesis. Atherosclerosis. 1995;113:247–258. doi: 10.1016/0021-9150(94)05452-o. [DOI] [PubMed] [Google Scholar]
- [21].Hatley ME, Srinivasan S, Reilly KB, Bolick DT, Hedrick CC. Increased production of 12/15 lipoxygenase eicosanoids accelerates monocyte/endothelial interactions in diabetic db/db mice. J. Biol. Chem. 2003;278:25369–25375. doi: 10.1074/jbc.M301175200. [DOI] [PubMed] [Google Scholar]
- [22].Xu ZG, Lanting L, Vaziri ND, Li Z, Sepassi L, Rodriguez-Iturbe B, Natarajan R. Upregulation of angiotensin II type 1 receptor, inflammatory mediators, and enzymes of arachidonate metabolism in obese Zucker rat kidney: reversal by angiotensin II type 1 receptor blockade. Circulation. 2005;111:1962–1969. doi: 10.1161/01.CIR.0000161831.07637.63. [DOI] [PubMed] [Google Scholar]
- [23].Yoshida H, Sasaki K, Hirowatari Y, Kurosawa H, Sato N, Furutani N, Tada N. Increased serum iron may contribute to enhanced oxidation of low-density lipoprotein in smokers in part through changes in lipoxygenase and catalase. Clin. Chim. Acta. 2004;345:161–170. doi: 10.1016/j.cccn.2004.03.018. [DOI] [PubMed] [Google Scholar]
- [24].O'Leary VJ, Darley-Usmar VM, Russell LJ, Stone D. Pro-oxidant effects of lipoxygenase-derived peroxides on the copper-initiated oxidation of low-density lipoprotein. Biochem. J. 1992;282:631–634. doi: 10.1042/bj2820631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Leopold JA, Loscalzo J. Oxidative risk for atherothrombotic cardiovascular disease. Free. Radic. Biol. Med. 2009;47:1673–1706. doi: 10.1016/j.freeradbiomed.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J. Physiol. 2004;555:589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug. Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free. Radic. Biol. Med. 2007;43:332–347. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shen J, Herderick E, Cornhill JF, Zsigmond E, Kim HS, Kühn H, Guevara NV, Chan L. Macrophage-mediated 15-lipoxygenase expression protects against atherosclerosis development. J. Clin. Invest. 1996;98:2201–2208. doi: 10.1172/JCI119029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kotla S, Singh NK, Heckle MR, Tigyi GJ, Rao GN. The transcription factor CREB enhances interleukin-17A production and inflammation in a mouse model of atherosclerosis. Sci. Signal. 2013;6:ra83. doi: 10.1126/scisignal.2004214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Potula HS, Wang D, Quyen DV, Singh NK, Kundumani-Sridharan V, Karpurapu M, Park EA, Glasgow WC, Rao GN. Src-dependent STAT-3-mediated expression of monocyte chemoattractant protein-1 is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. J. Biol. Chem. 2009;284:31142–31155. doi: 10.1074/jbc.M109.012526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Funk SD, Yurdagul A, Jr., Albert P, Traylor JG, Jr., Jin L, Chen J, Orr AW. EphA2 activation promotes the endothelial cell inflammatory response: a potential role in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012;32:686–695. doi: 10.1161/ATVBAHA.111.242792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yamamoto N, Takeshita K, Shichijo M, Kokubo T, Sato M, Nakashima K, Ishimori M, Nagai H, Li YF, Yura T, Bacon KB. The orally available spleen tyrosine kinase Inhibitor 2-[7-(3,4-dimethoxyphenyl)-imidazo[1,2-c]pyrimidin-5-ylamino]nicotinamide dihydrochloride (BAY 61-3606) blocks antigen-induced airway inflammation in rodents. J. Pharmacol. Exp. Ther. 2003;306:1174–1181. doi: 10.1124/jpet.103.052316. [DOI] [PubMed] [Google Scholar]
- [35].Buckbinder L, Crawford DT, Qi H, Ke HZ, Olson LM, Long KR, Bonnette PC, Baumann AP, Hambor JE, Grasser WA, 3rd, Pan LC, Owen TA, Luzzio MJ, Hulford CA, Gebhard DF, Paralkar VM, Simmons HA, Kath JC, Roberts WG, Smock SL, Guzman-Perez A, Brown TA, Li M. Proline-rich tyrosine kinase 2 regulates osteoprogenitor cells and bone formation, and offers an anabolic treatment approach for osteoporosis. Proc. Natl. Acad. Sci. USA. 2007;104:10619–24. doi: 10.1073/pnas.0701421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sandberg EM, Ma X, He K, Frank SJ, Ostrov DA, Sayeski PP. Identification of 1,2,3,4,5,6-hexabromocyclohexane as a small molecule inhibitor of jak2 tyrosine kinase autophosphorylation [correction of autophophorylation] J. Med. Chem. 2005;48:2526–2533. doi: 10.1021/jm049470k. [DOI] [PubMed] [Google Scholar]
- [37].Rosenson RS, Brewer HB, Jr., Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, Remaley AT, Rothblat GH, Tall AR, Yvan-Charvet L. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chava KR, Karpurapu M, Wang D, Bhanoori M, Kundumani-Sridharan V, Zhang Q, Ichiki T, Glasgow WC, Rao GN. CREB-mediated IL-6 expression is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2009;29:809–815. doi: 10.1161/ATVBAHA.109.185777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhao T, Wang D, Cheranov SY, Karpurapu M, Chava KR, Kundumani-Sridharan V, Johnson DA, Penn JS, Rao GN. A novel role for activating transcription factor-2 in 15(S)-hydroxyeicosatetraenoic acid-induced angiogenesis. J. Lipid. Res. 2009;50:521–533. doi: 10.1194/jlr.M800388-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Singh NK, Kundumani-Sridharan V, Rao GN. 12/15-Lipoxygenase gene knockout severely impairs ischemia-induced angiogenesis due to lack of Rac1 farnesylation. Blood. 2011;118:5701–5712. doi: 10.1182/blood-2011-04-347468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Podrez EA, Febbraio M, Sheibani N, Schmitt D, Silverstein RL, Hajjar DP, Cohen PA, Frazier WA, Hoff HF, Hazen SL. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J. Clin. Invest. 2000;105:1095–1108. doi: 10.1172/JCI8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, Silverstein RL, Hoff HF, Salomon RG, Hazen SL. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J. Biol. Chem. 2002;277:38517–38523. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- [43].Shashkin P, Dragulev B, Ley K. Macrophage differentiation to foam cells. Curr. Pharm. Des. 2005;11:3061–3072. doi: 10.2174/1381612054865064. [DOI] [PubMed] [Google Scholar]
- [44].Nagelin MH, Srinivasan S, Nadler JL, Hedrick CC. Murine 12/15-lipoxygenase regulates ATP-binding cassette transporter G1 protein degradation through p38-and JNK2-dependent pathways. J. Biol. Chem. 2009;284:31303–31314. doi: 10.1074/jbc.M109.028910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Nakashima I, Takeda K, Kawamoto Y, Okuno Y, Kato M, Suzuki H. Redox control of catalytic activities of membrane-associated protein tyrosine kinases. Arch. Biochem. Biophys. 2005;434:3–10. doi: 10.1016/j.abb.2004.06.016. [DOI] [PubMed] [Google Scholar]
- [46].Tonks NK. Redox redux: Revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- [47].Katsume A, Okigaki M, Matsui A, Che J, Adachi Y, Kishita E, Yamaguchi S, Ikeda K, Ueyama T, Matoba S, Yamada H, Matsubara H. Early inflammatory reactions in atherosclerosis are induced by proline-rich tyrosine kinase/reactive oxygen species-mediated release of tumor necrosis factor-alpha and subsequent activation of the p21Cip1/Ets-1/p300 system. Arterioscler. Thromb. Vasc. Biol. 2011;31:1084–1092. doi: 10.1161/ATVBAHA.110.221804. [DOI] [PubMed] [Google Scholar]
- [48].Hilgendorf I, Eisele S, Remer I, Schmitz J, Zeschky K, Colberg C, Stachon P, Wolf D, Willecke F, Buchner M, Zirlik K, Ortiz-Rodriguez A, Lozhkin A, Hoppe N, von zur Muhlen C, zur Hausen A, Bode C, Zirlik A. The oral spleen tyrosine kinase inhibitor fostamatinib attenuates inflammation and atherogenesis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011;31:1991–1999. doi: 10.1161/ATVBAHA.111.230847. [DOI] [PubMed] [Google Scholar]
- [49].Agrawal S, Febbraio M, Podrez E, Cathcart MK, Stark GR, Chisolm GM. Signal transducer and activator of transcription 1 is required for optimal foam cell formation and atherosclerotic lesion development. Circulation. 2007;115:2939–2947. doi: 10.1161/CIRCULATIONAHA.107.696922. [DOI] [PubMed] [Google Scholar]
- [50].Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400:378–382. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- [51].Chattopadhyay R, Dyukova E, Singh NK, Ohba M, Mobley JA, Rao GN. Vascular Endothelial Tight Junctions and Barrier Function Are Disrupted by 15(S)-Hydroxyeicosatetraenoic Acid Partly via Protein Kinase C{epsilon}-mediated Zona Occludens-1 Phosphorylation at Threonine 770/772. J. Biol. Chem. 2014;289:3148–3163. doi: 10.1074/jbc.M113.528190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Clària J, Lee MH, Serhan CN. Aspirin-triggered lipoxins (15-epi-LX) are generated by the human lung adenocarcinoma cell line (A549)-neutrophil interactions and are potent inhibitors of cell proliferation. Mol. Med. 1996;2:583–596. [PMC free article] [PubMed] [Google Scholar]
- [53].Poeckel D, Zemski Berry KA, Murphy RC, Funk CD. Dual 12/15- and 5-lipoxygenase deficiency in macrophages alters arachidonic acid metabolism and attenuates peritonitis and atherosclerosis in ApoE knock-out mice. J. Biol. Chem. 2009;284:21077–21089. doi: 10.1074/jbc.M109.000901. [DOI] [PMC free article] [PubMed] [Google Scholar]