

Abstract
Malfunction of autophagy, the process that mediates breakdown and recycling of intracellular components in lysosomes, has been linked to a variety of human diseases. As the number of pathologies associated with defective autophagy increases, emphasis has switched from the mere description of the status of autophagy in these conditions to a more mechanistic dissection of the autophagic changes. Understanding the reasons behind the autophagic defect, the immediate consequences of the autophagic compromise and how autophagy changes with the evolution of the disease has become a “must,” especially now that manipulation of autophagy is being considered as a therapeutic strategy. Here, we comment on some of the common themes that have emerged from such detailed analyses of the interplay between autophagy and disease conditions.
Introduction
The impaired ability to maintain organelle and protein homeostasis, or proteostasis, has been implicated as a common cause of numerous human diseases. Chaperones and the two main proteolytic systems that participate in this cellular quality control, the proteasome and the lysosomal system or autophagy, have become attractive targets in the treatment of protein conformation diseases. In the case of autophagy, the main topic of this review, its additional role in maintaining the cellular energetic balance has made autophagic failure relevant for human metabolic disorders, further increasing the interest of the biomedical community in this process. The first pharmacologic modulators of autophagy have made their debut in clinical trials for cancer, myopathies, genetic liver disorders and heart conditions (Database: ClinicalTrials.gov) and searches for genetic polymorphisms in autophagy-related genes (ATG) that could affect predisposition to metabolic diseases or neurodegeneration are under way. As the relevance of autophagy to human disease increases, further consideration as to what the autophagic changes are “telling us” about each disease becomes necessary. In this review, we comment on common themes concerning the relationship between autophagy and disease that we foresee will become important in the future implementation of therapies that target the autophagic process.
Autophagy: the basics
The degradation of intracellular components by lysosomes, or autophagy, occurs in a multi-step fashion that requires recognition of the substrate to be degraded (or cargo), delivery to lysosomes, degradation and recycling of the breakdown products. Depending on the molecular components involved in each of these steps, three types of autophagy have been identified to co-exist in most cell types (Fig. 1). In macroautophagy, cargo is sequestered inside double-membrane vesicles (autophagosomes) for delivery to lysosomes through vesicular fusion (Box 1) [1]. In microautophagy, cytosolic material is internalized for degradation in single-membrane vesicles that form through invaginations in the surface of lysosomes or late endosomes [2]. In contrast, vesicles are not necessary in chaperone-mediated autophagy (CMA) in which substrate proteins are identified by a cytosolic chaperone that delivers them to the surface of lysosomes for internalization through a translocation complex, formed by multimerization of the CMA receptor protein, LAMP-2A (Box 2) [3].
Figure 1. Mammalian autophagic pathways.
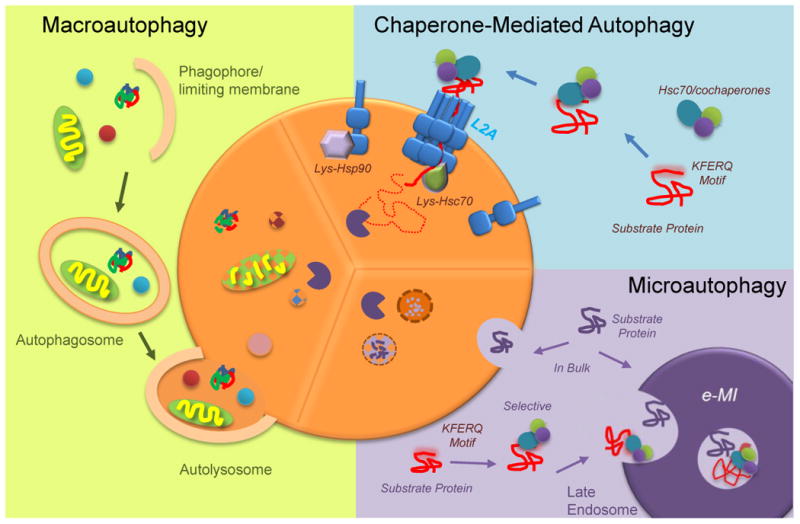
Scheme depicting the main autophagic pathways in mammals and the different steps of macroautophagy and chaperone-mediated autophagy (CMA), which constitute the main emphasis of this review. Hsc70: heat shock cognate protein of 70kDa; hsp90: heat shock protein of 90KDa; L2A: lysosome associated membrane protein type 2A; KFERQ motif: targeting motif; e-MI: endosomal microautophagy.
Box 1. Steps and effectors of macroautophagy.
Macroautophagy occurs through the following sequential steps orchestrated by proteins generally known as autophagy-related proteins or Atg:
Cargo recognition: In selective macroautophagy, the material sequestered for lysosomal degradation is identified by cargo-recognizing proteins. These adaptor molecules simultaneously bind the cargo (often post-translationally modified residues) and a component of the forming autophagosome membrane (LC3).
Initiation of autophagy and formation of the phagophore/isolation membrane: Assembly of autophagy (Atg) proteins for the formation of the autophagosome membrane is initiated by two kinase complexes, ULK1/Atg13/FIP200 and Beclin-1/Vps34/Atg14, and by the arrival of vesicles containing Atg9 to the site of formation. Additional Atgs associate to the kinase core complex, depending on the cellular location where autophagy is initiated, and modulate its activity. Phosphorylation of specific proteins and lipids in these areas mediate the elongation of the phagophore/isolation membrane through conjugation of Atg proteins with other Atgs and with specific lipid moieties. A series of ligases modify proteins undergoing conjugation and link them to the final product. Genetic manipulation of Atg7, the upstream ligase shared by the two conjugation pathways, is commonly used to experimentally abrogate macroautophagy activity.
Sealing of the membrane and autophagosome-lysosome fusion: Different SNARE proteins and small GTPases participate in the sealing of the autophagosome membrane, docking at the lysosome, and fusion. Motor proteins such as dynein and their associated modulators are also utilized for microtubule-dependent trafficking of autophagosomes.
Recycling of essential components: Permeases and reverse transporters in the lysosomal membrane mediate the efflux of degraded components into the cytosol. The products resulting from autophagic degradation can be reutilized in anabolic or catabolic processes.
Box 2. Molecular basis of chaperone-mediated autophagy.
Delivery of substrates to lysosomes by chaperone-mediated autophagy (CMA) is a multistep process. Cytosolic proteins that contain a pentapeptide motif inherent in their amino acid sequence are recognized by the chaperone hsc70 and delivered to the surface of lysosomes. In order to cross the lysosomal membrane, substrate proteins interact with the receptor protein Lysosome-Associated Membrane Protein type 2A (LAMP-2A). Binding of the substrates to the cytosolic tail of this single-spanning membrane protein promotes multimerization of LAMP-2A and the formation of an oligomeric complex required for translocation of proteins into the lysosomal lumen for degradation. A lysosome-resident form of hsp90 stabilizes LAMP-2A during this transition, and a second lysosome chaperone, lys-hsc70, completes substrate internalization.
CMA is maximally upregulated in response to stress (i.e. prolonged lack of nutrients, oxidative stress, and cellular insults resulting in protein damage). However, basal CMA activity is detectable in most cells. Cells compensate for loss of CMA by upregulating macroautophagy or the ubiquitin-proteasome system, but this compensation is insufficient under certain conditions, rendering CMA-incompetent cells more susceptible to stress. Interestingly, this cross-talk among proteolytic systems is multi-directional because cells with compromised macroautophagy exhibit upregulated CMA while both macroautophagy and CMA are upregulated in response to proteasome inhibition.
Most of the regulation of CMA takes place at the lysosome, where rates of substrate uptake are determined by the levels of LAMP-2A at the lysosomal membrane and of lys-hsc70 in the lumen. The ability of LAMP-2A to organize into a translocation complex is another rate-limiting step in CMA-mediated degradation. The signalling mechanisms that integrate the activating stimuli of CMA are a subject of current investigation.
Most connections between autophagy and disease stem from its role in quality control of the proteome and organelles and in the maintenance of the cellular energetic balance. Disruption of autophagy in post-mitotic tissues such as neurons and cardiomyocytes leads to accumulation of altered proteins and organelles, even in absence of any insult [4]. Activation of autophagy is also part of the cellular response to stressors that inflict protein or organelle damage (i.e. oxidative stress, ER stress, genetic mutations) and to challenges that require major adaptive changes in proteome and organelle content to assure cellular survival (i.e. nutrient and growth factor withdrawal, infection or hypoxia) [4].
During nutrient deprivation, autophagy breaks down proteins to replenish the pool of free amino acids and increase cellular ATP levels [5]. The discovery of lipophagy (macroautophagy degradation of lipid droplet triglycerides into free fatty acids [6]) and glycophagy (macro- and microautophagy degradation of glycogen stores into oligosaccharides and glucose [7]) have reinforced the contribution of autophagy to metabolic homeostasis. Lipophagy also exerts a protective function against lipotoxicity, and in fact, upregulation of the transcription factor EB (TFEB), which controls lysosomal biogenesis and activates macroautophagy, prevents diet-induced obesity and the metabolic syndrome [8, 9]. CMA can also modulate cellular energetics through the regulated degradation of enzymes involved in distinct metabolic pathways [10, 11].
Autophagy and disease
Alterations in autophagy occur in systemic diseases such as cancer [12], metabolic dysfunction [6] and vascular instability [13] and in organ-specific pathologies such as neurodegeneration [14], cardiomyopathies and myopathies [15, 16], non-alcoholic fatty liver disease [17] or Crohn's disease [18]. Next, we summarize some emerging themes in the relationship of autophagy and disease.
Where does autophagic function go awry?
The multi-step nature of autophagy makes it vulnerable to failure at different levels (Fig. 1). Identifying the step(s) affected in disease is important because of the distinct downstream consequences and therapeutic implications.
Pathologies affecting each of the steps in macroautophagy have been described (Fig. 2). Reduced ability to recognize cargo can originate from alterations in the degradation tags or in the adaptor molecules that bridge these tags and the autophagic machinery. For example, defective mitochondria turnover by mitophagy in familial Parkinson's disease (PD) has been linked to recessive mutations in parkin and PINK1, proteins responsible for mitochondrial priming for mitophagy [19]. Mutations in the adaptor p62 have been associated with Paget disease and amyotrophic lateral sclerosis (ALS) [20]. Abnormal interactions of pathogenic proteins with autophagy adaptors can also limit cargo recognition. For example, aberrant binding of pathogenic huntingtin to p62 prevents selective recognition of mitochondria, lipid droplets, and even cytosolic aggregates of the mutant protein in neurons from Huntington's disease (HD) patients [21]. Failure to selectively recognize and degrade energy stores also compromises the energetic balance of the affected neurons. Better understanding of cargo recognition effectors may link alterations in this step to other diseases. For example, mishandling of glycogen in cardiac pathologies has been recently attributed to defective glycophagy due to reduced expression of the glycophagy receptor, starch binding domain-containing protein 1 [22].
Figure 2. Autophagy-related human diseases.

Scheme of organ-specific (left) and systemic (right) human diseases in which alterations in autophagy are discussed in this review. Red dot indicates primary autophagy defects and green dot autophagy changes secondary or reactive to disease. Mutations (m), polymorphisms (p) and haplo-insufficiencies (h) described in molecular components of the autophagic pathways in the respective diseases are indicated in blue.
A growing number of diseases present with defects at the level of initiation and autophagosome formation (Fig. 2). In fact, monoallelic loss of the initiation complex component Beclin-1 was the first connection established between autophagy and cancer [23]. Mutations in autophagy genes involved in autophagosomal elongation such as Atg16L1[18] and WIPI4 [24] have been associated with Crohn's disease and neurological disorders, respectively, and polymorphisms in Atg5 with asthma and systemic lupus erythematosus [25, 26]. Further efforts should focus on discriminating whether disease originates from autophagic malfunction or from autophagy-independent functions of these Atg proteins.
Autophagic activity depends on the integration of inputs from multiple signaling pathways. Consequently, pathologies with primary alterations in the signaling molecules that participate in these pathways can result in defective autophagy initiation. Current efforts are focused on analyzing how extracellular signals that activate autophagy are integrated and transduced and how cellular intercommunication affects this process. Among the novel signaling transducers, the recently described regulation of nutrient-induced autophagy by primary cilia [27] raises the possibility that autophagy could be altered in common ciliopathies. Likewise, conditions that affect intercellular communication such as diseases with mutations in connexins may also impact autophagy in light of the recently identified inhibitory effect of connexins on autophagy activation [28].
Later autophagy steps such as autophagosome maturation (by lysosomal fusion), cargo degradation and recycling of breakdown products are also compromised in disease (Fig. 2). Mutations in motor and adaptor proteins that participate in autophagosome trafficking and conditions that disrupt the cytoskeleton network all impact macroautophagy. Defective autophagosome clearance can also be due to primary defects in the proteins that participate in autophagosome-lysosome fusion such as mitofusin 2, whose depletion in cardiomyocytes renders them susceptible to ischemia-reperfusion [29], UVRAG, mutated in human gastric cancer [30], EPG5, implicated in the systemic Vici syndrome [31] or LAMP-2, mutated in Danon disease patients [32]. Pathologies that prevent autophagosome clearance post-lysosomal fusion include most lysosomal storage disorders (LSD) caused by defects in lysosomal enzymes [33] and conditions that interfere with lysosomal acidification or membrane stability [34].
In the case of CMA, pathology can arise by defects in substrate targeting, translocation across the lysosomal membrane or luminal degradation (Fig. 1). Post-translational modifications in the CMA targeting motif in substrate proteins can alter their lysosomal delivery, such as for PKM2 in cancer cells [10]. More common are pathologies with defective CMA at the level of substrate translocation across the membrane. PD-related proteins α–synuclein, LRRK2, and UCH-L1 interfere with the assembly of the CMA translocation complex [35, 36], whereas in tauopathies, pathogenic tau remains stuck inside the translocation complex [37] (Fig. 2). Conditions that destabilize LAMP-2A at the lysosomal membrane, like dietary lipid challenges or aging, also affect translocation [38].
What are the downstream consequences of autophagy failure in disease?
Altered protein quality control, disrupted metabolic homeostasis, and inefficient stress response are common consequences of most types of autophagic failure. Other detrimental effects of disrupted macroautophagy vary depending on the site of autophagic blockage. For example, defects in macroautophagy initiation or cargo recognition lead to toxicity because of persistence of cargo in the cytosol. Failure to degrade lipid stores can lead to their toxic accumulation, and in fact defective lipophagy has been postulated to underlie the basis of fatty liver diseases [6]. Defective glycophagy would lead to cytosolic glycogen deposition [7], different from its intralysosomal accumulation in LSD such as Pompe disease. Accumulation of cargo inside autophagic vacuoles or lysosomes, although less toxic, also gradually alters cellular homeostasis in part due to a vesicular trafficking-jam and in part because of the failure to recycle the breakdown products of the sequestered material. When defective clearance persists, autophagosome membrane stability is often compromised, leading to toxicity from cytosolic leakage of enzymes and undegraded materials, as described in Alzheimer's Disease (AD) [14].
Defects in initiation of autophagy may benefit from treatments that increase autophagosome formation. However, this treatment would be ineffective when compromise occurs in the later macroautophagy steps, as it would only exacerbate the vesicular traffic-jam. Therapies should aim at repairing the specific defect, restoring cytoskeleton dynamics, facilitating autophagosome/lysosome fusion, or in case of primary defects in lysosomes, at recovering full degradative capacity. Interestingly, even in the presence of the original defect, expanding the lysosomal compartment, for example by expressing TFEB [39, 40] or enhancing the degradative capacity of lysosomes [41], has proven beneficial in neurodegenerative diseases.
To date, all of the described CMA defects affect substrate targeting or lysosomal translocation. Persistence of CMA substrates in the cytosol due to faulty targeting leads to toxicity in part from undesirable conformational changes (aggregation) and in part from loss of their specific cellular functions. Disease-related CMA substrates include proteins involved in cancer (HIF-1α [42], pyruvate kinase M2 [10] and mutant p53 [43]) and neurodegenerative diseases (huntingtin [44]). Pathogenic proteins that fail to translocate but bind tightly to the lysosomal membrane such as α-synuclein [35], LRRK2 [36] or tau [37], organize into oligomeric complexes that often disrupt lysosomal membrane dynamics and stability. Future studies are needed to elucidate if defective lysosomal proteolysis or accumulation of undegraded material as in the case of LSD could also negatively impact CMA on the long run.
What are the consequences of the biphasic autophagic response?
It is not unusual that studies on the same disease have reached opposing conclusions regarding the status of autophagy. Discrepancy may have arisen depending on the cellular conditions, the autophagic steps examined or the time during disease progression at which autophagy was analyzed. Autophagy often exhibits a biphasic response whereby activation occurs early in the pathogenesis as a protective mechanism, followed by a decline in autophagic function that becomes a contributing factor to disease progression. For example, although autophagic flux is compromised later in AD, at early stages, the affected neurons react by inducing autophagosome formation. This enhanced induction can contribute later on to neuronal toxicity as the newly-formed autophagosomes accumulate, but upregulation of autophagy early enough in the disease my offer a window of therapeutic opportunity [41].
Cancer is also a prime example of bi-phasic changes in autophagy. Whereas primary loss of autophagy predisposes to malignant transformation [45], autophagic activation may confer tumor cells a survival advantage in metabolically stressful environments or in response to anti-oncogenic therapeutics injury [12]. Understanding whether autophagy is pro- or anti-oncogenic in a particular stage is essential since inducing autophagy would be counterproductive in cells already employing this pathway as a pro-survival mechanism. In fact, in some cases, blockage of autophagy has shown promising anti-oncogenic effects [12]. However, the complex interplay between cancer and autophagy goes beyond mere time-course changes and is affected by many other factors. For example, a recent study on pancreatic adenocarcinoma revealed that the role of autophagy in tumor development depends on the status of the tumor suppressor protein, p53 [46]. In the presence of p53, blockage of autophagy prevents tumor progression, whereas cancer cells lacking p53 exhibit accelerated tumor formation by favoring activation of anabolic pathways. These types of findings add complexity to the implementation of therapies based on modulation of autophagy and highlights the need to understand the role of autophagy in the disease to assure that the outcome of these interventions is indeed anti-oncogenic.
Autophagic failure: initial insult or adding insult to injury?
The therapeutic success in diseases with associated alterations in autophagy will be contingent on the ability to discriminate whether the autophagic change is primary secondary or reactive to disease-related changes. Differentiating between primary and secondary defects is important to identify the best therapeutic target, whereas identifying changes in autophagy as primary or reactive often defines whether the therapy should drive autophagy upregulation or downregulation.
Genetic deletions, mutations and single-nucleotide polymorphisms (SNPs) in genes that participate in autophagy have been identified as a primary defect in a growing number of conditions. Besides the modifications in core autophagy genes described above, abnormalities in genes involved in the biogenesis of autophagy-related organelles can also lead to a primary defect in autophagy. For instance, mutations in presenilin-1 (PS1), that targets the proton pump to lysosomes, disrupts autophagic flux in AD [34], and mutations the ESCRT protein CHMP2 (charged multivesicular body protein) that modulates multivesicular body formation, explains the altered autophagy activity in ALS affected neurons [47] (Fig. 2).
Autophagy failure can also be secondary to disease-associated cellular changes. For example, the recently identified inhibitory effect of high-lipid content diets on macroautophagy and CMA [38, 48] explains how metabolic disorders that lead to increased intracellular lipids, such as obesity or fatty liver disease, may disrupt these two pathways. Despite the reactive activation of autophagy in the early stages of the metabolic condition as a defense against lipotoxicity, persistence of the lipid accumulation induces changes in the membrane lipids of autophagic compartments that reduce autophagic function. Similar membrane lipid changes are observed with age, implying that dietary changes could accelerate the age-related decline of macroautophagy and CMA.
In a growing number of conditions, autophagic toxicity is secondary to changes in substrates normally degraded by this pathway. For example, while proteins such as α-synuclein, LRRK2 and tau undergo degradation through CMA, pathogenic modifications of these proteins in PD or tauopathies lead to CMA toxicity due to their abnormal interaction with components of this autophagic pathway (Fig. 2). CMA becomes a “victim” of its own substrates and in fact, preventing the targeting of these proteins to the lysosomal compartment is sufficient to decrease lysosomal toxicity and restore CMA activity.
Conclusions
Our current understanding of the contribution of autophagy to disease has benefitted in recent years from the thorough molecular characterization of autophagic pathways, their regulation and new physiological roles. Although some of the changes in the context of disease are still anecdotal, they are already helping to catalogue the different types of autophagy-related pathologies. We predict that current sequencing efforts will lead to the identification of additional diseases with mutations in autophagy genes and will provide a better understanding of the relevance of SNPS and genetic variations identified in these genes. Future expansion in the field of microRNAs, rapidly increasing in importance in disease diagnosis and prognosis, is also anticipated. In fact some microRNAs have already been implicated in autophagy regulation and autophagy regulatory microRNA signatures have been identified in Crohn's disease [49], heart conditions [50], PD [51] and some types of cancer [52]. Although the number of available chemical modulators of autophagy is still rather limited, the recent better understanding of the contribution of autophagy to disease initiation and progression should help to develop in the near future effective interventions targeting autophagy for the treatment of disease.
Acknowledgments
Research in our group is supported by grants from the National Institutes of Health AG21904, AG031782, AG038072 ACTC, DK098408 and NS038370, awards from The Rainwaters Foundation and The Beatrice and Roy Backus Foundation and a generous gift from Robert and Renee Belfer. JLS is supported by T32-GM007288 and F30AG046109 grants
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
• of special interest
• • of outstanding interest
- 1.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM, Santambrogio L. Microautophagy of Cytosolic Proteins by Late Endosomes. Developmental Cell. 2011;20:131–139. doi: 10.1016/j.devcel.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22:407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol. 2013;8:105–137. doi: 10.1146/annurev-pathol-020712-163918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011;13:495–504. doi: 10.1016/j.cmet.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6• •.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. First evidence of contribution of macroautophagy to lipid metabolism by direct mobilization and breakdown of lipid droplets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kotoulas OB, Kalamidas SA, Kondomerkos DJ. Glycogen autophagy in glucose homeostasis. Pathol Res Pract. 2006;202:631–638. doi: 10.1016/j.prp.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 8• •.Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Jurgen Klisch T, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15:647–658. doi: 10.1038/ncb2718. Identification of the transcriptional regulation of lipophagy and first studies that showed upregulation of macroautophagy is protective against lipotoxicity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9• •.O'Rourke EJ, Ruvkun G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat Cell Biol. 2013;15:668–676. doi: 10.1038/ncb2741. First data supporting that the lysosomal enzymatic content is transcriptionally regulated to adapt to the arriving cargo. They provide examples of transcriptional upregulation of lysosomal lipases genes during lipophagy activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11•.Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, Cuervo AM. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med. 2011;3:109–117. doi: 10.1126/scitranslmed.3003182. Link between CMA and cancer. Most cancer cells have elevated CMA activity that is required to sustain their elevated glucose metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathew R, White E. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev. 2011;21:113–119. doi: 10.1016/j.gde.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryter SW, Lee SJ, Smith A, Choi AM. Autophagy in vascular disease. Proc Am Thorac Soc. 2010;7:40–47. doi: 10.1513/pats.200909-100JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 15.Xie M, Morales CR, Lavandero S, Hill JA. Tuning flux: autophagy as a target of heart disease therapy. Curr Opin Cardiol. 2011;26:216–222. doi: 10.1097/HCO.0b013e328345980a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. 2013;6:25–39. doi: 10.1242/dmm.010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider JL, Cuervo AM. Liver autophagy: much more than just taking out the trash. Nat Rev Gastroenterol Hepatol. 2013;11:187–200. doi: 10.1038/nrgastro.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18• •.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. First evidence of mutation in an ATG gene in a human disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 20.Rea SL, Majcher V, Searle MS, Layfield R. SQSTM1 mutations - Bridging Paget disease of bone and ALS/FTLD. Exp Cell Res. 2014 doi: 10.1016/j.yexcr.2014.01.020. [DOI] [PubMed] [Google Scholar]
- 21•.Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. First example of alterations in macroautophagy at the level of cargo recognition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reichelt ME, Mellor KM, Curl CL, Stapleton D, Delbridge LM. Myocardial glycophagy - a specific glycogen handling response to metabolic stress is accentuated in the female heart. J Mol Cell Cardiol. 2013;65:67–75. doi: 10.1016/j.yjmcc.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 23• •.Liang X, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. First connection between macroautophagy malfunction and a human disease. [DOI] [PubMed] [Google Scholar]
- 24.Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K, Kasai-Yoshida E, Sawaura N, Nishida H, Hoshino A, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 2013;45:445–449. 449–441. doi: 10.1038/ng.2562. [DOI] [PubMed] [Google Scholar]
- 25.Martin LJ, Gupta J, Jyothula SS, Butsch Kovacic M, Biagini Myers JM, Patterson TL, Ericksen MB, He H, Gibson AM, Baye TM, et al. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PLoS One. 2012;7:e33454. doi: 10.1371/journal.pone.0033454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou XJ, Lu XL, Lv JC, Yang HZ, Qin LX, Zhao MH, Su Y, Li ZG, Zhang H. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis. 2011;70:1330–1337. doi: 10.1136/ard.2010.140111. [DOI] [PubMed] [Google Scholar]
- 27.Pampliega O, Orhon I, Patel B, Sridhar S, Diaz-Carretero A, Beau I, Codogno P, Satir BH, Satir P, Cuervo AM. Functional interaction between autophagy and ciliogenesis. Nature. 2013;502:194–200. doi: 10.1038/nature12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bejarano E, Yueste A, Patel B, S RF, Spray D, Cuervo AM. Connexins modulate autophagosome biogenesis. Nat Cell Biol. 2014 doi: 10.1038/ncb2934. E-pub ahead of printing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao T, Huang X, Han L, Wang X, Cheng H, Zhao Y, Chen Q, Chen J, Cheng H, Xiao R, et al. Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J Biol Chem. 2012;287:23615–23625. doi: 10.1074/jbc.M112.379164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH, Yoo NJ. Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum Pathol. 2008;39:1059–1063. doi: 10.1016/j.humpath.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 31.Cullup T, Kho AL, Dionisi-Vici C, Brandmeier B, Smith F, Urry Z, Simpson MA, Yau S, Bertini E, McClelland V, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet. 2013;45:83–87. doi: 10.1038/ng.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32• •.Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs J, Oh S, Koga Y, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. Example of lysosomal storage disorder that led to alterations in autophagy by mutation of an autophagy-related protein. [DOI] [PubMed] [Google Scholar]
- 33.Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8:719–730. doi: 10.4161/auto.19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34•.Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. Identification of a primary defect in lysosomal acidification as the cause of altered autophagosome clearance in Alzheimer's disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35•.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. First connection of disruption of CMA in neurodegeneration. [DOI] [PubMed] [Google Scholar]
- 36.Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16:394–406. doi: 10.1038/nn.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodriguez-Navarro JA, Kaushik S, Koga H, Dall'armi C, Shui G, Wenk MR, Di Paolo G, Cuervo AM. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2012;109:E705–714. doi: 10.1073/pnas.1113036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ebrahimi-Fakhari D, Wahlster L. Restoring impaired protein metabolism in Parkinson's disease--TFEB-mediated autophagy as a novel therapeutic target. Mov Disord. 2013;28:1346. doi: 10.1002/mds.25601. [DOI] [PubMed] [Google Scholar]
- 40.Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A. 2013;110:E1817–1826. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–277. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferreira JV, Fofo H, Bejarano E, Bento CF, Ramalho JS, Girao H, Pereira P. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy. 2013;9:1349–1366. doi: 10.4161/auto.25190. [DOI] [PubMed] [Google Scholar]
- 43.Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, Pan L, Ince TA, Kroemer G, Brugge JS, et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013;27:1718–1730. doi: 10.1101/gad.220897.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, Qin ZH. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS One. 2012;7:e46834. doi: 10.1371/journal.pone.0046834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46••.Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, Mackay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. doi: 10.1038/nature12865. Evidence that the effect of autophagy in tumorogenesis is directly dependent on the status of the anti-oncogene p53. [DOI] [PubMed] [Google Scholar]
- 47.Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179:485–500. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhai Z, Wu F, Chuang AY, Kwon JH. miR-106b fine tunes ATG16L1 expression and autophagic activity in intestinal epithelial HCT116 cells. Inflamm Bowel Dis. 2013;19:2295–2301. doi: 10.1097/MIB.0b013e31829e71cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao J, Zhu X, He B, Zhang Y, Kang B, Wang Z, Ni X. MiR-204 regulates cardiomyocyte autophagy induced by ischemia-reperfusion through LC3-II. J Biomed Sci. 2011;18:35. doi: 10.1186/1423-0127-18-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alvarez-Erviti L, Seow Y, Schapira AH, Rodriguez-Oroz MC, Obeso JA, Cooper JM. Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson's disease. Cell Death Dis. 2013;4:e545. doi: 10.1038/cddis.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abraham D, Jackson N, Gundara JS, Zhao J, Gill AJ, Delbridge L, Robinson BG, Sidhu SB. MicroRNA profiling of sporadic and hereditary medullary thyroid cancer identifies predictors of nodal metastasis, prognosis, and potential therapeutic targets. Clin Cancer Res. 2011;17:4772–4781. doi: 10.1158/1078-0432.CCR-11-0242. [DOI] [PubMed] [Google Scholar]