

Abstract
Proteomics is inherently a systems science that studies not only measured protein and their expressions in a cell, but also the interplay of proteins, protein complexes, signaling pathways, and network modules. There is a rapid accumulation of Proteomics data in recent years. However, Proteomics data are highly variable, with results being sensitive to data preparation methods, sample condition, instrument types, and analytical method. To address this challenge in Proteomics data analysis, we review common approaches developed to incorporate biological function and network topological information. We categorize existing tools into four categories: tools with basic functional information and little topological features (e.g., GO category analysis), tools with rich functional information and little topological features (e.g., GSEA), tools with basic functional information and rich topological features (e.g., Cytoscape), and tools with rich functional information and rich topological features (e.g., PathwayExpress). We review the general application potential of these tools to Proteomics. In addition, we also review tools that can achieve automated learning of pathway modules and features, and tools that help perform integrated network visual analytics.
Keywords: pathway analysis, functional analysis, hybrid strategy, network modules, complex networks
1 Introduction
Proteomics, the collective study of all measured proteins in cells of a given condition, is inherently a systems science that requires the understanding of not only the independent parts—protein constituents and their expressions in a cell—but also the interplay of proteins, protein complexes, signaling pathways, and network modules as a whole for achieving biochemical functions. In 2001, Ideker et al. introduced an integrated approach to identify metabolic networks and build cellular pathway models, by using measurements from DNA microarrays, protein expressions, and protein interaction knowledge [1]. This work provides systems biology researchers with a practical example how biological networks could be used to perform integrative functional genomics data analysis. By gaining system-wide perspectives of protein functions, Proteomics promises to further study which subsets of proteins are essential in regulating specific biological process. In Proteomics analysis, the incorporating of prior knowledge how groups of proteins work in concert with each other or with other genes and metabolites has made it possible to unravel the complexity inherent in the analysis of cellular functions [2]. New network biology and systems biology techniques have emerged in recent Proteomics studies [3, 4] including cancer [5].
There has been a rapid accumulation of data due to advances in Proteomics technologies [2]. Proteomics data are often generated from high-throughput experimental platforms, e.g., two-dimensional (2D) gel, liquid chromatography coupled tandem mass spectrometers (LC-MS/MS), multiplexed immunoassays, and protein microarrays [6, 7]. These platforms can assay thousands of proteins simultaneously from complex biological samples [8] to measure the relative abundance of proteins or peptides in various biological conditions. More accurate quantitative measure of peptides could also be performed with isotopic labelling of proteins in two different samples [9]. Similar to Genomics, Proteomics studies have been widely used to extract functional and temporal signals identified in biological systems [10]. Popular experimental techniques to measure protein-protein interactions include the yeast two-hybrid (Y2H) system [11].
In contract to the recent accelerated application of next-generation sequencing (NGS) in biology, a primary hurdle that slows down Proteomics' applications is the Proteomics data's high variability, which makes it difficult to interpret Proteomics data analysis results biologically [12]. Possible sources of data variations arise from biological sample heterogeneity, sample preparation variance, protein separation variance, detection limits of various proteomics techniques, and pattern-matching peptide/protein identification or quantification inaccuracies from Proteomics data management software. The unusual high level of data noises inherent in Proteomics studies in contrast to those in DNA microarrays or NGS instruments have made Proteomics experiments difficult to repeat, and many statistical methods developed for Genomics applications ineffective. There are plenty of reviews that cover the computational challenges [13-15] and solutions to apply statistical machine learning approaches to the problem, e.g., with the use of support vector machines (SVM) [16], Markov clustering [17], ant colony optimization [18], and semi-supervised learning [19] techniques. The ultimate challenge, however, is how to extract functional and biological information from a long list of proteins identified or discovered from high-throughput Proteomic experiments, in order to provide biological insights into the underlying molecular mechanisms of different conditions [20]. Therefore, additional protein functional knowledge, e.g., the abundance of proteins, cellular locations, protein complexes, and gene/protein regulatory pathways, should be incorporated in the second phase of proteomics analysis in order to filter out noisy protein identifications missed in the first statistical analysis phase of Proteomics analysis.
Pathway and network analysis techniques can help address the challenge in interpreting Proteomics results. Analysis of proteomic data at the pathway level has become increasingly popular (Figure 1). For pathway analysis, we refer to data analysis that aims to identify activated pathways or pathway modules from functional proteomic data. Biological pathways can be viewed as signaling pathways, gene regulatory pathways, and metabolic pathways, all of which are curated carefully in reputable scientific publications. Pathway analysis can help organize a long list of proteins onto a short list of pathway knowledge maps, making it easy to interpret molecular mechanisms underlying these altered proteins or their expressions [20]. For network analysis, we refer to data analysis that build, overlay, visualize, and infer protein interaction networks from functional Proteomics and other systems biology data. Network analysis usually requires the use of graph theory, information theory, or Bayesian theory. Different from pathway analysis, network analysis aims to use comprehensive network wiring diagram derived both from prior experimental sources and new in silico prediction to gain systems-level biological meanings [21]. Many large knowledge bases on biological pathways and protein networks have been published, e.g., BioGRID [22], STRING [23], KEGG [24], Reactome [25], BioCarta [26], PID [27], HAPPI [28], HPD [29], and PAGED [30] databases.
Figure 1.
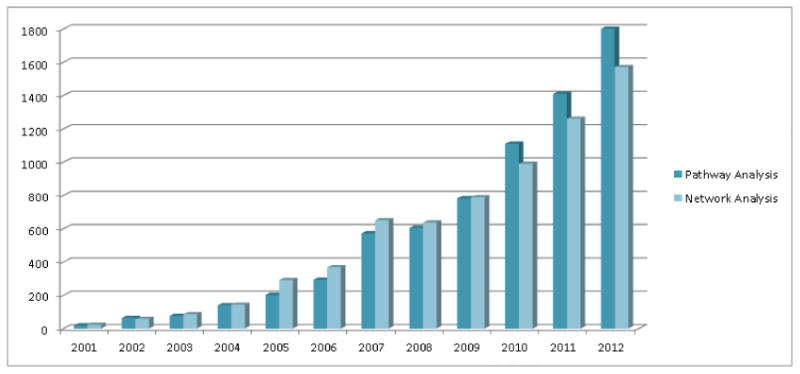
Trends of pathway and network analysis in Proteomics from decade publications (searched in Google Scholar with terms of [“pathway analysis” AND “Proteomics”], and [“network analysis” AND “Proteomics”]).
Compared to pathway and network analysis approaches applied in genomics, the advantages of the related researches in proteomics are listed below: 1) Pathway analysis for proteomic data can be directly interpreted in signaling pathways with signal proteins. 2) Network analysis for proteomic data can have direct evidences supported by protein-protein interaction data validated by in-vitro experiments. 3)Both pathway analysis and network analysis for proteomic data can be visualized in a functional protein network with transcriptional factors labeled, which are all measured indirectly in genomic studies.
2 Pathway and Network Analysis for Proteomics
Many pathway databases and pathway analysis software tools have become available in the last decade [20, 31], with some directly applicable to Proteomics [5, 32]. In Proteomics, statistically significant proteins identified from high-throughput Proteomic instruments often suffer from high false discovery rate [13], partly because the inherently high level of variance in Proteomics data can make it difficult to identify true biological signals [14]. To assess the biological significance of Proteomics results, additional information such as Gene Ontology (GO) and pathways is needed. While there are numerous approaches to incorporate biological pathway and network data into Proteomics data analysis, we categorize existing approaches into two major characteristics, one focusing on integration of “functional information” and the other focusing on integration of “topological information”. For functional information, we refer to functional descriptions that aggregate genes into common protein complexes, biological pathways, network modules, and other genes sets consisting of genes playing similar roles. For topological information, we refer to regulatory relationships that exist among genes, protein complexes, biological pathways, and biological network modules. In Figure 2, we organize the two independent characteristics as the x- and y- axis to categorize representative pathway and network analysis tools in a two-dimensional space. With this framework, we can further categorize existing pathway analysis tools into roughly four quadrants:
Figure 2.
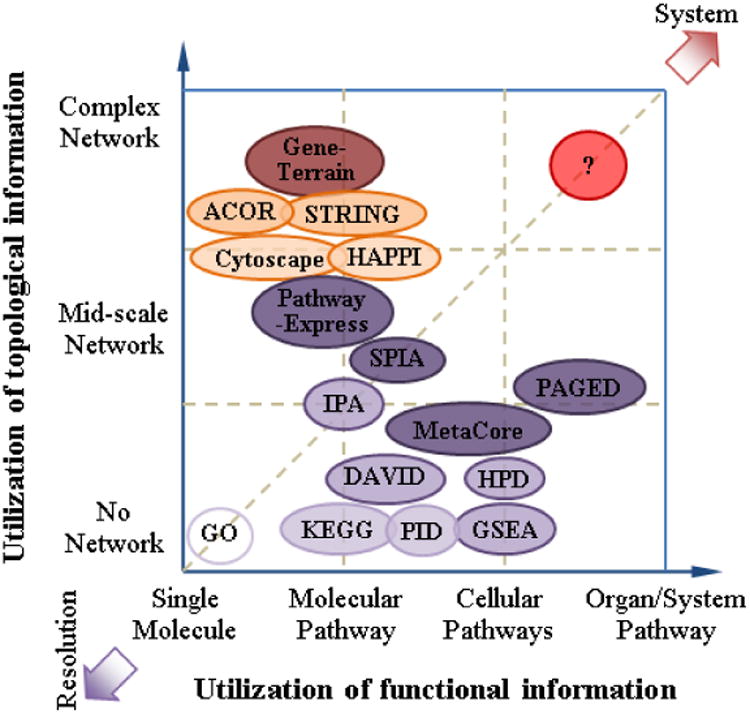
Conceptual plot of different pathway analysis tools according to the utilization of functional information and/or topological information (positions are NOT absolute).
Basic functional information and basic topological features (F-T-). An example is the uses of minimal additional information, e.g., GO categories, to interpret Proteomics results. Since the GO categories contain curated and known functions, and the interaction or regulation relationship information is not tested, the value for pathway and network analysis from the F-T- quadrant may be quite limited. We also consider the traditional feature selection method (e.g., linear programming based feature selection approach [33] or heterogeneous set identification [34]) in the F-T- quadrant, which is based on the classification algorithm and purely used the data itself. When facing simple problems that only require obtaining basic functional information from proteomic data, approaches in the F-T- quadrant will work very well.
Basic functional information but rich topological features (F-T+). An example is the use of protein interaction or gene regulatory networks to help prioritize top-ranked proteins retrieved from the Proteomics results. Since the protein-protein interaction or gene regulatory network contains the biological context, pathway and network analysis from the F-T+ quadrant can help reduce false discovery rate. A latest example is NOA (Network Ontology Analysis) [35]. If the applications are related to cascade regulation or signaling relationships, approaches from the F-T+ quadrant will be more suitable than the ones from the F-T- quadrant.
Rich functional information but basic topological features (F+T-). An example is the use of gene set knowledge and corresponding knowledge to characterize significant biological phenomena that are strongly associated with Proteomics results. Since the gene set information— including both characterized and uncharacterized pathway-related gene sets—can be quite comprehensive, integrated Proteomics data analysis using computational techniques such as the GSEA analysis from the quadrant can reveal significant biological insights. If the applications are related to complex functional identification, especially for protein biomarker discovery, approaches from the quadrant will be more suitable than the ones from the F-T-quadrant.
Rich functional information and rich topological features (F+T+). An example is the simultaneous use of both protein interaction/gene regulation information and curated gene set knowledge to build biological networks at different functional categorical levels (i.e., multiple biosystems scales). Since the multi-scale pathway interaction/regulation network can be complex, the F+T+ model can properly mimic the actual biological systems to provide the highest value to Proteomics researchers. Pathway-Express [36] is an exemplar tool showing how to move toward this new quadrant. Once we meet problems related to both cascade regulation/signaling relationships and complex functional identification, especially for complex disease biomarker discovery, approaches shown in the F+T+ quadrant could be considered as our first options.
1) Pathway analysis using protein functional category information
Many pathway analysis tools in the F-T- or the F-T+ quadrant use basic functional information, since these tools focus on protein functional annotation or basic “functional enrichment analysis” among an unordered set of proteins identified from Proteomics data analysis [37]. These approaches aim to identify proteins with statistical significance first and functional significance subsequently. For example, GoMiner [38] can organize lists of “interesting” genes/proteins for biological interpretation in the context of GO terms, which is at the single-molecule level. DAVID [39] provides a comprehensive set of functional annotation tools which can not only identify enriched biological themes, particularly GO terms, but also discover enriched functionally-related gene groups and visualize genes/proteins in pathway diagrams based on the famous pathway databases – KEGG [24] and BioCarta [26]. To provide broad pathway data coverage, the Human Pathway Database (HPD) [29] integrated KEGG [24], Reactome [25], BioCarta [26], and PID [27] databases ranges from molecular pathways to cellular pathways. The functional enrichment analysis of Proteomics results against these database resources is performed usually with an overlap cut-off score, e.g., as in the single enrichment analysis (SEA) [37]; therefore true signals that are marginally significant from statistical tests may be filtered out prematurely.
Pathway analysis tools moving from the F-T- quadrant to the quadrant is able to better integrate statistical significance from Proteomics data analysis into functional enrichment. Compared with SEA, gene set enrichment analysis (GSEA) [40] evaluates statistical significance of a ranked list of genes/proteins (i.e. gene sets) against one or more pathway data set. GSEA not only can detect group-wise statistically-significant genes and proteins, but also enriched pathway gene sets against a large database of gene sets previously characterized in functional genomic studies. To support GSEA, databases such as the Molecular Signature Database (MSigDB) [41], GeneSigDB [42], and PAGED [43] have been developed to integrate GO categories, pathways from KEGG [24], gene regulatory targets from TRANSFAC [44], micro-RNA targets, and curated gene sets that are co-expression signatures from literature. GSEA and comprehensive databases populated pathway modules can help streamline statistical and functional determination of groups of proteins identified from generally “noisy” Proteomics results.
2) Pathway analysis using network topological information
Moving from the F-T- quadrant to the F-T+ quadrant, tools take a different strategy to perform pathway analysis, i.e., to treat pathways and pathway models as a form of network data structure from which one may incorporate network topological information into the Proteomic data analysis. Here, we refer to biological pathways and biological pathway models interchangeably. In practice, however, biological pathways refer to signaling pathways, gene regulatory networks and metabolic pathways [45], whereas biological pathway models refer to computer representation of actual biological events that have been abstracted. Network representation of biological pathway models involve topologically connected molecules (e.g., genes, proteins, or metabolites) and molecular events (e.g., protein interactions, gene regulations, or metabolite reactions) that are carefully assembled into a graph. While there are 550 biological pathway data sources according to Pathguide (http://www.pathguide.org/), only approximately 10% of them provide pathway diagrammatic details suitable as pathway models; the remaining 90% may only be useful for functional category analysis described earlier. Cytoscape [46] is an open-source biological network analysis platform to visualize and analyze biological pathways based on network topological information. IPA from Ingenuity and MetaCore from GeneGo are commercially available to perform network and pathway analysis for manual pathway data analysis and modeling. However, manual examination of a given biological pathway structure is no longer scalable when it involves more than a few dozen nodes and several hundred edges in the network.
To address scalability issues, tools in the F-T+ quadrant must evaluate both statistical significance and topological significance with computational method. An example is Pathway-Express [36], which develops “impact analysis” techniques to prioritize biologically-significant genes/proteins with lower FDRs. Impact analysis measures network topological information as degree of connectivity and clustering coefficient and applies it as weight for given genes/proteins in the biological pathway to calculate an “impact factor” for the entire pathway. It further evaluates whether the impact factor obtained is significant due to a possible network perturbation event or a random chance. Separately, signaling pathway impact analysis (SPIA) combines both functional evidences from classical enrichment analysis and topological evidences represented as perturbation factor on a given pathway under a given condition [47]. Network analysis using partial network modules are also promising, e.g., developing pathway biomarkers from proteomic data [48] and breast cancer subtyping from plasma proteins [49]. In all, these pathway/network analysis tools integrates network topological information at a limited scale, either at the protein interaction network level or at the network module level.
3) Pathway analysis using a multi-scale hybrid strategy
To understand complex molecular mechanisms associated with a biological condition using Proteomics, a researcher must not only study specific proteins whose expressions are altered or specific pathways in which signaling cascades take place, but also understand how external and internal stimuli translates into coordinated changes of genes, proteins, metabolites, signaling network modules, pathways, and other functional components in a cell. This is why tools in the F+T+ quadrant must be developed. For example, the concept of “GO functional crosstalk network” was introduced in 2008, based on graph representations that use GO functional categories as nodes and enriched protein interactions between GO functional categories as edges [50]. In this work, researchers integrated network topological information and functional information together, resulting in enhanced characterization of complex ovarian cancer drug resistance development mechanism from Proteomics tandem mass spectrometry data. Similarly, pathway similarity networks can be built from heterogeneous pathway data as nodes and pathway-pathway similarity measurement as edges [29]. Pathway association networks (PAN) as a more special form of “GO functional crosstalk networks” can be built from heterogeneous pathway data as nodes and significant protein-protein interaction enrichments as edges [51]. The concept of PANs have already been successfully applied into complex disease modeling for cancer progression [52], Alzheimer's disease [53], and colorectal cancer [54]. Recently, a comprehensive approach to construct multi-edge gene-set networks based on co-memberships, protein interactions, and co-enrichment has also been proposed [55].
Tools in the F+T+ quadrant can benefit significantly from knowledge bases that build relationships between different molecular biosystem components, e.g., pathways, disease-associated gene sets, molecular signatures, microRNA and all their gene targets, and protein interaction network modules. Using molecular biosystem component similarity measures for human in PAGED, a PAN can be developed to serve as a system-level pathway model for interpretation of complex molecular profiling study results. In Figure 3, we demonstrate a workflow platform with which we apply multi-scale pathway analysis to the characterization of colorectal cancer MS-based proteomic data. The input LC-MS data comes from the cceHUB web portal (The Cancer Care Engineering project, hosted at https://ccehub.org/). This workflow utilizes both functional information from the PAGED [30] and topological information from the protein-protein interaction (PPI) database, such as HAPPI or STRING. The functional information validates Proteomics results obtained from LC-MS experiments of the colorectal cancer sample, while the final findings are subsequently examined in the integrated pathway model constructed from protein-protein interaction databases. In this study, we not only confirmed BRAF as a prognostic biomarker for colorectal cancer [56], but also discovered NNMT to be a potential biomarker worth experimental validations [57].
Figure 3.
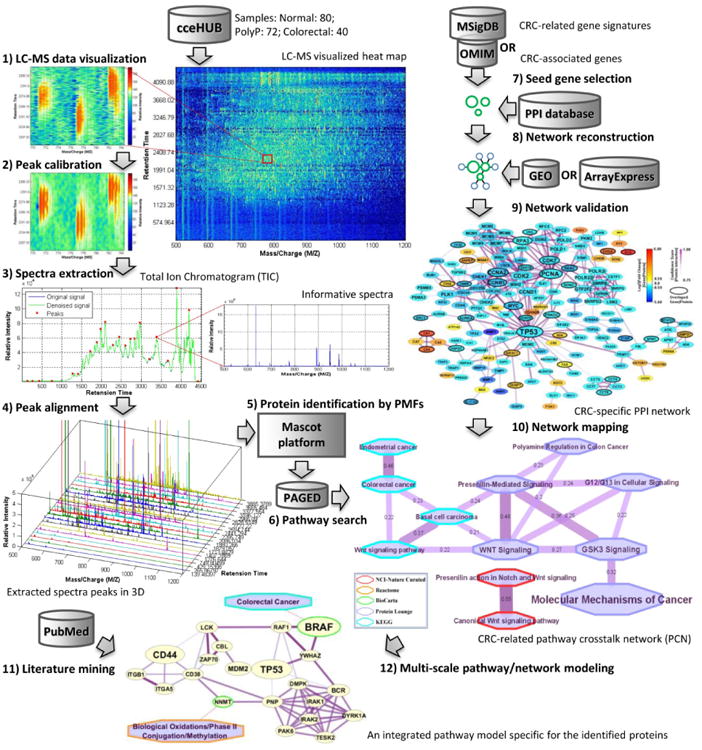
Illustration of multi-scale pathway analysis using colorectal cancer proteomic data as an example. The protein-protein interaction (PPI) database for the Step 8) could use STRING or HAPPI.
4) Automated learning of pathway modules and features
Functional and network information related to pathway models can be either extracted from large existing databases, or learned automatically from functional genomics and Proteomics data sets. There are two types of knowledge discovery tasks. The first is the discovery of pathway modules from pathway and network data relevant to Proteomics results. The second is the discovery of network topological features.
In the first type, “pathway module discovery”, one can assume that there is a close relationship between common protein function categories and proteins closely regulated in the same pathway or network [58]. Existing pathway knowledge or other functional information could also be used to validate newly-discovered pathway models or pathway modules. Hartwell et al. [59] define “network module” as an entity comprising of different types of interacting molecules with strong connections within each other but weak connections outside of the entity. Network modules may map to protein complexes or molecular pathways, consisting of a large number of molecules that co-regulate each other to perform particular cellular functions. Due to the difficulty by human curators to read hundreds of research articles and document molecular regulation details in biological networks, computerized techniques to identify network modules usually involve some form of automated graph clustering of the biological network data [60-62]
In the second type, “network feature discovery”, automated network-based learning of topological and functional information can be done with nonlinear dynamical modeling, when there is no absolute rank for each protein as the node and no clear cluster network module boundaries in the network [63]. Hence, traditional network analysis approaches, such as node ranking and graph clustering, are not directly applicable [64]. The lack of absolute rank or cluster boundaries is characteristic of scale-free biological networks and is also common in other nonlinear systems such as fractals (multi-scale self-similarity), chaos, and phase transitions [65]. Nonlinear dynamical modeling approach, e.g., ant colony optimization (ACO) [66], has already been applied to the analysis Proteomic data in 2007 [18]. An ACO-based network reordering (ACOR) algorithm has been show effective in analyzing complex networks to reveal fractal-like patterns in the studies of yeast lethal gene study [67], breast cancer [68], and Alzheimer's disease (AD) [69]. A recent study to classify AD and normal brain tissue samples showed that prediction based on the ACOR algorithm had better performance than even the best available approaches using either node ranking or graph clustering alone [70]. In contract, Proteomics biomarker results obtained from traditional network analysis approaches such as [71-73] reported that sometimes breast cancer metastasis predictions consisting of multiple genes cannot compete well in performance against optimized single-gene classifiers by comparison [74].
3 Network Analysis for Complex Protein Networks
Complex protein networks are often characterized by scale-free properties [63], i.e., their node distribution follow power laws. Such networks are highly robust to node communication errors, even with unrealistically high failure rates [75]. The ability of error tolerance not only appears in complex protein networks, but also has been found in many other types of scale-free networks, such as World-Wide Web (WWW), the Internet, social networks and cell networks [64]. This suggests that network modeling and analysis methods originally designed for complex social networks can be also applied to analyzing complex protein networks.
The motivation for network analysis specific for complex protein networks derives from complex disease (e.g. various cancers, Alzheimer's disease and type II diabetes etc.) network biomarker discovery, since there are thousands of genes/proteins respond to disease driving factors and drug sensitivity/resistance. As we all know, a complex disease is usually not one disease, but multiple subtypes of disease phenotypes. To discover hidden molecular mechanisms for early diagnosis, prognosis, and drug response, we have to deal with large-scale disease-specific protein networks with hierarchical functional relationships under different conditions, in order to develop tailored therapies for different subtypes of patients, which is the main goal of personalized medicine. Here we will introduce several cutting-edge works for modeling and analyzing large-scale complex protein networks, utilizing vast topological information and group functional information.
1) Network reordering using global topological information
A complex network with scale-free property may also have high-degree “inseparability”, which means that there is no “absolute rank” for each node or no “clear cluster” in the network. Hence, traditional network analysis approaches, such as node ranking and graph clustering, often failed when facing complex networks. Scale-free is an analogy to the situation where power laws arise and no single characteristic scale can be identified, which also happens in other nonlinear phenomena, such as fractals (multi-scale self-similarity), chaos, and phase transitions [65]. Based on this connection, nonlinear dynamical modeling may have great potential in analyzing complex protein networks. As a typical nonlinear dynamical modeling approach, ant colony optimization (ACO) [66], which has been already applied to analyzing MS-based proteomic data in 2007 [18], can be also employed for complex protein network analysis. An ACO-based network reordering (ACOR) algorithm was developed in 2009 to analyze complex networks and the results revealed fractal-like patterns in protein interaction networks for yeast lethal gene study [67], breast cancer (BRCA) research [68], and Alzheimer's disease (AD) diagnosis [69] respectively. These interesting patterns might be closely related to scale-free properties.
Different with traditional network analysis approaches only using local topological information, the ACOR algorithm can efficiently extract global topological information in a complex network, through assigning each node an order number - “relative rank” in “overlapped clusters”. In a recent case study on microarray classification for brain tissue samples of AD patients vs. normal controls, prediction based on the ACOR algorithm showed better performance than the one using either node ranking or graph clustering [70]. Interestingly, the prediction power of these traditional network analysis approaches is only at the same level with the one using random-ordering but still keeping node degree values – typical local topological information. Another case study on breast cancer metastasis prediction also showed that several most popular pathway or network-based approaches [71-73] even cannot compete with a simple, single genes based classifier in an extensive and critical comparison [74]. In Figure 4, we showed an intuitional comparison of the results respectively produced by conventional network-based gene ranking (similar to PageRank algorithm used by Google) [76], 2D hierarchical clustering [77] and ACOR [67-69], for analyzing a BRCA-related protein interaction network [68]. From this comparison, we can see directly that only ACOR approach can reorder the adjacency matrix of the BRCA-related protein network to a meaningful pattern, which has many clusters closely overlapped. All the evidences showed here directly point to an important conclusion – utilization of global topological information is the key of analyzing complex protein networks.
Figure 4.
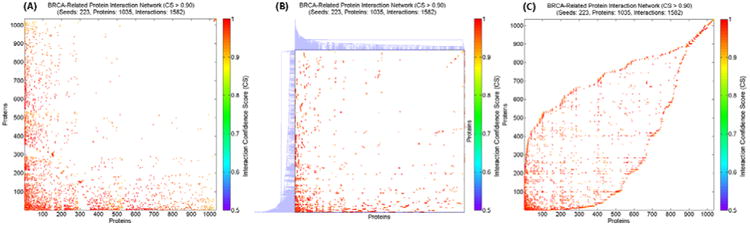
Re-ordered network adjacency matrices of a weighted BRCA-related protein interaction network with 1035 proteins and 1582 interaction, expanded in HAPPI from 223 breast cancer associated genes from OMIM. (A) The result ranked by GeneRank (similar to PageRank algorithm used by Google), (B) The result clustered by 2D hierarchical clustering in Matlab Bioinformatics Toolbox, and (C) The result reordered by Ant Colony Optimization Reordering (ACOR) algorithm. (CS: confidence score for protein interaction in the HAPPI database)
2) Visual analytics using both topological and functional information
A complex protein network usually consists of thousands proteins, which make the network layout looks like a messy hair ball on conventional network visualization platforms. An example of complex protein network visualization by Cytoscape can be seen in Figure 5. One way to overview complex networks is to visualize them at the functional level. A functional category crosstalk network was constructed based on protein interaction networks for ovarian cancer drug resistance study in 2007. This network was first shown as a matrix of interactions between related GO terms, also called GO-GO interactions [78], which took the advantages of both local topological information and group functional information. Another way to simplify complex network visualization is to use the concept of molecular network terrain. Molecular network terrain visualization grows from the work of Kim S.K., et al. in 2001 [79], who assembled data from C. Elegans DNA microarray experiments, and visualized grouped co-regulated genes in a three-dimensional (3D) expression map that displays correlations of gene expression profiles as distances in two dimensions and gene density in the third dimension. In a subsequent study, You, Q., et al. visualized an Alzheimer's disease (AD) specific protein interaction network as a 3D terrain, and successfully differentiated the three distinct stages of AD [80]. The visual analytics approach based on molecular network terrains could increase accuracy and noise endurance for sample molecular classifications, by utilizing both global topological information and group functional information.
Figure 5.
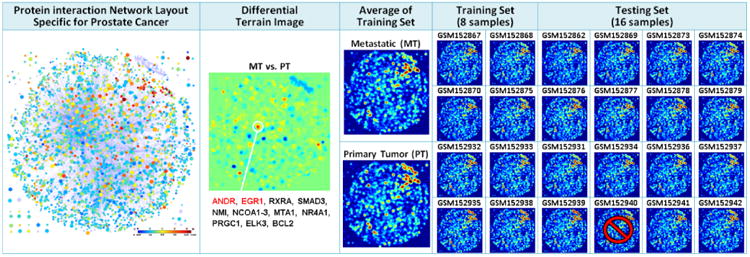
Prostate cancer microarray classification between primary prostate tumor (PT) samples and metastatic (MT) samples by using terrain-based visual analytics approach. The terrain model derived from a PC-specific protein interaction network containing 2637 proteins and 5772 interactions. 24 gene expression profiles (12 PT samples and 12 MT samples) are randomly selected from a microarray dataset GSE6919 in GEO. The only one PT sample classified incorrectly is marked.
As shown in Figure 5, the terrain-based classification approach exhibited amazing performance in a case study on prostate cancer (PC) microarray classification between primary prostate tumor (PT) samples and metastatic (MT) samples. We randomly selected 24 gene expression profiles (12 PT samples and 12 MT samples) from a microarray dataset (GSE6919 [81, 82]) in GEO. We only used 4 PT samples and 4 MT samples as training set, and used the left 16 samples as testing set. Although all the terrain images here look like the same, they can be easily distinguished by computer program. We applied a terrain model, derived from a PC-specific protein interaction network containing 2637 proteins and 5772 interactions (also shown in figure 5), and simply used the distance between a testing terrain image and average terrain image to determine its group for two-group classification. Although these metastatic samples derive from different organs, and are highly heterogeneous in expression, the left 8 MT samples are all correctly classified (100%), and the left 7 of 8 PT samples are also correctly classified (87.5%), which makes the total accuracy reach 93.75%. Moreover, as clearly shown in Figure 5, the differential terrain image between two groups identified a crucial gene clusters, including androgen receptor (ANDR) and early growth response protein (EGR1), which are all well-known, and have been validated to be closely related to PC metastasis previously [82]. This case study demonstrates again the power of using global topological information. Furthermore, it shows that utilization of group functional information (from network modules) not only can be an important supplement to pathway analysis, but also brings great convenience to the interpretation for complex network analysis.
4 Summary
Due to the data variability issues inherent in Proteomics measurements, statistical significance alone is insufficient to the evaluation of Proteomics results. We believe both pathway models' functional information and topological information should be integrated to make Proteomics data interpretation relevant to biological mechanism. With the availability of two types of information, one in protein functional categories and the other in network topological features, we can categorize pathway analysis tools available to Proteomics researchers today as falling into any one of the 2 × 2 quadrants as described in this review. GSEA enables molecular signature-based statistical significance testing, which integrates protein functional category information effectively with statistical testing of functional genomics or Proteomics results. Cytoscape enables network-based data analysis of biological data in situations where functional information may or may not be available. SPIA enables pathway-based statistical assessment by combining both functional annotation and local topological annotation of the network. Ultimately, future tools must support elucidation of complex molecular mechanisms suggested from Proteomics results from multi-scale network data and molecular signature data. A workflow with a hybrid strategy for multi-scale pathway analysis of LC-MS proteomic data was presented. New tools to extract and integrate gene set knowledge from public databases using PAGED and ACOR can be promising. Ultimately, the use of terrain-based visual analytics can be more fruitful, because it gives users inexperienced with network biology or systems biology analysis a rich user experience based on a visualization interface. However, there are still significant challenges in designing next-generation network/pathway analysis tools. In large complex gene regulatory networks and pathway association networks, network coverage can be poor. Accurate protein or protein group functional information at each network scale may be missing. Proposed findings of molecular mechanisms at the network module level can also be more challenging to validate experimentally than at the individual protein level. Nonetheless, the opportunity to discover novel complex mechanisms of biological processes will keep researchers in the field occupied for quite some time.
Table 1.
Selected pathway/network analysis resource that can benefit Proteomics data analysis.
| Name | Description | Link | Reference | Functional Info Using | Topological Info Using |
|---|---|---|---|---|---|
| GoMiner | Gene Ontology (GO) analysis for Omic data | http://discover.nci.nih.gov/gominer/ | [38] | Single molecule | Non |
| KEGG | Kyoto Encyclopedia of Genes and Genomes | http://www.genome.jp/kegg/ | [24] | Molecular pathway | Non |
| DAVID | The Database for Annotation, Visualization and Integrated Discovery | http://david.abcc.ncifcrf.gov/ | [39] | Molecular pathway | Small-scale |
| PID | Pathway Interaction Database | http://pid.nci.nih.gov/ | [27] | Cellular pathway | Non |
| HPD | Human Pathway Database | http://bio.informatics.iupui.edu/HPD | [29] | Cellular pathway | Small-scale |
| GESA | Gene Set Enrichment Analysis | http://www.broadinstitute.org/gsea/ | [40] | Cellular pathway | Small-scale |
| IPA | Ingenuity pathway analysis | http://www.ingenuity.com/ | N/A | Molecular pathway | Small-scale |
| MetaCore | Thomson Reuters pathway analysis and knowledge mining | http://thomsonreuters.com/metacore/ | N/A | Cellular pathway | Small-scale |
| Pathway-Express | A systems biology approach for pathway level impact analysis | http://vortex.cs.wayne.edu/projects.htm | [36] | Molecular pathway | Mid-Scale |
| SPIA | Signaling Pathway Impact Analysis | http://www.bioconductor.org/packages/2.12/bioc/html/SPIA.html | [47] | Molecular pathway | Mid-Scale |
| PAGED | An integrated Pathway And Gene Enrichment Database | http://bio.informatics.iupui.edu/PAGED | [30] | System pathway | Mid-Scale |
| HAPPI | Human Annotated and Predicted Protein Interaction database | http://bio.informatics.iupui.edu/HAPPI | [28] | Single molecule | Large-scale |
| STRING | Search Tool for the Retrieval of Interacting Genes/Proteins | http://string.embl.de/ | [23] | Single molecule | Large-scale |
| CytoScape | An open source platform for complex network analysis and visualization | http://www.cytoscape.org/ | [46] | Molecular pathway | Large-scale |
| ACOR | Ant Colony Optimization Reordering | N/A | [67-70] | Molecular pathway | Large-scale |
| Gene-Terrain | Terrain-based visual analysis for complex networks | N/A | [79], [80] | Network module | Large-scale |
Acknowledgments
This work is partly supported by Indiana Center for Systems Biology and Personalized Medicine (CSBPM) and Wenzhou Medical University.
Footnotes
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, Bumgarner R, Goodlett DR, Aebersold R, Hood L. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292(5518):929–934. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- 2.MacBeath G. Protein microarrays and proteomics. nature genetics. 2002;32:526–532. doi: 10.1038/ng1037. [DOI] [PubMed] [Google Scholar]
- 3.Bensimon A, Heck AJ, Aebersold R. Mass spectrometry-based proteomics and network biology. Annual review of biochemistry. 2012;81:379–405. doi: 10.1146/annurev-biochem-072909-100424. [DOI] [PubMed] [Google Scholar]
- 4.Sabidó E, Selevsek N, Aebersold R. Mass spectrometry-based proteomics for systems biology. Current opinion in biotechnology. 2012;23(4):591–597. doi: 10.1016/j.copbio.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 5.Goh WWB, Wong L. Networks in proteomics analysis of cancer. Current opinion in biotechnology. 2013 doi: 10.1016/j.copbio.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 6.Altelaar AM, Munoz J, Heck AJ. Next-generation proteomics: towards an integrative view of proteome dynamics. Nature Reviews Genetics. 2013;14(1):35–48. doi: 10.1038/nrg3356. [DOI] [PubMed] [Google Scholar]
- 7.Kingsmore SF. Multiplexed protein measurement: technologies and applications of protein and antibody arrays. Nature reviews Drug discovery. 2006;5(4):310–321. doi: 10.1038/nrd2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 9.Ong SE, Mann M. Mass spectrometry–based proteomics turns quantitative. Nature chemical biology. 2005;1(5):252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 10.Blagoev B, Ong SE, Kratchmarova I, Mann M. Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nature biotechnology. 2004;22(9):1139–1145. doi: 10.1038/nbt1005. [DOI] [PubMed] [Google Scholar]
- 11.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proceedings of the National Academy of Sciences. 2001;98(8):4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colinge J, Bennett KL. Introduction to computational proteomics. PLoS computational biology. 2007;3(7):e114. doi: 10.1371/journal.pcbi.0030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vitek O. Getting Started in Computational Mass Spectrometry–Based Proteomics. PLoS computational biology. 2009;5(5):e1000366. doi: 10.1371/journal.pcbi.1000366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noble WS, MacCoss MJ. Computational and statistical analysis of protein mass spectrometry data. PLoS computational biology. 2012;8(1):e1002296. doi: 10.1371/journal.pcbi.1002296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barla A, Jurman G, Riccadonna S, Merler S, Chierici M, Furlanello C. Machine learning methods for predictive proteomics. Briefings in bioinformatics. 2008;9(2):119–128. doi: 10.1093/bib/bbn008. [DOI] [PubMed] [Google Scholar]
- 16.Elias JE, Gibbons FD, King OD, Roth FP, Gygi SP. Intensity-based protein identification by machine learning from a library of tandem mass spectra. Nature biotechnology. 2004;22(2):214–219. doi: 10.1038/nbt930. [DOI] [PubMed] [Google Scholar]
- 17.Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440(7084):637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 18.Ressom HW, Varghese RS, Drake SK, Hortin GL, Abdel-Hamid M, Loffredo CA, Goldman R. Peak selection from MALDI-TOF mass spectra using ant colony optimization. Bioinformatics. 2007;23(5):619–626. doi: 10.1093/bioinformatics/btl678. [DOI] [PubMed] [Google Scholar]
- 19.Käll L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nature methods. 2007;4(11):923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 20.Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Computational Biology. 2012;8(2):e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu X, Chen JY. Molecular interaction networks: topological and functional characterizations. In: Alterovitz G, Benson R, Ramoni M, editors. Automation in Proteomics and Genomics: An Engineering Case-Based Approach. Wiley; 2009. [Google Scholar]
- 22.Chatr-aryamontri A, Breitkreutz BJ, Heinicke S, Boucher L, Winter A, Stark C, Nixon J, Ramage L, Kolas N, O'Donnell L. The BioGRID interaction database: 2013 update. Nucleic acids research. 2013;41(D1):D816–D823. doi: 10.1093/nar/gks1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C. STRING v9 1: protein-protein interaction networks, with increased coverage and integration. Nucleic acids research. 2013;41(D1):D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B. Reactome knowledgebase of human biological pathways and processes. Nucleic acids research. 2009;37(suppl 1):D619–D622. doi: 10.1093/nar/gkn863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishimura D. BioCarta. Biotech Software & Internet Report: The Computer Software Journal for Scient. 2001;2(3):117–120. [Google Scholar]
- 27.Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, Buetow KH. PID: the pathway interaction database. Nucleic acids research. 2009;37(suppl 1):D674–D679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Mamidipalli S, Huan T. HAPPI: an online database of comprehensive human annotated and predicted protein interactions. BMC genomics. 2009;10(Suppl 1):S16. doi: 10.1186/1471-2164-10-S1-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chowbina SR, Wu X, Zhang F, Li PM, Pandey R, Kasamsetty HN, Chen JY. HPD: an online integrated human pathway database enabling systems biology studies. BMC bioinformatics. 2009;10(Suppl 11):S5. doi: 10.1186/1471-2105-10-S11-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang H, Wu X, Sonachalam M, Mandape SN, Pandey R, MacDorman KF, Wan P, Chen JY. PAGED: a pathway and gene-set enrichment database to enable molecular phenotype discoveries. BMC bioinformatics. 2012;13(Suppl 15):S2. doi: 10.1186/1471-2105-13-S15-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramanan VK, Shen L, Moore JH, Saykin AJ. Pathway analysis of genomic data: concepts, methods, and prospects for future development. TRENDS in Genetics. 2012;28(7):323–332. doi: 10.1016/j.tig.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goh WW, Lee YH, Chung M, Wong L. How advancement in biological network analysis methods empowers proteomics. Proteomics. 2012;12(4-5):550–563. doi: 10.1002/pmic.201100321. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Wu QF, Chen C, Wu LY, Yan XZ, Yu SG, Zhang XS, Liang FR. Revealing metabolite biomarkers for acupuncture treatment by linear programming based feature selection. BMC systems biology. 2012;6(Suppl 1):S15. doi: 10.1186/1752-0509-6-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren X, Wang Y, Chen L, Zhang XS, Jin Q. ellipsoidFN: a tool for identifying a heterogeneous set of cancer biomarkers based on gene expressions. Nucleic acids research. 2013;41(4):e53–e53. doi: 10.1093/nar/gks1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Huang Q, Liu ZP, Wang Y, Wu LY, Chen L, Zhang XS. NOA: a novel Network Ontology Analysis method. Nucleic acids research. 2011;39(13):e87–e87. doi: 10.1093/nar/gkr251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C, Georgescu C, Romero R. A systems biology approach for pathway level analysis. Genome research. 2007;17(10):1537–1545. doi: 10.1101/gr.6202607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4(4):R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):P3. [PubMed] [Google Scholar]
- 40.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Culhane AC, Schwarzl T, Sultana R, Picard KC, Picard SC, Lu TH, Franklin KR, French SJ, Papenhausen G, Correll M, et al. GeneSigDB--a curated database of gene expression signatures. Nucleic Acids Res. 2010;38(Database issue):D716–725. doi: 10.1093/nar/gkp1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang H, Wu X, Sonachalam M, Mandape SN, Pandey R, MacDorman KF, Wan P, Chen JY. PAGED: a pathway and gene-set enrichment database to enable molecular phenotype discoveries. BMC Bioinformatics. 2012;13(Suppl 15):S2. doi: 10.1186/1471-2105-13-S15-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, Meinhardt T, Prüβ M, Reuter I, Schacherer F. TRANSFAC: an integrated system for gene expression regulation. Nucleic acids research. 2000;28(1):316–319. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bader GD, Cary MP, Sander C. Pathguide: a pathway resource list. Nucleic acids research. 2006;34(suppl 1):D504–D506. doi: 10.1093/nar/gkj126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim Js, Kim CJ, Kusanovic JP, Romero R. A novel signaling pathway impact analysis. Bioinformatics. 2009;25(1):75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang F, Chen J. Discovery of pathway biomarkers from coupled proteomics and systems biology methods. BMC genomics. 2010;11(Suppl 2):S12. doi: 10.1186/1471-2164-11-S2-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang F, Chen JY. Breast cancer subtyping from plasma proteins. BMC medical genomics. 2013;6(Suppl 1):S6. doi: 10.1186/1755-8794-6-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Agarwal P, Rajagopalan D. A global pathway crosstalk network. Bioinformatics. 2008;24(12):1442–1447. doi: 10.1093/bioinformatics/btn200. [DOI] [PubMed] [Google Scholar]
- 51.Wu X, Chen JY. Genomic Signal Processing and Statistics,(GENSIPS), 2012 IEEE International Workshop on 2012. IEEE; An evaluation for merging signaling pathways by using protein-protein interaction data; pp. 203–206. [Google Scholar]
- 52.Edelman EJ, Guinney J, Chi JT, Febbo PG, Mukherjee S. Modeling cancer progression via pathway dependencies. PLoS computational biology. 2008;4(2):e28. doi: 10.1371/journal.pcbi.0040028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu ZP, Wang Y, Zhang XS, Chen L. Identifying dysfunctional crosstalk of pathways in various regions of Alzheimer's disease brains. BMC systems biology. 2010;4(Suppl 2):S11. doi: 10.1186/1752-0509-4-S2-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pradhan MP, Nagulapalli K, Palakal MJ. Cliques for the identification of gene signatures for colorectal cancer across population. BMC systems biology. 2012;6(Suppl 3):S17. doi: 10.1186/1752-0509-6-S3-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parikh JR, Xia Y, Marto JA. Multi-Edge Gene Set Networks Reveal Novel Insights into Global Relationships between Biological Themes. PloS one. 2012;7(9):e45211. doi: 10.1371/journal.pone.0045211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yokota T, Ura T, Shibata N, Takahari D, Shitara K, Nomura M, Kondo C, Mizota A, Utsunomiya S, Muro K. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. British journal of cancer. 2011;104(5):856–862. doi: 10.1038/bjc.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roeβler M, Rollinger W, Palme S, Hagmann ML, Berndt P, Engel AM, Schneidinger B, Pfeffer M, Andres H, Karl J. Identification of nicotinamide N-methyltransferase as a novel serum tumor marker for colorectal cancer. Clinical cancer research. 2005;11(18):6550–6557. doi: 10.1158/1078-0432.CCR-05-0983. [DOI] [PubMed] [Google Scholar]
- 58.Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nature Reviews Genetics. 2011;12(1):56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402(6761 Suppl):C47–52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 60.Dittrich MT, Klau GW, Rosenwald A, Dandekar T, Müller T. Identifying functional modules in protein–protein interaction networks: an integrated exact approach. Bioinformatics. 2008;24(13):i223–i231. doi: 10.1093/bioinformatics/btn161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He J, Li C, Ye B, Zhong W. Efficient and accurate greedy search methods for mining functional modules in protein interaction networks. BMC Bioinformatics. 2012;13(Suppl 10):S19. doi: 10.1186/1471-2105-13-S10-S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pereira-Leal JB, Enright AJ, Ouzounis CA. Detection of functional modules from protein interaction networks. Proteins: Structure, Function, and Bioinformatics. 2004;54(1):49–57. doi: 10.1002/prot.10505. [DOI] [PubMed] [Google Scholar]
- 63.Barabási AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nature Reviews Genetics. 2004;5(2):101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 64.Barabási AL. Scale-free networks: a decade and beyond. Science. 2009;325(5939):412–413. doi: 10.1126/science.1173299. [DOI] [PubMed] [Google Scholar]
- 65.Strogatz SH. Exploring complex networks. Nature. 2001;410(6825):268–276. doi: 10.1038/35065725. [DOI] [PubMed] [Google Scholar]
- 66.Dorigo M, Birattari M. Encyclopedia of Machine Learning. Springer; Ant colony optimization; pp. 2010pp. 36–39. [Google Scholar]
- 67.Wu X, Pandey R, Chen JY. Engineering in Medicine and Biology Society, 2009 EMBC 2009 Annual International Conference of the IEEE: 2009. IEEE; Network topological reordering revealing systemic patterns in yeast protein interaction networks; pp. 6954–6957. [DOI] [PubMed] [Google Scholar]
- 68.Wu X, Harrison SH, Chen JY. Pattern Discovery in Breast Cancer Specific Protein Interaction Network. Summit on translational bioinformatics. 2009;2009:1. [PMC free article] [PubMed] [Google Scholar]
- 69.Wu X, Huan T, Pandey R, Zhou T. Finding fractal patterns in molecular interaction networks: a case study in Alzheimer's disease. International Journal of Computational Biology and Drug Design. 2009;2(4):340–352. doi: 10.1504/IJCBDD.2009.030765. [DOI] [PubMed] [Google Scholar]
- 70.Wu X, Huang H, Sonachalam M, Reinhard S, Shen J, Pandey R, Chen JY. Reordering based integrative expression profiling for microarray classification. BMC bioinformatics. 2012;13(Suppl 2):S1. doi: 10.1186/1471-2105-13-S2-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chuang HY, Lee E, Liu YT, Lee D, Ideker T. Network-based classification of breast cancer metastasis. Molecular systems biology. 2007;3(1) doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee E, Chuang HY, Kim JW, Ideker T, Lee D. Inferring pathway activity toward precise disease classification. PLoS computational biology. 2008;4(11):e1000217. doi: 10.1371/journal.pcbi.1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Taylor IW, Linding R, Warde-Farley D, Liu Y, Pesquita C, Faria D, Bull S, Pawson T, Morris Q, Wrana JL. Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nature biotechnology. 2009;27(2):199–204. doi: 10.1038/nbt.1522. [DOI] [PubMed] [Google Scholar]
- 74.Staiger C, Cadot S, Kooter R, Dittrich M, Müller T, Klau GW, Wessels LF. A critical evaluation of network and pathway-based classifiers for outcome prediction in breast cancer. PloS one. 2012;7(4):e34796. doi: 10.1371/journal.pone.0034796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Albert R, Jeong H, Barabási AL. Error and attack tolerance of complex networks. Nature. 2000;406(6794):378–382. doi: 10.1038/35019019. [DOI] [PubMed] [Google Scholar]
- 76.Morrison JL, Breitling R, Higham DJ, Gilbert DR. GeneRank: using search engine technology for the analysis of microarray experiments. BMC bioinformatics. 2005;6(1):233. doi: 10.1186/1471-2105-6-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bar-Joseph Z, Gifford DK, Jaakkola TS. Fast optimal leaf ordering for hierarchical clustering. Bioinformatics. 2001;17(Suppl 1):S22. doi: 10.1093/bioinformatics/17.suppl_1.s22. [DOI] [PubMed] [Google Scholar]
- 78.Chen JY, Yan Z, Shen C, Fitzpatrick DP, Wang M. A systems biology approach to the study of cisplatin drug resistance in ovarian cancers. Journal of bioinformatics and computational biology. 2007;5(02a):383–405. doi: 10.1142/s0219720007002606. [DOI] [PubMed] [Google Scholar]
- 79.Kim SK, Lund J, Kiraly M, Duke K, Jiang M, Stuart JM, Eizinger A, Wylie BN, Davidson GS. A gene expression map for Caenorhabditis elegans. Science. 2001;293(5537):2087–2092. doi: 10.1126/science.1061603. [DOI] [PubMed] [Google Scholar]
- 80.You Q, Fang S, Chen JY. GeneTerrain: visual exploration of differential gene expression profiles organized in native biomolecular interaction networks. Information Visualization. 2010;9(1):1–12. [Google Scholar]
- 81.Yu YP, Landsittel D, Jing L, Nelson J, Ren B, Liu L, McDonald C, Thomas R, Dhir R, Finkelstein S. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. Journal of Clinical Oncology. 2004;22(14):2790. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 82.Chandran U, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, Michalopoulos G, Becich M, Monzon F. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC cancer. 2007;7(1):64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]