

Abstract
Genome-wide association studies (GWAS) and whole-exome sequencing (WES) generate massive amounts of genomic variant information, and a major challenge is to identify which variations drive disease or contribute to phenotypic traits. Because the majority of known disease-causing mutations are exonic non-synonymous single nucleotide variations (nsSNVs), most studies focus on whether these nsSNVs affect protein function. Computational studies show that the impact of nsSNVs on protein function reflects sequence homology and structural information and predict the impact through statistical methods, machine learning techniques, or models of protein evolution. Here, we review impact prediction methods and discuss their underlying principles, their advantages and limitations, and how they compare to and complement one another. Finally, we present current applications and future directions for these methods in biological research and medical genetics.
Keywords: functional impact prediction methods, disease causing SNV (single nucleotide variation), single nucleotide polymorphism prioritization, missense variant classification, non-synonymous protein mutations
Introduction
Accurate prediction of SNV impact is an important challenge
Since making its first clinical diagnosis in 2009,1 whole exome sequencing has been on the rise for both individual patient diagnosis and large-scale projects, in keeping with decreasing production costs (Fig. 1). Our capacity to obtain sequencing information has expanded so quickly that it now far out-paces Moore's doubling law for computing power.5 Whereas targeted gene sequencing and Genome Wide Association Studies (GWAS) at predetermined loci used to be the cutting edge,6,7 new studies aim to identify single nucleotide variations (SNVs) in all genes and to analyze their association with disease.8 There are now thousands of sequenced exomes encompassing phenotypes both rare (e.g., Joubert syndrome,9 myofibrillar myopathy10), and relatively common (e.g., cancer11–16 and epilepsy17,18). Many of these exome projects have been catalogued and made available for analysis through the Database of Genotypes and Phenotypes (dbGaP),19,20 and multi-center efforts like the NHLBI Exome Sequencing Project21 are actively gathering more data. With this influx of information, researchers are now limited not by a lack of material, but instead by the challenges of processing and interpreting this wealth of information. With more candidate SNVs to evaluate than ever before, accurate methods that predict the effect of SNVs are crucial to ensure that research focuses on those variations that are most likely to cause disease.
Figure 1.
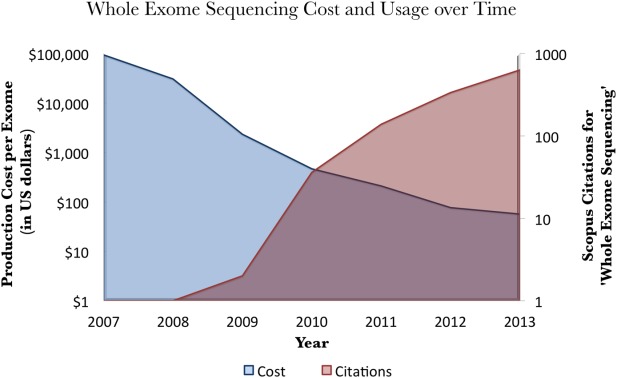
Production cost and usage of whole exome sequencing over time. As the cost of exome sequencing (blue) decreases, the number of articles containing the phrase “whole exome sequencing” (red) increases. The number of articles is found via Scopus.2 The production cost is defined by the National Human Genome Research Institute3 and includes the costs of labor, sequencing instruments, and data processing, but not quality control, technology development, or data analysis. As of April 2014, the production cost for an exome on the Illumina or SOLiD platform at 30-fold coverage was $49.20, although the actual cost to the consumer is considerably higher, with costs advertised in the range of $700 to $2000 per sample.4
Most tools focus on coding SNVs rather than other SNVs
Decoding the relationship between genotype and phenotype is a major challenge in genetics. In humans there are more than four million DNA differences between two random individuals.22,23 Because additions and deletions typically have stronger impact,24–26 and are selected against more often, ∼80% of these differences are single nucleotide variations (SNVs).27–29 Over the entire human population, an estimated 81%30 to 93%31 of human genes contain at least one SNV. Although only a small fraction of variants are non-synonymous single nucleotide variations (nsSNVs), about 10,000 are found between two random individuals,27–29 and over 85% of known disease associations are culled from this important class of mutations.1 For this reason, methods for predicting the impact of SNVs have historically focused on the high-yield category of non-synonymous coding SNVs. The existence of disease-associated synonymous mutations32,33 and nocoding variations with effects on lincRNA,34 miRNA,35,36 and promoters37,38 has produced interest in other types of mutations as well, but different tools will be needed to analyze these types of variations and such tools are comparatively still new and untested.39–41
Most nsSNVs affect protein function but in distinct ways
nsSNVs may affect folding,42,43 binding affinity,44,45 expression,46 post-translational modification,47,48 and other protein features. However, not all nsSNVs impact protein function. Some variations may produce no perceivable changes to the protein, in which case the mutation may not be pathogenic. On the other hand, purifying selection should eliminate over time the mutations that are most deleterious to fitness. A telltale signal is a decreased ratio of non-synonymous to synonymous mutations compared to a model of neutral mutation theory.49 Importantly, not all non-synonymous mutations are under the same strength of purifying selection. An analysis of exomes from the 1000 Genomes Project,50 in accordance with simulations51 and with Fisher's geometric model52 showed that the number of the nsSNVs retained in the human population decreases exponentially as the impact on fitness increases. The same analysis also showed that the exponential decrease becomes steeper for nsSNVs with higher allelic frequency, reflecting that the more common mutations have been selected against stronger constraints. This demonstrates the complexity of the genotype-to-phenotype relationship and implies that a binary classification of a mutation into deleterious or neutral, although very convenient, may be too simplistic.53
Goal of the review
Predictors of the impact of nsSNVs are useful for associating variants to phenotypic traits and diseases, but they should be used cautiously and with an understanding of the benefits and pitfalls of using each method. However, researchers attempting to understand the field may feel overwhelmed by the plethora of available predictors to consider. Here we classify current predictors of functional impact by their underlying theory and we discuss the fundamental principles, assumptions, strengths, and limitations of each type of method. Finally, we speculate on the future directions of variant prioritization and review applications for nsSNV impact prediction in guided mutagenesis studies, the identification of disease-causing nsSNVs, the association of genes to diseases, and the prediction of polygenic phenotypes from whole exome data.
Predicting SNV Impact
While many features have been used to predict the impact of nsSNVs, there are two major features that are commonly used in bioinformatics tools: structure and sequence homology.
Structural metric of nsSNV impact
Some of the first methods to predict the impact of nsSNVs were based solely on structure.54,55 They assumed that deleterious nsSNVs destabilize the folding of proteins and therefore aimed to estimate the free energy change of folding (ΔΔG) due to a mutation. Roughly three quarters of amino acid substitutions that result in Mendelian diseases do affect protein stability, proving the value of this assumption.56,57 Impacting protein stability typically implies local or total unfolding of the protein, but occasionally deleterious aggregates like amyloid fibrils58,59 may form. Rarely, single mutations have been known to cause a switch between stable folds.60 To avoid the computational expense of physical models like Molecular Dynamics simulations, most methods use statistical (PopMuSiC-2.0,61 SDM54) or empirical (FoldX/SNPeffect,62,63 Dmutant55) effective energy functions. These methods typically require a structure for the region of the protein under investigation, although some methods can use sequence information alone.64 Originally, SDM, a knowledge-based approach, used environment-dependent amino acid substitution with propensity tables and considered a structure's main-chain conformations, solvent accessibilities, hydrogen bonds, and disulfide bonds.54 Later methods used this information to help calculate basic potentials, low-order and high-order coupling terms, volume terms, and solvent accessibility terms for comprehensive scoring functions that can be weighted through training with machine learning techniques61 or direct fitting to empirical data.63,65 Other structural components that are taken into account include small-molecule binding sites, protein–protein interactions, entropy optimization, and Van der Waals and torsional clashes.63,66
These structure-based methods give insight about the local environment of the mutation. Variants on the surface are, in general, more likely to be neutral than variants in the core,67 indicating that disease-associated mutations often affect intrinsic structural features of proteins.68 However, surface mutations at important protein–protein interaction sites are more likely to be disease-associated.69 Using the structure also has the advantage of accounting for the interactions between amino acid residues that are close in three-dimensional space but far apart in the protein's sequence. Loss or gain of disulfide, electrostatic or hydrophobic interactions that affect protein stability or aggregation are examples of interaction changes that the use of 3-D structure can help identify.70,71
Unfortunately, even with a deposition rate outpacing PubMed article submission72 and after recently reaching the milestone of 100,000 structures,73 it is still a relatively small fraction of all proteins that can be found in the protein data bank. For example, in a recent study on epilepsy disorders66 only 18/68 of the proteins of interest had partial structures. For the remaining proteins, only 22% of the mutations could be mapped onto a predicted structure from theoretical models based on homology of known structures.66 For a larger perspective, only 7.6% of 57,525 nsSNVs from the Humsavar database could be mapped to structures.74 This percentage increased to 60.4% when Phyre2 homology models75 were included,74 but still the proportion of unaddressed SNVs was large. Another pitfall is that the PDB may contain structures, often flagged with a warning,76 that have unresolved concerns regarding geometry, stereochemistry, or solvent, and that contribute to inconsistency in the quality of the available structures.77 Overall, structural information has its greatest value in nsSNV impact prediction in cases where a complete and robust protein structure is available and where the protein has few homologs, compromising the prediction accuracy of methods that rely heavily on homology.78,79
Evolutionary metrics of nsSNV impact
A complementary approach to determine the impact of nsSNVs is based on evolutionary principles. At first, substitution matrices like BLOSUM6280 were used to classify a nsSNV as impactful or not81 by the similarity of an amino acid substitution as judged by the interchanges between homologous proteins. This type of substitution matrix was originally designed for database searching and pairwise alignment82 and then repurposed to predict nsSNV impact. When used as a standalone prediction tool, BLOSUM62 matrices over-predict non-conservative substitutions,83 and many early methods demonstrated their feasibility by showing improvements in accuracy over BLOSUM62 predictions.83,84 While BLOSUM62 uses a non-specific substitution profile, many homology-based methods now assess amino acid substitution profiles in a more sophisticated and family specific manner.
Homology-based methods typically assume that the overrepresented substitutions in a protein family are neutral on protein function and that the underrepresented ones are deleterious25,83,84 (Fig. 2). This implies two hypotheses: that each substitution has an independent effect on protein function (no epistasis) and that all homologs have identical function (the fitness landscape is constant).83,84 The prediction accuracy is significantly affected upon violation of these hypotheses and most methods attempt to minimize this problem by optimizing the sequence selection to mostly orthologous proteins, thereby minimizing changes in the fitness landscape.25 Although non-native alignments can sometimes improve the accuracy of a method,85 customizing the sequence alignment in a rational way requires a great deal of knowledge and finesse.
Figure 2.
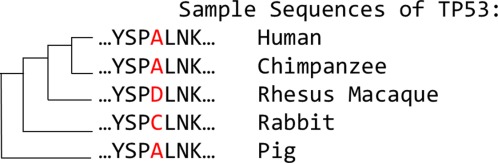
TP53 sequences from different species and variations in their amino acids. Some homology-based methods would predict that the human sequence would tolerate a substitution of alanine to aspartic acid or to cysteine at the highlighted position. Other methods account for the conservation of a position, concluding that the highlighted position would likely tolerate more substitutions than other positions.
At the most basic level, the early homology methods (SIFT,83 Panther84) judge the impact of nsSNVs by scoring the substitution frequency amongst homologues. To improve upon this simple principle, SIFT normalized the probabilities of all possible amino acid substitutions and Panther uses a Hidden Markov Model.84 The next generation of methods (A-GVGD,86 MAPP87) score the observed frequency of biochemical properties in each position of the alignment, such as the volume, polarity, hydropathy and charge, and how they differ from the properties of the substituted amino acid. These methods then conclude that a residue is deleterious for protein function when it does not comply with the protein family's substitution profile.86,87
More recent implementations of homology have combined homology information together with substitution matrices. Provean uses an alignment-based score that measures the change in sequence similarity of the query sequence with each of its homologs, before and after the introduction of the mutation.25 The similarity is estimated by using the BLOSUM62 matrix, and it can provide predictions for multiple amino acid substitutions, insertions, and deletions. Alternatively, the Evolutionary Action method models the genotype-to-phenotype relationship with an equation stating that the impact of a mutation is a product of the functional importance of the mutated residue and of the amino acid similarity of the substitution.50 The functional importance is approximated by the Evolutionary Trace method88,89 and the amino acid similarity by substitution matrices that depend on the functional importance of the residues and optionally on their structural features. Overall, the abundance of such methods highlights the ability of homology to accurately predict the impact of nsSNVs independently from other features.
Homology has been a steadfast component of nsSNV impact prediction, whether by itself or in combination with structural information, but there are several limitations to its predictive power. In particular, the lack of available homologous sequences may result in lower prediction accuracy.87 For example, the Provean method uses 100–200 homologous sequences on average, but when their number drops below 50, the accuracy is lower.25 Another caveat is that the selection of sequences must be balanced to represent sufficiently deep evolution of the protein family without being biased to distant phylogenetic branches that have evolved to retain functions that are specific only to that branch. When the Provean method was tested on sequence alignments derived by using the UniProtKB/Swiss-Prot instead of the NCBI NR database, the accuracy dropped by 7% and this was attributed to the lack of orthologous and distantly related sequences.25 Furthermore, the choice of the alignment has a major effect on the accuracy of a method. When each of four alignments was used as input to SIFT, A-GVGD, PolyPhen-2, and Xvar (now MutationAssessor), their accuracy varied widely, with A-GVGD being extremely sensitive to, and PolyPhen-2 being more robust to, changes in alignment.85 Interestingly, the native alignments of each method did not necessarily give the best predictions for that method.85 Overall, sequence homology can be applied to nsSNV impact prediction with great success if there are sufficient homologues in broad and deep branches of the phylogeny.
Integrative machine learning approaches
Several methods predict the impact of nsSNVs using both structure and homology, along with other types of information such as function annotation and biochemical properties. To combine key features, these methods use supervised machine learning techniques that integrate disparate data types through nonlinear relationships and handle outliers and noise more readily than linear approaches.64 Supervised learning requires training with large numbers of known phenotype associations in order to deduce these complex relationships.64,90,91 Ultimately, they classify the data into categories91 like deleterious or neutral, and they may provide a confidence score for each prediction. Commonly used machine learning techniques include Support Vector Machines,5,64,71,92–96 Naive Bayes,97,98 Neural Networks,90,99 Random Forests,100,101 and Decision Trees.93,102
Perhaps the most well-known impact predictor that uses machine-learning is PolyPhen2, which uses a naive Bayes classifier on substitution events in homologs, structural parameters, function annotation, and physicochemical features.98 Typical training features include amino acid substitution profiles or homology derived scores,71,94,98 biophysical properties of the substitution (volume,71,92,98 hydropathy,71,92,94 and charge71,92,94), structure information (secondary structure,94 solvent accessibility,92,98 and crystallographic B-factors98), function annotation,92,98,103 local environment information (neighbors in sequence or space),64,93,94,104 statistical potentials,64 aggregation property,62,105 and intrinsically disordered regions.105 Recently, SuSPect74 even incorporated a network of protein-protein interactions from the STRING database106 into its analysis.
Machine learning methods aim to identify and use non-redundant features that are highly correlated to accurate classification.107 However, optimizing the selection of features may cause predictions to be less accurate for those proteins dependent on “atypical” features. For example, disruption of intrinsic disorder, a rarely used feature, is critical for predicting the impact of mutations in the tumor suppressor APC.108 Determining which features contain the most relevant information and the least amount of noise has been a constant challenge, and several methods integrate predictions of existing methods with other methods (Condel),109 or with additional features, (SNAP99 and MutPred110), in order to increase the accuracy. At the publication time of this review, there is no consensus for a “best” set of features to predict the impact of SNVs, with different combinations working for different methods and datasets. The features considered by each method are detailed in Supporting Information Table1.
Table I.
SNP Impact Predictors
| Server | Year | Input | URL | Pubmed ID |
|---|---|---|---|---|
| Structural | ||||
| SDM | 1997 | PDB ID | http://www-cryst.bioc.cam.ac.uk/∼sdm/sdm.php | 9051729 |
| Dmutant | 2002 | PDB ID | http://sparks.informatics.iupui.edu/hzhou/mutation.html(Unavailable) | 12381853 |
| PoPMuSiC | 2009 | PDB ID | http://dezyme.com/ | 19654118 |
| SDS | 2014 | - | Cannot automate | 24795746 |
| Homology | ||||
| SIFT | 2001 | Protein identifier, SNP IDs, or alignment | http://sift.jcvi.org/ | 11337480 |
| Panther | 2003 | Sequence | http://www.pantherdb.org/tools/csnpScoreForm.jsp | 12952881 |
| MAPP | 2005 | Alignment and phylogenetic tree | http://mendel.stanford.edu/SidowLab/downloads/MAPP/index.html | 15965030 |
| A-GVGD | 2006 | Alignment | http://agvgd.iarc.fr/agvgd_input.php | 16014699 |
| mutationassessor (xvar) | 2011 | Protein identifier or chrom. location | http://mutationassessor.org/ | 21727090 |
| Provean | 2012 | Sequence or chrom. location | http://provean.jcvi.org/index.php | 23056405 |
| Evolutionary action | 2014 | Protein identifier | http://mammoth.bcm.tmc.edu/EvolutionaryAction/ | |
| Hybrid | ||||
| PolyPhen | 2002 | Protein identifier or sequence | http://genetics.bwh.harvard.edu/pph/ | 12202775 |
| LogR.E-value | 2004 | Site is down for maintenance | http://lpgws.nci.nih.gov/cgi-bin/GeneViewer.cg | 14751981 |
| nsSNPAnalyzer | 2005 | Sequence (requires available PDB structure) | http://snpanalyzer.uthsc.edu/ | 15980516 |
| SNPeffect | 2005 | Sequence, PDB ID, UniProt ID | http://snpeffect.switchlab.org/menu | 15608254 |
| LS-SNP | 2005 | SNP, protein or pathway identifier | http://modbase.compbio.ucsf.edu/LS-SNP/ | 15827081 |
| MUpro | 2005 | Protein sequence, structure (optional) | mupro.proteomics.ics.uci.edu | 16372356 |
| pmut | 2005 | Sequence (on demand version) or PDB ID (precalculated version) | http://mmb2.pcb.ub.es:8080/PMut/ | 15879453 |
| PhD-SNP | 2006 | Protein identifier or sequence | http://snps.biofold.org/phd-snp/phd-snp.html | 16895930 |
| SNPs3D | 2006 | SNP identifier | http://www.snps3d.org/ | 16551372 |
| Parepro | 2007 | Alignment | http://www.mobioinfor.cn/parepro/index.htm | 18005451 |
| SAPRED | 2007 | Sequence and PDB files | http://sapred.cbi.pku.edu.cn/ (Login required) | 17384424 |
| Imutant 3.0 | 2007 | Sequence or PDB ID | http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi | 18387208 |
| SNAP | 2007 | Sequence | http://rostlab.org/services/snap/submit | 17526529 |
| AUTO-MUTE | 2010 | PDB ID | http://proteins.gmu.edu/automute/AUTO-MUTE_nsSNPs.html | 20573719 |
| Mutation Taster | 2010 | Transcript, gene, or ORF | http://www.mutationtaster.org | 20676075 |
| PolyPhen2 | 2010 | Protein or SNP identifier or sequence | http://genetics.bwh.harvard.edu/pph2/ | 20354512 |
| Condel | 2011 | Protein identifier, mutation, homology tree | No server, but can get PERL pipeline scripts and then download each tool | 21457909 |
| CADD | 2014 | VCF file | http://cadd.gs.washington.edu/score | 24487276 |
| VarMod | 2014 | Sequence | http://www.wasslab.org/varmod/ | 24906884 |
| SuSPect | 2014 | Sequence or VCF | http://www.sbg.bio.ic.ac.uk/suspect/index.html | 24810707 |
Another limitation of the machine learning methods is that they may rely on asymmetric training sets that may misrepresent population characteristics.104 For example, if a Gaussian distribution was randomly sampled, one might obtain by chance a few more samples on one side of the curve (Supporting Information Fig. 1). Using this skewed distribution in a machine learning technique underfits the data and can cause false predictions defeating the purpose of the learning process.91,111 However, this “generalization error” can at least be minimized by mathematical models.112 Equally problematic, if a method is over-trained on a dataset, noise will be built in and the performance of the model will drop.91,111,112 Finally, using machine learning methods to predict the impact of mutations that differ fundamentally from the training data may require retraining and revalidating the tool. For example, using SuSPect, which was initially trained only on human SNVs, to predict the impact of mutations in non-human proteins dropped the AUC by about 10%.74
Availability and Comparisons
A summary of well-known current methods to predict the impact of nsSNV is provided in Table1, and a more detailed version of this table exists in the Supporting Information Table1. The majority of these methods are freely available to the research community through web servers or through downloadable files for local use. Using them often requires basic to advanced bioinformatics skills, as presented in Karchin 2008.113 At its most basic, a user has to input just an identifier of the protein of interest or its sequence, and in some cases the specific amino acid substitution as well. To better assist users, many methods allow submitting large number of prediction requests at a time, and others give an option to input user-curated sequence alignments of the protein family.
New tools determine their accuracy by applying their method to various sets of nsSNVs whose impact is known and measuring how well they are able to distinguish harmful mutations from benign ones. Ambitious mutagenesis work on a particular protein is one way these validation sets are developed. For example, 4041 mutations of the E. coli LacI protein,114,115 336 mutations of HIV-1 protease,116 2015 mutations of bacteriophage T4 lysozyme,117 and 2314 mutations of the human p53 protein118 have been assayed for functional effect and catalogued. Many tools, including SIFT,83 MutationAssessor,65 Provean,25 MAPP,87 and EA,50 compare to one or more of these classic datasets. Another type of validation set comes from reference human SNVs that have been classified as disease-associated variants (deleterious) or common polymorphisms (presumed benign). These datasets include VariBench,119 HGMD,120 and the “human polymorphisms and disease mutations” set available from the UniProtKB/Swiss-Prot database,121 each of which contains tens of thousands of missense variants. This type of validation set has the advantage of being human-specific and encompassing many proteins, but relies on the accuracy of annotations in the databases and can only consider SNV impact in a binary fashion. On the other hand, validation sets from mutagenesis studies are more limited in scope but involve functional assays that consider impact on a continuous scale.
The performance of different methods to predict the impact of mutations is typically compared with the area under the curve (AUC) of the receiver operating characteristic (ROC) plots. An ROC plots the true positive rate against the false positive rate and demonstrates the trade-off between sensitivity and specificity. The AUC quantifies the success of this trade-off. A perfect prediction would result in a vertical line (infinite slope) at the origin and an AUC of 1, in contrast to a completely random prediction that would result in a line with a slope of 1 and an AUC of 0.5. Other measures to evaluate the ability of prediction methods to prioritize the impact of mutations include the balanced accuracy, which is the average of the sensitivity and specificity,25 the F1 score, which is the harmonic mean of precision and recall,122 the Matthews correlation coefficient (MMC),93 the Spearman's rank correlation coefficient,123 the Kendall tau rank correlation coefficient,124 and the scale-dependent metric root-mean-square deviation (RMSD).61
It is important to be cautious when attempting to objectively compare methods, and only new, unpublished data should be included in a validation set in order to keep the methods on equal footing. Otherwise, machine learning methods that have used part of the validation data in their training may appear to be more accurate than they really are. When available, comparisons that are performed by independent researchers are preferable.53,85 In one such study, the performance of four commonly used methods (SIFT, Align-GVGD, PolyPhen-2, and Xvar which is now called MutationAssessor) was compared for 267 well-characterized human missense mutations in the BRCA1, MSH2, MLH1, and TP53 genes.85 All four algorithms performed similarly, with an AUC of about 80%, but the predictions by each algorithm were often discordant even when each one was provided the same input alignment.85 Thus, while these methods perform similarly in their overall accuracy, their predictions are different,85 a phenomenon that is documented for other tools as well125 and suggests complementarity.109 There are also independent third-party challenges that use unpublished data to assess the ability of methods to predict the functional impact of mutations on proteins, including the critical assessment of genome interpretation (CAGI),126 in which competing groups evaluate genetic variants blindly and have their predictions judged against experimental results on a variety of measures. Most often, no single method outperforms all others in every one of these diverse measures of quality; nevertheless an average rank can be calculated for each method over all of the quality measures. In Figure 3, we plotted the average ranks of impact predictors in two of the CAGI challenges, where we participated with predictions made by the evolutionary action method (simply Action). The identities of methods other than our own will remain anonymous until the CAGI community publishes comprehensive results.
Figure 3.
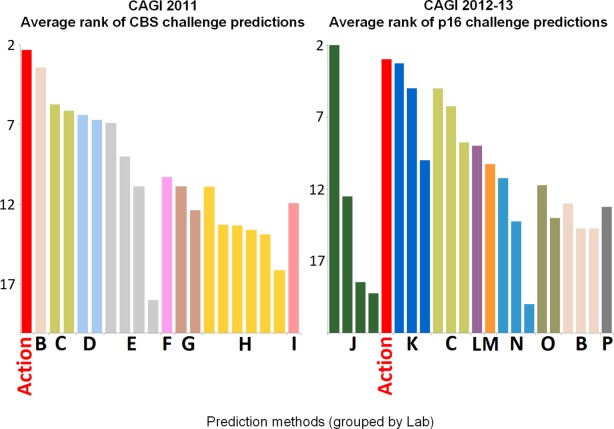
Average rank of predictions in two CAGI challenges from the competitions of 2011 and 2012–13. The Cystathionine beta-Synthase (CBS) challenge of 2011 asked predictors to submit the effect of 84 variants in the function of CBS at two different cofactor concentrations,127 which were assessed by nine measures for each concentration (precision, recall, accuracy, harmonic mean F1, Spearman's rank, Student's t test, RMSD, RMSD over z scores, and AUC). The p16 challenge of 2012–13 asked predictors to submit evaluations of how 10 variants of the p16 protein impact its ability to block cell proliferation,128 which were assessed by four measures (AUC, RMSD, Kendall tau, and the number of correct predictions within a range of 10%). A total of 16 participants (color-coded) to one or both challenges submitted one or multiple predictions (20 predictions in 2011 and 22 predictions in 2012–13). The number shown on the vertical axis is an average rank so that in order to have a rank of one, the prediction would need to rank first in all of the evaluation measures that were used. Conversely, the worst a prediction could do would be to be last in every evaluation measure, leading to an average rank equal to the total number of prediction sets in that challenge. Besides Action, only the participants B and C submitted predictions in both challenges. The Evolutionary Action method can be found at: http://mammoth.bcm.tmc.edu/EvolutionaryAction/.
A way to estimate the popularity of the impact prediction methods is to look at the number of citations per method over years since publication (Fig. 4). It is clear that certain methods, such as PolyPhen, SIFT, Panther, and Dmutant, have made a lasting impression on the field, and that the methods featured in Table1 have such a large variety in their number of citations that only a logarithmic scale can adequately capture the spread. While this data does not relate to the accuracy of the method or to the applicability of the method to a dataset, it reflects the scientific community's perception of the method.
Figure 4.
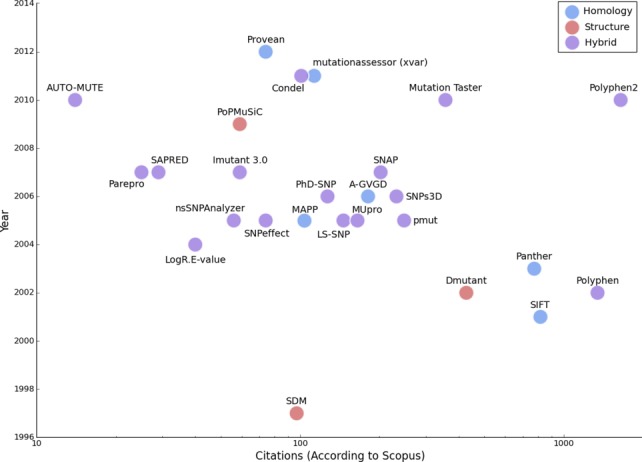
The total number of citations since each method was published, on a logarithmic scale, according to Scopus2 for methods published before 2014. The methods are colored by the type of information they use as seen in the figure legend. The older and well-established methods of PolyPhen, DMutant, SIFT, and Panther are at the bottom right, in contrast to the new and less-known, methods at the top left, while an abundance of methods are clustered at the center of the graph. Of particular interest is PolyPhen2, which despite its recent release, it is currently the most cited of any method.
In summary, one may choose an impact prediction method not only based on its accuracy against a variety of benchmark datasets, but also based on the strengths and limitations of the method in the context of the data at hand. The availability of a structure, the number of available homologs, the convenience of a predictor (web server or local installation), and the ability to submit multiple requests with various formats (vcf files or lists of single amino acid variants) may all affect the preference of a user in practice. In general, the confidence of a prediction is higher when multiple methods are in agreement,129 so studies often use the results from multiple methods to bolster evidence for pathogenicity.42,130,131 To this end, metaservers that compile the results from multiple methods are often time-saving, and several are noted in Supporting Information Table1 along with the original methods.
Applications
Typically, SNV impact prediction methods are used to associate amino acid variations to loss of protein function or to risk of diseases. An increasing number of studies use the predicted impact in a variety of applications, and have reported that SNV impact predictions match experimental findings.130,132,133 Such applications include guiding mutagenesis,134,135 identifying disease associated genes in both Mendelian and common diseases,1,136–139 separating disease-causing variants from linkage disequilibrium variants,140 identifying somatic mutations that drive cancer,65,141,142 and predicting the overall phenotype of an organism.143 These applications highlight the value of SNV impact prediction and the need for further improvement.
Guided mutagenesis
Predictions of impact may guide mutagenesis studies that aim to uncover functional sites or fine-tune the activity of proteins. Rather than using laborious random mutagenesis and screening to identify functional residues,144,145 site-directed mutagenesis studies146 may be efficiently guided by computational predictions with high rates of success.134,147–149 Besides selecting strongly deleterious mutations that knock out protein function, often it is desirable to select mutations with an intermediate impact in order to redirect the protein activity.135 Methods like EA, which yield prediction on a continuous scale rather than in binary categories, are appropriate to engineer functional proteins that deviate variably from the wild-type phenotype.135
nsSNV disease association
nsSNV impact predictors can also aid in untangling disease etiology. Although thousands of associations have been made between nsSNVs and risk of various diseases through GWAS and catalogued in databases like HGMD,120 dbSNP,150 ENSEMBL,151 and UniProt,152 it is often unclear if the nsSNV itself is causative or merely linked to the disease-causing variant. In addition, predisposing nsSNVs usually account for a small fraction of the predicted genetic risk of the complex diseases, a major issue known as “missing” heritability.153–155 Current theories suggest that common diseases are caused by either common variants with small to modest effects155 or by multiple rare variants.156 In both cases the statistical power is limited by either the linkage disequilibrium or the low population frequency, respectively. nsSNV impact predictions may be used to distinguish the most deleterious nsSNVs from those that are merely in strong linkage disequilibrium with a causative nsSNV,157 or identify deleterious rare nsSNVs that occur on genes that are associated with the disease.158–160
Identifying genes that cause diseases
Another use of impact predictors is to discover genes associated with genetic disorders.161 In these studies, exome sequencing of unrelated patients with the disorder is conducted under the hypothesis that these exomes will be enriched in mutations that impact the function of a causative gene. The predicted impact of SNVs on protein function may then be used to associate new genes with the studied disorder, such as the genes FRAS1 and FREM2 with Congenital Abnormalities of the Kidney and Urinary Tract (CAKUT),136 the DHODH gene with the Miller syndrome,137 the SLC26A3 gene with Bartter syndrome,1 the TGM6 gene with spinocerebellar ataxias,138 and the VCP gene with Amyotrophic Lateral Sclerosis (ALS).139 With more exome sequencing studies on the way, there is much potential for the widespread use of mutation impact predictors in the clinical setting, given their continuous improvement and almost immediate access to results.
Identifying cancer driver mutations
The search for cancer-associated mutations also benefits from predictions of the functional impact. This is a particularly challenging problem, since although cancer-causing mutations may be inherited,162,163 most often they are acquired in somatic cells during tumor development.164,165 The average number of nonsynonymous somatic mutations in a tumor varies widely by cancer type, ranging from as low as four in pediatric rhabdoid cancer to as high as the thousands in colorectal cancer with microsatellite instability.166 Some of these mutations, called drivers, disrupt or further activate the function of proteins to promote cancer, while the rest confer no selective tumor growth advantage and are called passengers.167 Predicting the impact of the variants found by exome sequencing of numerous tumors can help in identifying the genes that are associated with each cancer type.168–170 Moreover, nsSNV impact can provide clinical information. For example, even when only the TP53 gene is under consideration, predicting the impact of head and neck tumor mutations can stratify patient survival into statistically significant groups.171
Several nsSNV impact predictors have specifically applied their method to cancer gene discovery, including CanPredict,141 MutationAssessor,65 and SNPs3D.142 CanPredict is a Random Forest classifier, trained on 800 cancerous and 200 non-cancerous mutations, that uses SIFT172 and Pfam-based scores173 to predict impact, and Gene Ontology174 to predict cancer association. This method identified as cancer-associated several novel germline variants that were not present in controls, suggesting they are markers for increased cancer risk.141 The MutationAssessor method predicted the impact of over 10,000 nsSNVs from the COSMIC database,175 which combined with the total number of mutations in a gene and the frequency of each mutation in different tumors, ranked genes for cancer association, recovering known drivers (TP53, PTEN, etc) and suggesting many others.65 The SNPs3D method, consisting of two SVMs based on protein stability and homology respectively, was applied to about 2000 somatic mutations from colorectal and breast cancer to find that virtually all mutations in known cancer genes are predicted to impact protein function and therefore can be detected by nsSNV impact prediction methods.142 These methods produced intriguing novel predictions and may foreshadow wider use of nsSNV impact predictions to elucidate cancer mechanisms.
Predicting the phenotypic behavior of single organisms by integrating the impact of multiple mutations
Although a simple, clinically useful pipeline to reliably annotate all likely phenotypes from a human genome is not yet possible,176 predicting phenotypic variation from genome sequences has made significant advances in model organisms like yeast and has illustrated the centrality of SNV impact prediction to these efforts.143 Genome-scale reverse genetic screens in model organisms have produced thorough, if not complete, sets of genes associated with a variety of phenotypes, aiding the prediction process and allowing for proof-of-concept experiments that apply to human genotype-to-phenotype research.177 One such study used gene sets for 115 phenotypes described by the Saccharomyces Genome Database (SGD)178 and considered how the mutational load in the protein-coding regions of these gene sets varied by yeast strain. The study applied a nsSNV effect predictor, SIFT,172 to determine the probability of damage for non-synonymous mutations. The overall phenotypic effect was calculated with an additive model that combined the SIFT scores with heuristic rules that evaluated premature stop codons and insertions and deletions.178 The actual phenotypic responses of the strains were experimentally determined and they were predicted by the genotype with an ROC AUC value of 0.76.143 These results offer hope that in the future SNV impact prediction methods may be similarly applied to integrate the impact of multiple mutations in the human genome as the genes known to be associated with a phenotype become more complete.179
Future Directions
What are the future challenges the field of SNV impact prediction needs to address?
Context-dependence
Despite steady progress in predicting the impact of non-synonymous coding variations, there remains a myriad of challenges for determining how the phenotype of an individual organism is affected by a specific SNV. For example, it is important to know whether and how the phenotypic impact is mitigated by zygosity,180,181 epistasis,182,183 mosaicism,184 gender,185,186 environment,187 epigenetics,188,189 or other unknown factors affecting penetrance and expressivity.190 The recently launched “Resilience Project”191 aims precisely to identify the factors that buffer disease in apparently healthy patients that carry high-risk disease variants.192 As our understanding of these factors expands, we may be able to incorporate this information on a large scale and provide personalized impact predictions.
Impact of protein function loss on phenotype
A necessary intermediate step in integrating genetic information is to understand the phenotypic association of each protein and its impact on the overall fitness of a species. For example, a SNV in a gene may render the protein nonfunctional, but this loss of protein function can, depending on the role of that protein, be fairly neutral to the organism193 or have observable consequences,194,195 including lethality.196 An additional complication comes from the redundant function of proteins or pathways, resulting in no noticeable phenotypic change when losing the function of only one involved gene.197,198 SNV impact prediction does not yet make any a priori assumptions about gene importance, but when the gene involved in the phenotype is well established, it can stratify patient outcomes171 and disease severity.50 Large-scale projects like the NIH Knockout Mouse Project (KOMP)196 and particularly systematic surveys of incidental human knockouts199–201 promise to shed light on the relative importance of the genes, their role in diseases, and the gene redundancy within a genome, presenting an opportunity for a leap forward in variant prioritization.
Noncoding regions
Finally, evidence that more than 80% of the human genome may display some functionality202 suggests that there are important limitations in exclusively analyzing exome sequencing data. Consequently, SNV impact prediction is beginning to branch into noncoding regions of the genome. Two recent tools, mrSNP39 and MicroSNiPer40 attempt to identify SNVs in 3'UTR regions that disrupt miRNA binding, and RNAsnp41 predicts the effect of SNVs on the local structure of noncoding RNAs. Future tools will hopefully expand upon this work and may also begin to predict how non-coding SNVs alter methylation patterns and other epigenetic changes.203,204 With the discovery that SNVs in noncoding regions are sometimes disease associated,34–38 additional methods to deal with these variants will likely arise over time to tackle this problem.
Developing computational methods to estimate the functional impact of SNVs is crucial to understanding the genotype–phenotype relationship, and their importance to research and clinical practice will only grow as sequencing costs plummet further. Already many nsSNV impact prediction methods find broad applications to guided mutagenesis and to the identification of disease causing variants and genes. There are already a plethora of tools available and many new ones complicate the choice of which to use. This review explored current predictors of functional impact in light of the strengths and limitations of the fundamental principles they apply. Factors such as tool availability, public usage, and, most importantly, accuracy must be carefully weighed and understood in the context of the target dataset. In the future, the technical improvements and the availability of new sequence and SNV data should help the computational methods to predict the impact of SNVs with even higher accuracy.
Acknowledgments
The authors gratefully acknowledge support from the National Institutes of Health (GM079656 and GM066099) and from the National Science Foundation (DBI-1062455 and CCF-0905536).
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information Figure 1.
Supplementary Information Table 1.
References
- Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scopus—document search. Available at: < http://www.scopus.com/ >. Accessed 2014.
- DNA Sequencing Costs. Available at: < http://www.genome.gov/sequencingcosts/ >. Accessed on June 15, 2014.
- Whole exome seq—compare prices and order services—science exchange. Available at: < https://www.scienceexchange.com/services/whole-exome- seq >. Accessed on June 24, 2014.
- Hayden EC. Technology: the $1,000 genome. Nature. 2014;507:294–295. doi: 10.1038/507294a. [DOI] [PubMed] [Google Scholar]
- Amos CI. Successful design and conduct of genome-wide association studies. Hum Mol Genet. 2007:R220–R225. doi: 10.1093/hmg/ddm161. ;16 Spec No.2: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seng KC, Seng CK. The success of the genome-wide association approach: a brief story of a long struggle. Eur J Hum Genet. 2008;16:554–564. doi: 10.1038/ejhg.2008.12. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurusaki Y, Kobayashi Y, Hisano M, Ito S, Doi H, Nakashima M, Saitsu H, Matsumoto N, Miyake N. The diagnostic utility of exome sequencing in Joubert syndrome and related disorders. J Hum Genet. 2013;58:113–115. doi: 10.1038/jhg.2012.117. [DOI] [PubMed] [Google Scholar]
- Schessl J, Bach E, Rost S, Feldkirchner S, Kubny C, Müller S, Hanisch FG, Kress W, Schoser B. Novel recessive myotilin mutation causes severe myofibrillar myopathy. Neurogenetics. 2014;15:151–156. doi: 10.1007/s10048-014-0410-4. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke SL, Shlien A, Marshall J, Pipinikas CP, Martincorena I, Tubio JM, Li Y, Menzies A, Mudie L, Ramakrishna M, Yates L, Davies H, Bolli N, Bignell GR, Tarpey PS, Behjati S, Nik-Zainal S, Papaemmanuil E, Teixeira VH, Raine K, O'Meara S, Dodoran MS, Teague JW, Butler AP, Iacobuzio-Donahue C, Santarius T, Grundy RG, Malkin D, Greaves M, Munshi N, Flanagan AM, Bowtell D, Martin S, Larsimont D, Reis-Filho JS, Boussioutas A, Taylor JA, Hayes ND, Janes SM, Futreal PA, Stratton MR, McDermott U, Campbell PJ, ICGC Breast Cancer Group. Processed pseudogenes acquired somatically during cancer development. Nat Commun. 2014;5:3644. doi: 10.1038/ncomms4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, Lopez-Otin C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdes-Mas R, van Buuren MM, van't Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome I, Consortium IBC, Consortium IM-S, PedBrain I, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Cancer Genome Consortium. Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, Bhan MK, Calvo F, Eerola I, Gerhard DS, Guttmacher A, Guyer M, Hemsley FM, Jennings JL, Kerr D, Klatt P, Kolar P, Kusada J, Lane DP, Laplace F, Youyong L, Nettekoven G, Ozenberger B, Peterson J, Rao TS, Remacle J, Schafer AJ, Shibata T, Stratton MR, Vockley JG, Watanabe K, Yang H, Yuen MM, Knoppers BM, Bobrow M, Cambon-Thomsen A, Dressler LG, Dyke SO, Joly Y, Kato K, Kennedy KL, Nicolas P, Parker MJ, Rial-Sebbag E, Romeo-Casabona CM, Shaw KM, Wallace S, Wiesner GL, Zeps N, Lichter P, Biankin AV, Chabannon C, Chin L, Clement B, de Alava E, Degos F, Ferguson ML, Geary P, Hayes DN, Hudson TJ, Johns AL, Kasprzyk A, Nakagawa H, Penny R, Piris MA, Sarin R, Scarpa A, Shibata T, van de Vijver M, Futreal PA, Aburatani H, Bayes M, Botwell DD, Campbell PJ, Estivill X, Gerhard DS, Grimmond SM, Gut I, Hirst M, Lopez-Otin C, Majumder P, Marra M, McPherson JD, Nakagawa H, Ning Z, Puente XS, Ruan Y, Shibata T, Stratton MR, Stunnenberg HG, Swerdlow H, Velculescu VE, Wilson RK, Xue HH, Yang L, Spellman PT, Bader GD, Boutros PC, Campbell PJ, Flicek P, Getz G, Guigo R, Guo G, Haussler D, Heath S, Hubbard TJ, Jiang T, Jones SM, Li Q, Lopez-Bigas N, Luo R, Muthuswamy L, Ouellette BF, Pearson JV, Puente XS, Quesada V, Raphael BJ, Sander C, Shibata T, Speed TP, Stein LD, Stuart JM, Teague JW, Totoki Y, Tsunoda T, Valencia A, Wheeler DA, Wu H, Zhao S, Zhou G, Stein LD, Guigo R, Hubbard TJ, Joly Y, Jones SM, Kasprzyk A, Lathrop M, Lopez-Bigas N, Ouellette BF, Spellman PT, Teague JW, Thomas G, Valencia A, Yoshida T, Kennedy KL, Axton M, Dyke SO, Futreal PA, Gerhard DS, Gunter C, Guyer M, Hudson TJ, McPherson JD, Miller LJ, Ozenberger B, Shaw KM, Kasprzyk A, Stein LD, Zhang J, Haider SA, Wang J, Yung CK, Cros A, Liang Y, Gnaneshan S, Guberman J, Hsu J, Bobrow M, Chalmers DR, Hasel KW, Joly Y, Kaan TS, Kennedy KL, Knoppers BM, Lowrance WW, Masui T, Nicolas P, Rial-Sebbag E, Rodriguez LL, Vergely C, Yoshida T, Grimmond SM, Biankin AV, Bowtell DD, Cloonan N, deFazio A, Eshleman JR, Etemadmoghadam D, Gardiner BB, Kench JG, Scarpa A, Sutherland RL, Tempero MA, Waddell NJ, Wilson PJ, McPherson JD, Gallinger S, Tsao MS, Shaw PA, Petersen GM, Mukhopadhyay D, Chin L, DePinho RA, Thayer S, Muthuswamy L, Shazand K, Beck T, Sam M, Timms L, Ballin V, Lu Y, Ji J, Zhang X, Chen F, Hu X, Zhou G, Yang Q, Tian G, Zhang L, Xing X, Li X, Zhu Z, Yu Y, Yu J, Yang H, Lathrop M, Tost J, Brennan P, Holcatova I, Zaridze D, Brazma A, Egevard L, Prokhortchouk E, Banks RE, Uhlen M, Cambon-Thomsen A, Viksna J, Ponten F, Skryabin K, Stratton MR, Futreal PA, Birney E, Borg A, Borresen-Dale AL, Caldas C, Foekens JA, Martin S, Reis-Filho JS, Richardson AL, Sotiriou C, Stunnenberg HG, Thoms G, van de Vijver M, van't Veer L, Calvo F, Birnbaum D, Blanche H, Boucher P, Boyault S, Chabannon C, Gut I, Masson-Jacquemier JD, Lathrop M, Pauporte I, Pivot X, Vincent-Salomon A, Tabone E, Theillet C, Thomas G, Tost J, Treilleux I, Calvo F, Bioulac-Sage P, Clement B, Decaens T, Degos F, Franco D, Gut I, Gut M, Heath S, Lathrop M, Samuel D, Thomas G, Zucman-Rossi J, Lichter P, Eils R, Brors B, Korbel JO, Korshunov A, Landgraf P, Lehrach H, Pfister S, Radlwimmer B, Reifenberger G, Taylor MD, von Kalle C, Majumder PP, Sarin R, Rao TS, Bhan MK, Scarpa A, Pederzoli P, Lawlor RA, Delledonne M, Bardelli A, Biankin AV, Grimmond SM, Gress T, Klimstra D, Zamboni G, Shibata T, Nakamura Y, Nakagawa H, Kusada J, Tsunoda T, Miyano S, Aburatani H, Kato K, Fujimoto A, Yoshida T, Campo E, Lopez-Otin C, Estivill X, Guigo R, de Sanjose S, Piris MA, Montserrat E, Gonzalez-Diaz M, Puente XS, Jares P, Valencia A, Himmelbauer H, Quesada V, Bea S, Stratton MR, Futreal PA, Campbell PJ, Vincent-Salomon A, Richardson AL, Reis-Filho JS, van de Vijver M, Thomas G, Masson-Jacquemier JD, Aparicio S, Borg A, Borresen-Dale AL, Caldas C, Foekens JA, Stunnenberg HG, van't Veer L, Easton DF, Spellman PT, Martin S, Barker AD, Chin L, Collins FS, Compton CC, Ferguson ML, Gerhard DS, Getz G, Gunter C, Guttmacher A, Guyer M, Hayes DN, Lander ES, Ozenberger B, Penny R, Peterson J, Sander C, Shaw KM, Speed TP, Spellman PT, Vockley JG, Wheeler DA, Wilson RK, Hudson TJ, Chin L, Knoppers BM, Lander ES, Lichter P, Stein LD, Stratton MR, Anderson W, Barker AD, Bell C, Bobrow M, Burke W, Collins FS, Compton CC, DePinho RA, Easton DF, Futreal PA, Gerhard DS, Green AR, Guyer M, Hamilton SR, Hubbard TJ, Kallioniemi OP, Kennedy KL, Ley TJ, Liu ET, Lu Y, Majumder P, Marra M, Ozenberger B, Peterson J, Schafer AJ, Spellman PT, Stunnenberg HG, Wainwright BJ, Wilson RK, Yang H. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi4K Consortium. Epi4K: gene discovery in 4,000 genomes. Epilepsia. 2012;53:1457–1467. doi: 10.1111/j.1528-1167.2012.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu YF, Madou MR, Marson AG, Mefford HC, Esmaeeli Nieh S, O'Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin Thio L, Venkat A, Vining EP, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, Winawer MR. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tryka KA, Hao L, Sturcke A, Jin Y, Wang ZY, Ziyabari L, Lee M, Popova N, Sharopova N, Kimura M, Feolo M. NCBI's database of genotypes and phenotypes: dbGaP. Nucleic Acids Res. 2014;42:D975–D979. doi: 10.1093/nar/gkt1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, Popova N, Pretel S, Ziyabari L, Lee M, Shao Y, Wang ZY, Sirotkin K, Ward M, Kholodov M, Zbicz K, Beck J, Kimelman M, Shevelev S, Preuss D, Yaschenko E, Graeff A, Ostell J, Sherry ST. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, Leal SM, Gabriel S, Rieder MJ, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante CD, Bamshad MJ, Akey JM. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, Shendure J, Drmanac R, Jorde LB, Hood L, Galas DJ. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, Goode DL, Kvikstad E, Albers CA, Zhang ZD, Mu XJ, Ananda G, Howie B, Karczewski KJ, Smith KS, Anaya V, Richardson R, Davis J 1000 Genomes Project Consortium. MacArthur DG, Sidow A, Duret L, Gerstein M, Makova KD, Marchini J, McVean G, Lunter G. The origin, evolution, and functional impact of short insertion-deletion variants identified in 179 human genomes. Genome Res. 2013;23:749–761. doi: 10.1101/gr.148718.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, McGuire AL, Zhang F, Stankiewicz P, Halperin JJ, Yang C, Gehman C, Guo D, Irikat RK, Tom W, Fantin NJ, Muzny DM, Gibbs RA. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362:1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Levy S, Huang J, Stockwell TB, Walenz BP, Li K, Axelrod N, Busam DA, Strausberg RL, Venter JC. Genetic variation in an individual human exome. PLoS Genet. 2008;4:e1000160. doi: 10.1371/journal.pgen.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Ju YS, Park H, Kim S, Lee S, Yi JH, Mudge J, Miller NA, Hong D, Bell CJ, Kim HS, Chung IS, Lee WC, Lee JS, Seo SH, Yun JY, Woo HN, Lee H, Suh D, Lee S, Kim HJ, Yavartanoo M, Kwak M, Zheng Y, Lee MK, Park H, Kim JY, Gokcumen O, Mills RE, Zaranek AW, Thakuria J, Wu X, Kim RW, Huntley JJ, Luo S, Schroth GP, Wu TD, Kim H, Yang KS, Park WY, Kim H, Church GM, Lee C, Kingsmore SF, Seo JS. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–1015. doi: 10.1038/nature08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehne B, Lewis CM, Schlitt T. From SNPs to genes: disease association at the gene level. PloS One. 2011;6:e20133. doi: 10.1371/journal.pone.0020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti A. To a future of genetic medicine. Nature. 2001;409:822–823. doi: 10.1038/35057281. [DOI] [PubMed] [Google Scholar]
- Lazrak A, Fu L, Bali V, Bartoszewski R, Rab A, Havasi V, Keiles S, Kappes J, Kumar R, Lefkowitz E, Sorscher EJ, Matalon S, Collawn JF, Bebok Z. The silent codon change I507-ATC->ATT contributes to the severity of the DeltaF508 CFTR channel dysfunction. FASEB J. 2013;27:4630–4645. doi: 10.1096/fj.13-227330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Kim H, Verhaak RG. Silent mutations make some noise. Cell. 2014;156:1129–1131. doi: 10.1016/j.cell.2014.02.037. [DOI] [PubMed] [Google Scholar]
- Ning S, Wang P, Ye J, Li X, Li R, Zhao Z, Huo X, Wang L, Li F, Li X. A global map for dissecting phenotypic variants in human lincRNAs. Eur J Hum Genet. 2013;21:1128–1133. doi: 10.1038/ejhg.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Wang R, Zhang J, Li W, Gao C, Liu J, Wang J. Identification of miR-423 and miR-499 polymorphisms on affecting the risk of hepatocellular carcinoma in a large-scale population. Genet Test Mol Biomark. 2014;18:516–524. doi: 10.1089/gtmb.2013.0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PW, Huang YC, Hsieh CY, Hua KT, Huang YT, Chiang TH, Chen JS, Huang PM, Hsu HH, Kuo SW, Kuo ML, Lee JM. Association of miRNA-related genetic polymorphisms and prognosis in patients with esophageal squamous cell carcinoma. Ann Surg Oncol. 2014 doi: 10.1245/s10434-014-3709-3. PMID: 24770678. [DOI] [PubMed] [Google Scholar]
- Han Q, Zhang Y, Li W, Fan H, Xing Q, Pang S, Yan B. Functional sequence variants within the SIRT1 gene promoter in indirect inguinal hernia. Gene. 2014;546:1–5. doi: 10.1016/j.gene.2014.05.058. [DOI] [PubMed] [Google Scholar]
- De Castro-Oros I, Perez-Lopez J, Mateo-Gallego R, Rebollar S, Ledesma M, Leon M, Cofan M, Casasnovas JA, Ros E, Rodriguez-Rey JC, Civeira F, Pocovi M. A genetic variant in the LDLR promoter is responsible for part of the LDL-cholesterol variability in primary hypercholesterolemia. BMC Med Genom. 2014;7:17. doi: 10.1186/1755-8794-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveci M, Catalyürek UV, Toland AE. mrSNP: software to detect SNP effects on microRNA binding. BMC Bioinform. 2014;15:73. doi: 10.1186/1471-2105-15-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barenboim M, Zoltick BJ, Guo Y, Weinberger DR. MicroSNiPer: a web tool for prediction of SNP effects on putative microRNA targets. Hum Mutat. 2010;31:1223–1232. doi: 10.1002/humu.21349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabarinathan R, Tafer H, Seemann SE, Hofacker IL, Stadler PF, Gorodkin J. RNAsnp: efficient detection of local RNA secondary structure changes induced by SNPs. Hum Mutat. 2013;34:546–556. doi: 10.1002/humu.22273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira LG, Pereira LC, Drummond PR, De Mesquita JF. Structural and functional analysis of human SOD1 in amyotrophic lateral sclerosis. PloS One. 2013;8:e81979. doi: 10.1371/journal.pone.0081979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saranko H, Tordai H, Telbisz A, Ozvegy-Laczka C, Erdos G, Sarkadi B, Hegedus T. Effects of the gout-causing Q141K polymorphism and a CFTR DeltaF508 mimicking mutation on the processing and stability of the ABCG2 protein. Biochem Biophys Res Commun. 2013;437:140–145. doi: 10.1016/j.bbrc.2013.06.054. [DOI] [PubMed] [Google Scholar]
- Duning K, Wennmann DO, Bokemeyer A, Reissner C, Wersching H, Thomas C, Buschert J, Guske K, Franzke V, Flöel A, Lohmann H, Knecht S, Brand SM, Pöter M, Rescher U, Missler M, Seelheim P, Pröpper C, Boeckers TM, Makuch L, Huganir R, Weide T, Brand E, Pavenstädt H, Kremerskothen J. Common exonic missense variants in the C2 domain of the human KIBRA protein modify lipid binding and cognitive performance. Translat Psych. 2013;3:e272. doi: 10.1038/tp.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg H, Rowntree TJ, Tan SL, Drickamer K, Weis WI, Taylor ME. Common polymorphisms in human langerin change specificity for glycan ligands. J Biol Chem. 2013;288:36762–36771. doi: 10.1074/jbc.M113.528000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraksingh RR, Snyder MP. Impacts of variation in the human genome on gene regulation. J Mol Biol. 2013;425:3970–3977. doi: 10.1016/j.jmb.2013.07.015. [DOI] [PubMed] [Google Scholar]
- Yates CM, Sternberg MJ. The effects of non-synonymous single nucleotide polymorphisms (nsSNPs) on protein-protein interactions. J Mol Biol. 2013;425:3949–3963. doi: 10.1016/j.jmb.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Reimand J, Bader GD. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol Syst Biol. 2013;9:637. doi: 10.1038/msb.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Fu YX, Hewett-Emmett D, Boerwinkle E. Investigating single nucleotide polymorphism (SNP) density in the human genome and its implications for molecular evolution. Gene. 2003;312:207–213. doi: 10.1016/s0378-1119(03)00670-x. [DOI] [PubMed] [Google Scholar]
- Katsonis P, Lichtarge O. A formal perturbation relationship between genotype and phenotype determines the action of protein coding variations. Genome Res. 2014 doi: 10.1101/gr.176214.114. PMID: 25217195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. The genetic theory of adaptation: a brief history. Nat Rev Genet. 2005;6:119–127. doi: 10.1038/nrg1523. [DOI] [PubMed] [Google Scholar]
- Fisher RA. The genetical theory of natural selection. Oxford: The Clarendon Press; 1930. p. 272. [Google Scholar]
- Valdmanis PN, Verlaan DJ, Rouleau GA. The proportion of mutations predicted to have a deleterious effect differs between gain and loss of function genes in neurodegenerative disease. Human Mutat. 2009;30:E481–E489. doi: 10.1002/humu.20939. [DOI] [PubMed] [Google Scholar]
- Topham CM, Srinivasan N, Blundell TL. Prediction of the stability of protein mutants based on structural environment-dependent amino acid substitution and propensity tables. Protein Eng. 1997;10:7–21. doi: 10.1093/protein/10.1.7. [DOI] [PubMed] [Google Scholar]
- Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11:2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue WW, Froese DS, Brennan PE. The role of protein structural analysis in the next generation sequencing era. Top Curr Chem. 2014;336:67–98. doi: 10.1007/128_2012_326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Moult J. SNPs, protein structure, and disease. Hum Mutat. 2001;17:263–270. doi: 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]
- Pepys MB, Hawkins PN, Booth DR, Vigushin DM, Tennent GA, Soutar AK, Totty N, Nguyen O, Blake CC, Terry CJ. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature. 1993;362:553–557. doi: 10.1038/362553a0. [DOI] [PubMed] [Google Scholar]
- Pifer PM, Yates EA, Legleiter J. Point mutations in Abeta result in the formation of distinct polymorphic aggregates in the presence of lipid bilayers. PLoS One. 2011;6:e16248. doi: 10.1371/journal.pone.0016248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander PA, He Y, Chen Y, Orban J, Bryan PN. A minimal sequence code for switching protein structure and function. Proc Natl Acad Sci USA. 2009;106:21149–21154. doi: 10.1073/pnas.0906408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehouck Y, Grosfils A, Folch B, Gilis D, Bogaerts P, Rooman M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics. 2009;25:2537–2543. doi: 10.1093/bioinformatics/btp445. [DOI] [PubMed] [Google Scholar]
- Reumers J, Schymkowitz J, Ferkinghoff-Borg J, Stricher F, Serrano L, Rousseau F. SNPeffect: a database mapping molecular phenotypic effects of human non-synonymous coding SNPs. Nucleic Acids Res. 2005;33:D527–D532. doi: 10.1093/nar/gki086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins. 2006;62:1125–1132. doi: 10.1002/prot.20810. [DOI] [PubMed] [Google Scholar]
- Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preeprem T, Gibson G. SDS, a structural disruption score for assessment of missense variant deleteriousness. Front Genet. 2014;5:82. doi: 10.3389/fgene.2014.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P, Li Z, Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. J Mol Biol. 2005;353:459–473. doi: 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Bork P. Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends Genet. 2000;16:198–200. doi: 10.1016/s0168-9525(00)01988-0. [DOI] [PubMed] [Google Scholar]
- Wang X, Wei X, Thijssen B, Das J, Lipkin SM, Yu H. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat Biotech. 2012;30:159–164. doi: 10.1038/nbt.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E, Altman RB. Improving the prediction of disease-related variants using protein three-dimensional structure. BMC Bioinform. 2011;12(Suppl 4):S3. doi: 10.1186/1471-2105-12-S4-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinform. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Coimbatore Narayanan B, Costanzo LD, Dutta S, Ghosh S, Hudson BP, Lawson CL, Peisach E, Prlic A, Rose PW, Shao C, Yang H, Young J, Zardecki C. Trendspotting in the Protein Data Bank. FEBS Lett. 2013;587:1036–1045. doi: 10.1016/j.febslet.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PDB Reaches a New Milestone: 100,000+ Entries. Available at: < http://www.wwpdb.org/news/news_2014.html#13-May-2014 >. Accessed on June 21, 2014.
- Yates CM, Filippis I, Kelley LA, Sternberg MJ. SuSPect: enhanced prediction of single amino acid variant (SAV) phenotype using network features. J Mol Biol. 2014;426:2692–2701. doi: 10.1016/j.jmb.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- The Worldwide Protein Data Bank. Available at: < http://www.wwpdb.org/policy.html >. Accessed on June 22, 2014.
- Domagalski MJ, Zheng H, Zimmerman MD, Dauter Z, Wlodawer A, Minor W. The quality and validation of structures from structural genomics. Methods Mol Biol. 2014;1091:297–314. doi: 10.1007/978-1-62703-691-7_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Cui Y. Prediction of the phenotypic effects of non-synonymous single nucleotide polymorphisms using structural and evolutionary information. Bioinformatics. 2005;21:2185–2190. doi: 10.1093/bioinformatics/bti365. [DOI] [PubMed] [Google Scholar]
- Saunders CT, Baker D. Evaluation of structural and evolutionary contributions to deleterious mutation prediction. J Mol Biol. 2002;322:891–901. doi: 10.1016/s0022-2836(02)00813-6. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Performance evaluation of amino acid substitution matrices. Proteins. 1993;17:49–61. doi: 10.1002/prot.340170108. [DOI] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane CR, Lim EP, Kalyanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley GQ, Lander ES. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. Proc Natl Acad Sci USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks S, Wheeler DA, Plon SE, Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum Mutat. 2011;32:661–668. doi: 10.1002/humu.21490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A, Thomas A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- Mihalek I, Res I, Lichtarge O. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J Mol Biol. 2004;336:1265–1282. doi: 10.1016/j.jmb.2003.12.078. [DOI] [PubMed] [Google Scholar]
- Ferrer-Costa C, Gelpi JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- Sommer C, Gerlich DW. Machine learning in cell biology—teaching computers to recognize phenotypes. J Cell Sci. 2013;126:5529–5539. doi: 10.1242/jcs.123604. [DOI] [PubMed] [Google Scholar]
- Karchin R, Diekhans M, Kelly L, Thomas DJ, Pieper U, Eswar N, Haussler D, Sali A. LS-SNP: large-scale annotation of coding non-synonymous SNPs based on multiple information sources. Bioinformatics. 2005;21:2814–2820. doi: 10.1093/bioinformatics/bti442. [DOI] [PubMed] [Google Scholar]
- Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. 2006;22:2729–2734. doi: 10.1093/bioinformatics/btl423. [DOI] [PubMed] [Google Scholar]
- Tian J, Wu N, Guo X, Guo J, Zhang J, Fan Y. Predicting the phenotypic effects of non-synonymous single nucleotide polymorphisms based on support vector machines. BMC Bioinform. 2007;8:450. doi: 10.1186/1471-2105-8-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappalardo M, Wass MN. VarMod: modelling the functional effects of non-synonymous variants. Nucleic Acids Res. 2014;42:W331–W336. doi: 10.1093/nar/gku483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Baets G, Van Durme J, Reumers J, Maurer-Stroh S, Vanhee P, Dopazo J, Schymkowitz J, Rousseau F. SNPeffect 4.0: on-line prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Res. 2012;40:D935–D939. doi: 10.1093/nar/gkr996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Zhou M, Cui Y. nsSNPAnalyzer: identifying disease-associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res. 2005;33:W480–W482. doi: 10.1093/nar/gki372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masso M, Vaisman II. AUTO-MUTE: web-based tools for predicting stability changes in proteins due to single amino acid replacements. Protein Eng Des Sel. 2010;23:683–687. doi: 10.1093/protein/gzq042. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford RJ, Edmonson MN, Nguyen C, Buetow KH. Large-scale analysis of non-synonymous coding region single nucleotide polymorphisms. Bioinformatics. 2004;20:1006–1014. doi: 10.1093/bioinformatics/bth029. [DOI] [PubMed] [Google Scholar]
- Capriotti E, Fariselli P, Rossi I, Casadio R. A three-state prediction of single point mutations on protein stability changes. BMC Bioinform. 2008;9:S6. doi: 10.1186/1471-2105-9-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZQ, Zhao SQ, Gao G, Liu XQ, Langlois RE, Lu H, Wei L. Finding new structural and sequence attributes to predict possible disease association of single amino acid polymorphism (SAP) Bioinformatics. 2007;23:1444–1450. doi: 10.1093/bioinformatics/btm119. [DOI] [PubMed] [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath U, De Jong K, Shehu A. Effective automated feature construction and selection for classification of biological sequences. PloS One. 2014;9:e99982. doi: 10.1371/journal.pone.0099982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minde DP, Anvarian Z, Rüdiger SG, Maurice MM. Messing up disorder: how do missense mutations in the tumor suppressor protein APC lead to cancer? Mol Cancer. 2011;10:101. doi: 10.1186/1476-4598-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, Mooney SD, Radivojac P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25:2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastanlar Y, Ozuysal M. Introduction to machine learning. Methods Mol Biol. 2014;1107:105–128. doi: 10.1007/978-1-62703-748-8_7. [DOI] [PubMed] [Google Scholar]
- Lemm S, Blankertz B, Dickhaus T, Muller KR. Introduction to machine learning for brain imaging. Neuroimage. 2011;56:387–399. doi: 10.1016/j.neuroimage.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Karchin R. Next generation tools for the annotation of human SNPs. Brief Bioinform. 2009;10:35–52. doi: 10.1093/bib/bbn047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suckow J, Markiewicz P, Kleina LG, Miller J, Kisters-Woike B, Müller-Hill B. Genetic studies of the Lac repressor. XV: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J Mol Biol. 1996;261:509–523. doi: 10.1006/jmbi.1996.0479. [DOI] [PubMed] [Google Scholar]
- Markiewicz P, Kleina LG, Cruz C, Ehret S, Miller JH. Genetic studies of the lac repressor. XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as "spacers" which do not require a specific sequence. J Mol Biol. 1994;240:421–433. doi: 10.1006/jmbi.1994.1458. [DOI] [PubMed] [Google Scholar]
- Loeb DD, Swanstrom R, Everitt L, Manchester M, Stamper SE, Hutchison CA. Complete mutagenesis of the HIV-1 protease. Nature. 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- Rennell D, Bouvier SE, Hardy LW, Poteete AR. Systematic mutation of bacteriophage T4 lysozyme. J Mol Biol. 1991;222:67–88. doi: 10.1016/0022-2836(91)90738-r. [DOI] [PubMed] [Google Scholar]
- Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- Sasidharan Nair P, Vihinen M. VariBench: a benchmark database for variations. Hum Mutat. 2013;34:42–49. doi: 10.1002/humu.22204. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Stenson PD, Chuzhanova NA. The human gene mutation database (HGMD) and its exploitation in the study of mutational mechanisms. Curr Protoc Bioinform. 2006;12:1.13.1–1.13.20. doi: 10.1002/0471250953.bi0113s12. [DOI] [PubMed] [Google Scholar]
- Magrane M UniProt Consortium. UniProt Knowledgebase: a hub of integrated protein data. Database. 2011;2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, Lathe W, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Hoffman A, Wu X, Zhang H, Zhang Y, Leaderer D, Zheng T. Correlating observed odds ratios from lung cancer case-control studies to SNP functional scores predicted by bioinformatic tools. Mutat Res. 2008;639:80–88. doi: 10.1016/j.mrfmmm.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giollo M, Martin AJ, Walsh I, Ferrari C, Tosatto SC. NeEMO: a method using residue interaction networks to improve prediction of protein stability upon mutation. BMC Genom. 2014;15:S7. doi: 10.1186/1471-2164-15-S4-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellana S, Mazza T. Congruency in the prediction of pathogenic missense mutations: state-of-the-art web-based tools. Brief Bioinform. 2013;14:448–459. doi: 10.1093/bib/bbt013. [DOI] [PubMed] [Google Scholar]
- 2014 6/24/14. Critical Assessment of Genome Interpretation |. Available at: < https://genomeinterpretation.org/ >. Accessed 2014 6/24/14.
- Mayfield JA, Davies MW, Dimster-Denk D, Pleskac N, McCarthy S, Boydston EA, Fink L, Lin XX, Narain AS, Meighan M, Rine J. Surrogate genetics and metabolic profiling for characterization of human disease alleles. Genetics. 2012;190:1309–1323. doi: 10.1534/genetics.111.137471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaini MC, Minervini G, Elefanti L, Ghiorzo P, Pastorino L, Tognazzo S, Agata S, Quaggio M, Zullato D, Bianchi-Scarrá G, Montagna M, D'Andrea E, Menin C, Tosatto SC. CDKN2A unclassified variants in familial malignant melanoma: combining functional and computational approaches for their assessment. Hum Mutat. 2014;35:828–840. doi: 10.1002/humu.22550. [DOI] [PubMed] [Google Scholar]
- Chan PA, Duraisamy S, Miller PJ, Newell JA, McBride C, Bond JP, Raevaara T, Ollila S, Nyström M, Grimm AJ, Christodoulou J, Oetting WS, Greenblatt MS. Interpreting missense variants: comparing computational methods in human disease genes CDKN2A, MLH1, MSH2, MECP2, and tyrosinase (TYR) Hum Mutat. 2007;28:683–693. doi: 10.1002/humu.20492. [DOI] [PubMed] [Google Scholar]
- Jin ZB, Mandai M, Yokota T, Higuchi K, Ohmori K, Ohtsuki F, Takakura S, Itabashi T, Wada Y, Akimoto M, Ooto S, Suzuki T, Hirami Y, Ikeda H, Kawagoe N, Oishi A, Ichiyama S, Takahashi M, Yoshimura N, Kosugi S. Identifying pathogenic genetic background of simplex or multiplex retinitis pigmentosa patients: a large scale mutation screening study. J Med Genet. 2008;45:465–472. doi: 10.1136/jmg.2007.056416. [DOI] [PubMed] [Google Scholar]
- Francis C, Prapa S, Abdulkareem N, John S, Buchan R, Barton P, Jr, Jahangiri M, Athanassios Gatzoulis M, Pepper J, Cook SA. 95 Identification of likely pathogenic variants in patients with bicuspid aortic valve: correlation of complex genotype with a more severe aortic phenotype. Heart. 2014;100:A55–A56. [Google Scholar]
- Luoma LM, Deeb TM, Macintyre G, Cox DW. Functional analysis of mutations in the ATP loop of the Wilson disease copper transporter, ATP7B. Hum Mutat. 2010;31:569–577. doi: 10.1002/humu.21228. [DOI] [PubMed] [Google Scholar]
- Mitui M, Nahas SA, Du LT, Yang Z, Lai CH, Nakamura K, Arroyo S, Scott S, Purayidom A, Concannon P, Lavin M, Gatti RA. Functional and computational assessment of missense variants in the ataxia-telangiectasia mutated (ATM) gene: mutations with increased cancer risk. Hum Mutat. 2009;30:12–21. doi: 10.1002/humu.20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adikesavan AK, Katsonis P, Marciano DC, Lua R, Herman C, Lichtarge O. Separation of recombination and SOS response in Escherichia coli RecA suggests LexA interaction sites. PLoS Genet. 2011;7:e1002244. doi: 10.1371/journal.pgen.1002244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GJ, Yao R, Lichtarge O, Wensel TG. Evolution-guided discovery and recoding of allosteric pathway specificity determinants in psychoactive bioamine receptors. Proc Natl Acad Sci USA. 2010;107:7787–7792. doi: 10.1073/pnas.0914877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saisawat P, Tasic V, Vega-Warner V, Kehinde EO, Günther B, Airik R, Innis JW, Hoskins BE, Hoefele J, Otto EA, Hildebrandt F. Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kid Intl. 2012;81:196–200. doi: 10.1038/ki.2011.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JL, Yang X, Xia K, Hu ZM, Weng L, Jin X, Jiang H, Zhang P, Shen L, Guo JF, Li N, Li YR, Lei LF, Zhou J, Du J, Zhou YF, Pan Q, Wang J, Wang J, Li RQ, Tang BS. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain. 2010;133:3510–3518. doi: 10.1093/brain/awq323. [DOI] [PubMed] [Google Scholar]
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez-Lage M, Falcone D, Hernandez DG, Arepalli S, Chong S, Schymick JC, Rothstein J, Landi F, Wang YD, Calvo A, Mora G, Sabatelli M, Monsurrò MR, Battistini S, Salvi F, Spataro R, Sola P, Borghero G, Galassi G, Scholz SW, Taylor JP, Restagno G, Chiò A, Traynor BJ. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligo MA, Bonatti F, Guidugli L, Aretini P, Galli A. A yeast recombination assay to characterize human BRCA1 missense variants of unknown pathological significance. Hum Mutat. 2009;30:123–133. doi: 10.1002/humu.20817. [DOI] [PubMed] [Google Scholar]
- Kaminker JS, Zhang Y, Waugh A, Haverty PM, Peters B, Sebisanovic D, Stinson J, Forrest WF, Bazan JF, Seshagiri S, Zhang Z. Distinguishing cancer-associated missense mutations from common polymorphisms. Cancer Res. 2007;67:465–473. doi: 10.1158/0008-5472.CAN-06-1736. [DOI] [PubMed] [Google Scholar]
- Shi Z, Moult J. Structural and functional impact of cancer-related missense somatic mutations. J Mol Biol. 2011;413:495–512. doi: 10.1016/j.jmb.2011.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelier R, Semple JI, Garcia-Verdugo R, Lehner B. Predicting phenotypic variation in yeast from individual genome sequences. Nat Genet. 2011;43:1270–1274. doi: 10.1038/ng.1007. [DOI] [PubMed] [Google Scholar]
- Croyle ML, Woo AL, Lingrel JB. Extensive random mutagenesis analysis of the Na+/K+-ATPase alpha subunit identifies known and previously unidentified amino acid residues that alter ouabain sensitivity-implications for ouabain binding. Eur J Biochem/FEBS. 1997;248:488–495. doi: 10.1111/j.1432-1033.1997.00488.x. [DOI] [PubMed] [Google Scholar]
- Hill J, Andrew PW, Mitchell TJ. Amino acids in pneumolysin important for hemolytic activity identified by random mutagenesis. Infect Immun. 1994;62:757–758. doi: 10.1128/iai.62.2.757-758.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell RA, Sabo DL, Bandle EF, Weissmann C. Site-directed mutagenesis: effect of an extracistronic mutation on the in vitro propagation of bacteriophage Qbeta RNA. Proc Natl Acad Sci USA. 1975;72:367–371. doi: 10.1073/pnas.72.1.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonde MM, Yao R, Ma JN, Madabushi S, Haunso S, Burstein ES, Whistler JL, Sheikh SP, Lichtarge O, Hansen JL. An angiotensin II type 1 receptor activation switch patch revealed through evolutionary trace analysis. Biochem Pharmacol. 2010;80:86–94. doi: 10.1016/j.bcp.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribes-Zamora A, Mihalek I, Lichtarge O, Bertuch AA. Distinct faces of the Ku heterodimer mediate DNA repair and telomeric functions. Nat Struct Mol Biol. 2007;14:301–307. doi: 10.1038/nsmb1214. [DOI] [PubMed] [Google Scholar]
- Rababa'h A, Craft JW, Jr, Wijaya CS, Atrooz F, Fan Q, Singh S, Guillory AN, Katsonis P, Lichtarge O, McConnell BK. Protein kinase A and phosphodiesterase-4D3 binding to coding polymorphisms of cardiac muscle anchoring protein (mAKAP) J Mol Biol. 2013;425:3277–3288. doi: 10.1016/j.jmb.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersey PJ, Allen JE, Christensen M, Davis P, Falin LJ, Grabmueller C, Hughes DS, Humphrey J, Kerhornou A, Khobova J, Langridge N, McDowall MD, Maheswari U, Maslen G, Nuhn M, Ong CK, Paulini M, Pedro H, Toneva I, Tuli MA, Walts B, Williams G, Wilson D, Youens-Clark K, Monaco MK, Stein J, Wei X, Ware D, Bolser DM, Howe KL, Kulesha E, Lawson D, Staines DM. Ensembl genomes 2013: scaling up access to genome-wide data. Nucleic Acids Res. 2014;42:D546–D552. doi: 10.1093/nar/gkt979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh LS. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32:D115–D119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- Schork NJ, Murray SS, Frazer KA, Topol EJ. Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jitoku D, Hattori E, Iwayama Y, Yamada K, Toyota T, Kikuchi M, Maekawa M, Nishikawa T, Yoshikawa T. Association study of Nogo-related genes with schizophrenia in a Japanese case-control sample. Am J Med Genet. 2011;B5:581–592. doi: 10.1002/ajmg.b.31199. [DOI] [PubMed] [Google Scholar]
- Zhao N, Jiang M, Han W, Bian C, Li X, Huang F, Kong Q, Li J. A novel mutation in TNNT3 associated with Sheldon–Hall syndrome in a Chinese family with vertical talus. Eur J Med Genet. 2011;54:351–353. doi: 10.1016/j.ejmg.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Ribeiro LA, Bertolacini CD, Quiezi RG, Richieri-Costa A. A novel heterozygous missense mutation G316D of SIX3 gene in a Brazilian patient with holoprosencephaly-like phenotype and Langerhans cell histiocytosis. Clin Dysmorphol. 2011;20:160–162. doi: 10.1097/MCD.0b013e32834116ae. [DOI] [PubMed] [Google Scholar]
- McGee TL, Seyedahmadi BJ, Sweeney MO, Dryja TP, Berson EL. Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J Med Genet. 2010;47:499–506. doi: 10.1136/jmg.2009.075143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku CS, Naidoo N, Pawitan Y. Revisiting Mendelian disorders through exome sequencing. Hum Genet. 2011;129:351–370. doi: 10.1007/s00439-011-0964-2. [DOI] [PubMed] [Google Scholar]
- Knudson AG. Hereditary cancer, oncogenes, and antioncogenes. Cancer Res. 1985;45:1437–1443. [PubMed] [Google Scholar]
- Eng C, Hampel H, de la Chapelle A. Genetic testing for cancer predisposition. Annu Rev Med. 2001;52:371–400. doi: 10.1146/annurev.med.52.1.371. [DOI] [PubMed] [Google Scholar]
- Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, Wooster R. The COSMIC (catalogue of somatic mutations in cancer) database and website. Br J Cancer. 2004;91:355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordóñez GR, Bignell GR, Ye K, Alipaz J, Bauer MJ, Beare D, Butler A, Carter RJ, Chen L, Cox AJ, Edkins S, Kokko-Gonzales PI, Gormley NA, Grocock RJ, Haudenschild CD, Hims MM, James T, Jia M, Kingsbury Z, Leroy C, Marshall J, Menzies A, Mudie LJ, Ning Z, Royce T, Schulz-Trieglaff OB, Spiridou A, Stebbings LA, Szajkowski L, Teague J, Williamson D, Chin L, Ross MT, Campbell PJ, Bentley DR, Futreal PA, Stratton MR. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamborero D, Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Kandoth C, Reimand J, Lawrence MS, Getz G, Bader GD, Ding L, Lopez-Bigas N. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep. 2013;3:2650. doi: 10.1038/srep02650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Lopez-Bigas N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012;40:e169. doi: 10.1093/nar/gks743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, Ridge JA, Goodwin J, Kenady D, Saunders J, Westra W, Sidransky D, Koch WM. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007;357:2552–2561. doi: 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford RJ, Edmonson MN, Nguyen C, Buetow KH. Large-scale analysis of non-synonymous coding region single nucleotide polymorphisms. Bioinformatics. 2004;20:1006–1014. doi: 10.1093/bioinformatics/bth029. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. The catalogue of somatic mutations in cancer (COSMIC) Curr Protoc Hum Genet. 2008;57:10.11.1–10.11.26. doi: 10.1002/0471142905.hg1011s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg Y. Building a genome analysis pipeline to predict disease risk and prevent disease. J Mol Biol. 2013;425:3993–4005. doi: 10.1016/j.jmb.2013.07.038. [DOI] [PubMed] [Google Scholar]
- Lehner B. Genotype to phenotype: lessons from model organisms for human genetics. Nat Rev Genet. 2013;14:168–178. doi: 10.1038/nrg3404. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Saccharomyces genome database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–D705. doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofree M, Shen JP, Carter H, Gross A, Ideker T. Network-based stratification of tumor mutations. Nat Methods. 2013;10:1108–1115. doi: 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi IS, Yeo GS, Keogh JM, Aminian S, Jebb SA, Butler G, Cheetham T, O'Rahilly S. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest. 2000;106:271–279. doi: 10.1172/JCI9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlden H, Laura M, Wavrant-De Vrieze F, Blake J, Wood N, Reilly MM. Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type 2. Neurology. 2008;71:1660–1668. doi: 10.1212/01.wnl.0000319696.14225.67. [DOI] [PubMed] [Google Scholar]
- Moore JH, Williams SM. Epistasis and its implications for personal genetics. Am J Hum Genet. 2009;85:309–320. doi: 10.1016/j.ajhg.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlborg O, Haley CS. Epistasis: too often neglected in complex trait studies? Nat Rev Genet. 2004;5:618–625. doi: 10.1038/nrg1407. [DOI] [PubMed] [Google Scholar]
- Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet. 2013;14:307–320. doi: 10.1038/nrg3424. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Pan L, Abney M, Ober C. The sex-specific genetic architecture of quantitative traits in humans. Nat Genet. 2006;38:218–222. doi: 10.1038/ng1726. [DOI] [PubMed] [Google Scholar]
- Ober C, Loisel DA, Gilad Y. Sex-specific genetic architecture of human disease. Nat Rev Genet. 2008;9:911–922. doi: 10.1038/nrg2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdar W, Solberg LC, Gauguier D, Cookson WO, Rawlins JN, Mott R, Flint J. Genetic and environmental effects on complex traits in mice. Genetics. 2006;174:959–984. doi: 10.1534/genetics.106.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsson HT, Fallin MD, Feinberg AP. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 2004;20:350–358. doi: 10.1016/j.tig.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Rakyan VK, Preis J, Morgan HD, Whitelaw E. The marks, mechanisms and memory of epigenetic states in mammals. Biochem J. 2001;356:1–10. doi: 10.1042/0264-6021:3560001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Rifkin SA, Andersen E, van Oudenaarden A. Variability in gene expression underlies incomplete penetrance. Nature. 2010;463:913–918. doi: 10.1038/nature08781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Resilience Project. Available at: < http://www.resilienceproject.me >. Accessed on June 21, 2014.
- Friend SH, Schadt EE. Translational genomics. Clues from the resilient. Science. 2014;344:970–972. doi: 10.1126/science.1255648. [DOI] [PubMed] [Google Scholar]
- Balakirev ES, Ayala FJ. Pseudogenes: are they “junk” or functional DNA? Ann Rev Genet. 2003;37:123–151. doi: 10.1146/annurev.genet.37.040103.103949. [DOI] [PubMed] [Google Scholar]
- Apweiler R, Armstrong R, Bairoch A, Cornish-Bowden A, Halling PJ, Hofmeyr JH, Kettner C, Leyh TS, Rohwer J, Schomburg D, Steinbeck C, Tipton K. A large-scale protein-function database. Nat Chem Biol. 2010;6:785. doi: 10.1038/nchembio.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp A, Zollner A, Maier D, Albermann K, Hani J, Mokrejs M, Tetko I, Guldener U, Mannhaupt G, Munsterkotter M, Mewes HW. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 2004;32:5539–5545. doi: 10.1093/nar/gkh894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgin E. Mouse library set to be knockout. Nature. 2011;474:262–263. doi: 10.1038/474262a. [DOI] [PubMed] [Google Scholar]
- Nowak MA, Boerlijst MC, Cooke J, Smith JM. Evolution of genetic redundancy. Nature. 1997;388:167–171. doi: 10.1038/40618. [DOI] [PubMed] [Google Scholar]
- Ulitsky I, Shamir R. Pathway redundancy and protein essentiality revealed in the Saccharomyces cerevisiae interaction networks. Mol Syst Biol. 2007;3:104. doi: 10.1038/msb4100144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Tyler-Smith C. Loss-of-function variants in the genomes of healthy humans. Hum Mol Genet. 2010;19:R125–R130. doi: 10.1093/hmg/ddq365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, Albers CA, Zhang ZD, Conrad DF, Lunter G, Zheng H, Ayub Q, DePristo MA, Banks E, Hu M, Handsaker RE, Rosenfeld JA, Fromer M, Jin M, Mu XJ, Khurana E, Ye K, Kay M, Saunders GI, Suner MM, Hunt T, Barnes IH, Amid C, Carvalho-Silva DR, Bignell AH, Snow C, Yngvadottir B, Bumpstead S, Cooper DN, Xue Y, Romero IG 1000 Genomes Project Consortium. Wang J, Li Y, Gibbs RA, McCarroll SA, Dermitzakis ET, Pritchard JK, Barrett JC, Harrow J, Hurles ME, Gerstein MB, Tyler-Smith C. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser J. The hunt for missing genes. Science. 2014;344:687–689. doi: 10.1126/science.344.6185.687. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, Li K, Murty VV, Schupf N, Vilain E, Morris M, Haghighi F, Tycko B. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008;40:904–908. doi: 10.1038/ng.174. [DOI] [PubMed] [Google Scholar]
- Zhang D, Cheng L, Badner JA, Chen C, Chen Q, Luo W, Craig DW, Redman M, Gershon ES, Liu C. Genetic control of individual differences in gene-specific methylation in human brain. Am J Hum Genet. 2010;86:411–419. doi: 10.1016/j.ajhg.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information Figure 1.
Supplementary Information Table 1.