

Abstract
Discovering ways to control the activity of matrix metalloproteinases (MMPs), zinc-dependent enzymes capable of degrading extracellular matrix proteins, is an important field of cancer research. We report here a novel strategy for assembling MMP inhibitors on the basis of oligopeptide ligands by exploring the pattern known as the zinc finger motif. Advanced molecular modeling tools were used to characterize the structural binding motifs of experimentally tested MMP inhibitors, as well as those of newly proposed peptidomimetics, in their zinc-containing active sites. The results of simulations based on the quantum mechanics/molecular mechanics (QM/MM) approach and Car–Parrinello molecular dynamics with QM/MM potentials demonstrate that, upon binding of Regasepin1, a known MMP-9 inhibitor, the Zn2+(His3) structural element is rearranged to the Zn2+(Cys2His2) zinc finger motif, in which two Cys residues are borrowed from the ligand. Following consideration of the crystal structure of MMP-2 with its inhibitor, the oligopeptide APP-IP, we proposed a new peptidomimetic with two replacements in the substrate, Tyr3Cys and Asp6Cys. Simulations show that this peptide variant blocks an enzyme active site by the Zn2+(Cys2His2) zinc finger construct. Similarly, a natural substrate of MMP-2, Ace-Gln-Gly ∼ Ile-Ala-Gly-Nme, can be converted to an inhibiting compound by two replacements, Ile by Cys and Gly by the d isomer of Cys, favoring formation of the zinc finger motif.
Introduction
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases that play essential roles in various processes in the extracellular matrix.1 Finding efficient ways to control the MMP activity of gelatinases MMP-2 and MMP-9, in particular, is an important field of cancer research.2 The inhibitory power of small organic compounds containing zinc-binding groups (carboxylate, thiolate, phosphinyl, hydroxamate) is due to the ability of these groups to chelate the catalytic zinc ion and block the active site. These compounds are characterized by a high affinity for the Zn2+ ion but also often by a low specificity for certain types of MMPs, since all MMPs possess similar active sites.1−3
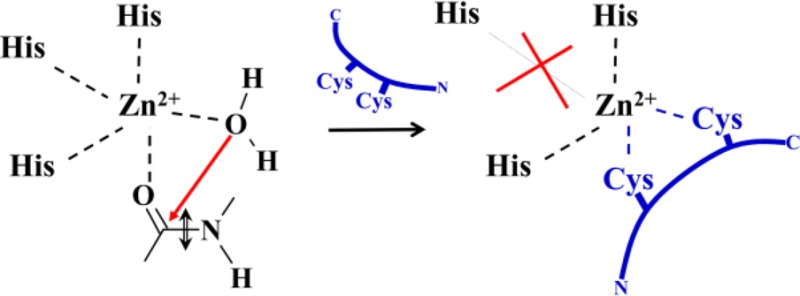
To enhance the specificity of potential MMP inhibitors, oligopeptide-based substances containing amino acid residues capable of interacting with the zinc ion must be considered, particularly Glu and Asp.4 Together with the histidine residues from an enzyme, these residues cooperate to perfectly match the typical coordination shells of Zn2+, whereas other residues interact with the various binding sites of an enzyme that favor the specific affinities of peptidomimetics. One successful attempt in this direction was described by Hu et al.,5 who discovered Regasepin1, a heptapeptide with inhibitory properties and a high selectivity to MMP-9. However, they did not explain the origin of the inhibitory power of Regasepin1, primarily because of a lack of structural data. Aiming to clarify this issue, we considered a complex of MMP-9 with Regasepin1 by using advanced molecular modeling methods and discovered that, upon binding of this peptide to the enzyme active site, a prominent zinc finger motif pattern can be recognized. This distinctive structural motif of the Cys2His2 class, with a tetrahedral coordination of the zinc ion, is typical for multiple protein families of cellular and transition factor types.6 In proteins, a zinc finger serves to stabilize the fold and to form domains that bind specific parts of nucleic acids or other proteins. Unlike these constructs, the zinc finger motif in the Regasepin1–MMP-9 complex is formed “on the fly”: an initial Zn2+(His3) structural element in the enzyme active site is rearranged to a more stable Zn2+(Cys2His2) pattern in the complex, in which two Cys residues are borrowed from the ligand. The inhibitory power of Regasepin1 may be attributable to this particular structural feature.
Motivated by this finding, we deliberately constructed novel in silico variants of peptidomimetics potentially capable of blocking gelatinase A (MMP-2) by forming the zinc finger motif via modifying the Zn2+(His3) pattern in the enzyme to Zn2+(Cys2His2) in the enzyme–ligand complexes. Specifically, we designed and computationally characterized a mutated variant of the known inhibitor APP-IP,7 as well as a novel candidate constructed by mutating a native substrate of MMP-2.
Materials and Methods
One of the problems in computational characterization of zinc-dependent proteins by classical molecular mechanics (MM) or molecular dynamics (MD) methods is due to a large positive charge on the Zn2+ cation. Although fairly rigid structural fragments can be described by classical force fields, the labile coordination shells surrounding the catalytic Zn2+ ion in these enzymes must be described via quantum methodology. Modern quantum chemical density functional theory (DFT) approaches provide valuable support to studies of metalloenzymes (e.g., the recent review by Blomberg, et al).8 Structures of the zinc finger patterns and chemical reactions with molecular clusters mimicking zinc finger constructs have been modeled in only a few DFT calculations.9−12 Development and practical implementation of multilevel quantum mechanical/molecular mechanical (QM/MM) methods13,14 allows one to enhance simulations, in particular targeting structures of zinc-containing proteins,15 and chemical transformations in MMP active sites.16−19 Including larger portions of the protein provides better models to study binding patterns and transformation of inhibitors that are not necessarily interacting directly with metal ions and their immediate ligation sphere.
We apply here the following strategy. First, appropriate model systems were constructed by motifs of the relevant crystal structures from the Protein Data Bank archive, and atomic coordinates were optimized in QM/MM calculations. Second, trajectories from the Car–Parrinello molecular dynamics with QM/MM potentials for model systems obtained at the first stage were analyzed.
For our QM/MM calculations, we used the NWChem program package.20 The QM subsystems were treated in the DFT/6-31G** approximation with the functional BB1K21 designed for simulations of chemical kinetics. The MM subsystems were considered with the AMBER force field parameters. Hydrogen capping atoms were added to the truncated bonds on the border of QM and MM regions. The convergence criteria were as follows: 10–6 a.u. for the total energy difference; 1.5 × 10–5 a.u. and 1.0 × 10–5 a.u. for maximal gradient and gradient root-mean-square; and 6.0 × 10–5 a.u. and 4.0 × 10–5 a.u. for maximum and root-mean-square Cartesian step, respectively. Calculations were performed in the electronic embedding scheme22 without cutoff for electrostatic and van der Waals interactions.
Calculations with the Car–Parrinello molecular dynamics with QM/MM potentials for the MMP–oligopeptide complexes were carried out using the Quickstep module of the CP2K program package.23,24 Forces and energies in the QM subsystems (constructed similarly to the corresponding QM/MM simulations) were computed using the density functional theory with the Gaussian and plane wave basis sets (DFT/GPW) approximation.25 The BLYP functional with the empirical dispersion corrections26 and the QZV2P basis set with Goedecker–Teter–Hutter pseudopotentials27 were applied. The multigrid approach24 with the cutoff of the finest grid level of 350 Ry was utilized. The MM part was treated with the CHARMM force field parameters. Each model system was solvated in the large rectangular water box. Sets of MD simulations were performed in the NVT ensemble at T = 300 K with the periodic boundary conditions using the Verlet integrator with a time step of 0.5 fs.
To compare predictions of the Car–Parrinello molecular dynamics with QM/MM potentials and those of the classical MD, we also performed trajectory calculations for the MMP9-Regasepin1 complex by using NAMD28 with the CHARMM force field parameters.
A theoretical assignment of the protonation state of polar residues requires calculation of pKa values of titrable groups in proteins, which presents a difficult task (see, e.g., ref (29)). Attempts to use automated procedures such as PROPKA30 primarily lead to conclusions consistent with a customary assignment, assuming that usually Lys and Arg are positively charged and Glu and Asp are negatively charged inside the protein matrix. We applied this rule at the stage of constructing molecular models and manually checked the hydrogen bond networks in local environments of polar groups: every electronegative atom should be saturated by hydrogen bonds. The initial assignment of protons may be altered in simulations if the corresponding molecular groups are included to quantum subsystems.
The specific atoms assigned to the QM parts are shown in Figures 1–3 in the Results section, below. The initial coordinates of the model systems were taken from the corresponding crystal structures specified below for each particular simulation.
Figure 1.

Equilibrium geometry configurations obtained for (a) MMP-9–Inhpro and (b) MMP-9–Regasepin1 complexes; distances are shown in angstroms (Å). For the MMP-9–Regasepin1 complex, the distances in parentheses were obtained in QM/MM minimization with the BLYP/GPW-QZV2P and CHARMM approaches. Panel c illustrates variations in the distance between the nitrogen atom of His1 and the Zn2+ along the MD trajectory in the MMP-9–Regasepin1 complex. Here and in Figures 2 and 3, the green, blue, red, white, and light blue colors correspond to carbon, nitrogen, oxygen, hydrogen, and zinc atoms, respectively.
Figure 3.
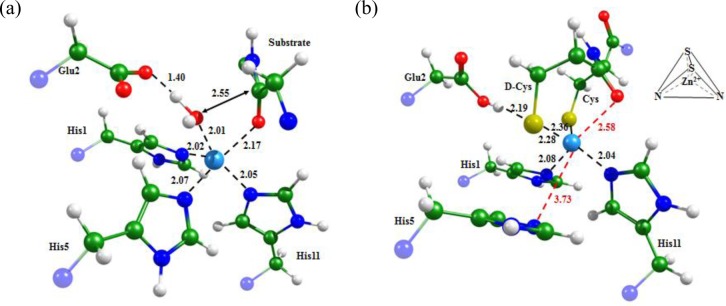
Equilibrium geometry configurations for (a) the ES complex of MMP-2 with a natural substrate and (b) the complex of MMP-2 with Inhd-Cys. Distances are given in angstroms.
Results
The Origin of the Inhibitory Power of Regasepin1
The sulfur atom of the cysteine in the prodomain of MMP (proMMP) has been shown to inactivate the catalytic zinc-containing domain.1 After proteolytic activation, a part of proMMP, the oligopeptide, which contains a cysteine, simultaneously dissociates from the active site.31 We constructed a complex composed of the MMP-9 catalytic domain and the remaining part of proMMP, Met-Arg-Thr-Pro-Arg-Cys-Gly-Val-Pro-Asp-Leu-Gly-Arg (Inhpro), on the basis of the corresponding crystal structure of proMMP-932 (PDB ID: 1L6J). The inhibitor Regasepin1, Pro-Arg-Cys-Bip-Cys-Gly-Glu (Bip stands for biphenylalanine), has a primary structure similar to Inhpro but acts as an inhibitor of MMP-9. We constructed the MMP-9–Regasepin1 complex by modifying the amino acids in the MMP-9–Inhpro construct. Equilibrium geometry configurations of both complexes were computed using the QM/MM approach. To justify the use of QM(BLYP/GPW-QZV2P)/MM(CHARMM) approximation in subsequent molecular dynamics simulations, we compared the structures of the MMP-9–Regasepin1 complex optimized in two approaches: (QM(BB1K/6-31G**)/MM(AMBER) and QM(BLYP/GPW-QZV2P)/MM(CHARMM). Only slight differences were noted (Figure 1b). The Car–Parrinello molecular dynamics simulations were initiated from the QM/MM optimized structures.
In Figure 1, we show the equilibrium geometry configurations obtained for the MMP-9–Inhpro (panel a) and MMP-9–Regasepin1 (panel b) complexes. Here, the numbering of the amino acid residues in the ligand refers to the structure PDB ID: 1L6J. According to calculation results, the QM/MM optimized minimum energy structures corresponds to the unprotonated Cys99–protonated Glu2 pair. An initial manual assignment assumed protonated Cys99 in the ligand and unrpotonated Glu2 in the protein. Both QM/MM and molecular dynamics simulations resulted in the conclusion that His1 is shifted out of the initial coordination shell of zinc in the MMP-9–Regasepin1 complex, as shown in Figure 1 (panel c) and Table 1.
Table 1. Key Distances (Å) Obtained in QM/MM Optimization and the Mean Values along Car–Parrinello Molecular Dynamics Trajectories for MMP-9–Inhpro and MMP-9–Regasepin1 Complexesa.
| Glu2H-Cys99S | Glu2O-Cys99S | Zn2+-Cys99S | Zn2+-His1N | Zn2+-His5N | Zn2+-His11N | Zn2+-Cys101S | ||
|---|---|---|---|---|---|---|---|---|
| MMP-9–Inhpro | opt | 2.24 | 3.21 | 2.28 | 2.01 | 1.96 | 1.99 | |
| X-ray | 2.95 | 2.32 | 2.26 | 2.05 | 2.21 | |||
| MD | 2.22 (0.19) | 3.21 (0.17) | 2.28 (0.06) | 2.10 (0.09) | 2.05 (0.07) | 2.04 (0.06) | ||
| MMP-9–Regasepin1 | opt | 2.12 | 3.11 | 2.29 | 3.14 | 2.00 | 1.99 | 2.40 |
| MD | 2.24 | 3.23 | 2.34 | 3.57 | 2.09 | 2.10 | 2.43 | |
| 10 ps | (0.16) | (0.14) | (0.07) | (0.26) | (0.07) | (0.07) | (0.09) | |
| MD | 2.25 | 3.23 | 2.33 | 3.59 | 2.08 | 2.10 | 2.44 | |
| 60 ps | (0.18) | (0.16) | (0.07) | (0.37) | (0.07) | (0.07) | (0.12) | |
For the latter, results of 10 and 60 ps simulations are presented. Rows with MD data show the mean values and standard deviations (in parentheses).
Table 1 summarizes the key distances obtained in QM/MM optimization and Car–Parrinello molecular dynamics simulations of MMP-9–Inhpro and MMP-9–Regasepin1 complexes. In the MMP-9–Regasepin1 complex, His1 escapes from the coordination sphere of Zn2+, and this shell becomes more flexible (Figure 1c), as indicated by the large corresponding standard deviation (Table 1). The distance between the nitrogen atom of His1 and the Zn2+ is shorter in the QM/MM optimization compared with the mean value obtained in MD simulations, but both values definitely show the absence of the coordination bond. Other coordination bond lengths are similar, showing no other disturbances in the MMP-9 active site. Additional stabilization of the bound MMP-9–Regasepin1 structure is due to the stacking interactions between the newly introduced biphenylalanine and Tyr179 of the catalytic domain.
As illustrated in Figure 1b, His1 is replaced by Cys101 from the ligand in the MMP-9–Regasepin1 complex. The metal ion Zn2+ has tetrahedral coordination in both cases: in the MMP-9–Inhpro complex, zinc forms coordination bonds with three nitrogen atoms of histidines in MMP-9 and with the sulfur of Cys99 in Inhpro. In Regasepin1, Val101 is changed to Cys, thus providing an additional coordinating group. Since Zn2+ prefers coordinating with the more polarizable sulfur atom (rather than with nitrogen), Cys101 substitutes His1 in the zinc shell. The newly formed coordination sphere includes two cysteines from the inhibitor and two histidines from the enzyme, leading to the classical, stable Cys2His2 zinc finger motif.
Importantly, the mean values and standard deviations from MD trajectories corresponding to 10 and 60 ps lengths are practically the same (last rows of Table 1), indicating that the structural motif of the zinc coordination sphere is stable. Taking into account that running the Car–Parrinello molecular dynamics with QM/MM potentials and a fairly large quantum subsystem is very expensive, here we can rely on the data from relatively short trajectories initiated from the QM/MM optimized structures.
To understand the importance of quantum mechanics-based calculations, we also performed classical MD simulations for the MMP-9-Regasepin1 complex by using two different sets of partial charges on atoms for the active site. One set uses the CHARMM force field parameters, and another set, the electrostatic potential (ESP) charges, is obtained from the electron density distribution calculated with the BB1K/6-31G** approximation. These computational data (summarized in the Supporting Information) demonstrate that the results of classical MD strongly depend on the choice of the partial charges on atoms and predict unreasonable structures of the zinc coordination shell.
Introduction of Zinc Finger Motif Enhances Inhibitory Properties of APP-IP
We started from the known X-ray structure (PDB ID: 3AYU(33)) of the inhibitor APP-IP complexed with the Glu121Ala mutant of MMP-2. This ordering of amino acid residues corresponds to the X-ray structure PDB ID: 3AYU; however, in Figure 2 and the text, we mark this residue as number 2. To restore the native sequence in MMP-2, we introduced Glu at position 121 (or position 2 in Figure 2) and made point mutations in APP-IP, aiming to facilitate formation of the zinc finger motif upon complexation with MMP-2. In the MMP-2–APP-IP complex, Asp6 of APP-IP coordinates Zn2+. We modified this residue to cysteine (Asp6Cys) because its side chain has a similar volume and can perfectly coordinate Zn2+. We also mutated Tyr3Cys to introduce an additional residue capable of entering the coordination sphere of zinc, naming the resulting oligopeptide APP-IPZnF.
Figure 2.

Equilibrium geometry configurations obtained for the (a) MMP-2–APP-IP and (b) MMP-2–APP-IPZnF complexes. Graphs showing distances (Å) between atoms along MD trajectories: (c) corresponds to the distances between the nitrogen of His5 and the Zn2+ in the MMP-2–APP-IPZnF complex; (d) corresponds to the distances between Asp6OD1 (blue) or Asp6OD2 (red) and the Zn2+ in the MMP-2–APP-IP complex. See also Table 2.
Figure 2 shows equilibrium geometry configurations obtained for the MMP-2–APP-IP and MMP-2–APP-IPZnF complexes. Zn2+ is coordinated by the Asp6 carboxylic group of APP-IP and by three histidines from MMP-2. The MMP-2–APP-IPZnF complex has a tetrahedral Zn2+ coordination sphere formed by two cysteines from the proposed oligopeptide and two histidines from the enzyme. The resulting Cys2His2 pattern corresponds to a stable zinc finger motif.
Table 2. Key Distances (Å) Obtained in QM/MM Optimization and in MD Simulations of MMP-2–APP-IP and MMP-2–APP-IPZnF Complexesa.
| Glu2H-Asp6OD2 | Glu2O- Asp6OD2 | Zn2+- Asp6OD2 | Zn2+- Asp6OD1 | Zn2+-His1N | Zn2+-His5N | Zn2+-His11N | ||
|---|---|---|---|---|---|---|---|---|
| MMP-2-APP-IP | opt | 1.71 | 2.19 | 2.19 | 2.08 | 2.02 | 2.03 | 2.03 |
| X-ray | 2.30 | 2.17 | 2.05 | 2.11 | 2.08 | |||
| MD | 1.71 (0.15) | 2.53 (0.31) | 2.53 (0.31) | 2.19 (0.26) | 2.03 (0.05) | 2.08 (0.06) | 2.03 (0.05) | |
| Glu2H-Cys6S | Glu2O- Cys6S | Zn2+- Cys6S | Zn2+- Cys3S | Zn2+-His1N | Zn2+-His5N | Zn2+-His11N | ||
|---|---|---|---|---|---|---|---|---|
| MMP-2-APP-IPZnF | opt | 2.07 | 3.05 | 2.31 | 2.33 | 2.02 | 3.55 | 2.03 |
| MD | 2.10 (0.15) | 3.10 (0.13) | 2.37 (0.09) | 2.37 (0.13) | 2.14 (0.08) | 3.37 (0.29) | 2.11 (0.08) | |
Rows with MD data show the mean values and standard deviations (in parentheses).
Zinc Finger Motif Turns a Natural MMP-2 Substrate into an Inhibitor
We started from the enzyme–substrate (ES) complex of the MMP-2 catalytic domain with the model substrate, Ace-Gln-Gly ∼ Ile-Ala-Gly-Nme, obtained previously,19 and suggested specific modifications to facilitate formation of a zinc finger motif. We modified the Gly ∼ Ile scissile fragment with a d-Cys–Cys dipeptide and constructed a species we named Inhd-Cys. We chose the d-isomer because we wanted the side chain of the amino acid replacing Gly to face toward the active site of the enzyme, and the side chain of the l-isomer would orient away from the active site.
Figure 3 shows equilibrium geometry configurations obtained for the ES19 complex and the complex of the MMP-2 catalytic domain and the suggested inhibitor Inhd-Cys. Upon binding, Inhd-Cys perturbs the coordination sphere of Zn2+ and substitutes one of the coordinating histidines (His5) of the enzyme with a cysteine from the oligopeptide because the Zn2+ ion prefers to form bonds with the sulfur atom rather than with nitrogen or oxygen. Moreover, d-Cys displaces a catalytic water molecule from the active site, thus eliminating the possibility of an enzyme-catalyzed reaction. In addition, these amino acid modifications lead to elongation of the coordination bond between the oxygen atom of the peptide group of the oligopeptide and the Zn2+ion. This distance is crucial in the ES complex because Zn2+ polarizes the carbonyl group of the peptide fragment to facilitate nucleophilic attack and helps to stabilize the tetrahedral intermediate formed in the first stage of the hydrolysis.19 In the enzyme–Inhd-Cys complex, this structural parameter is not important.
Discussion
Although exploring properties of the well-known Zn2+(CysxHis4-x) structural pattern in proteins continues to attract attention (e.g., see recent papers34−36), to the best of our knowledge, the idea of utilizing formation of the zinc finger motif upon MMP–ligand complexation to assemble oligopeptide-based MMP inhibitors is presented here for the first time. We use modern molecular modeling tools, including the QM/MM method, to support such a hypothesis. Other QM/MM-based theoretical approaches to model ligand binding sites of MMP-9 beyond the zinc finger construct are described in the literature.37,38
The MMP catalytic motif is composed of three permanent Zn2+ ligands (His) and a catalytic water molecule, which is very labile. Usually, inhibitors based on small-molecule organic compounds1,3 provide only one additional (albeit more stable) coordination bond over that from the reactive water molecule. In the strategy described in this work, two additional coordination bonds, arising from the ligand, can be introduced to the catalytic zinc. The Zn2+(Cys2His2) pattern formed in the enzyme–ligand complex in place of an initial Zn2+(His3) construct in the enzyme active site should provide additional stabilization to the complex9 and therefore enhance the inhibition power of a peptidomimetic. Correspondingly, introducing cysteine residues into the substrate fragment responsible for binding with the zinc-containing pocket favors production of MMP inhibitors. Keeping the other residues found in natural substrates that are responsible for recognizing binding sites in the enzyme should enhance the specificity of the ligand.
In this work, we examined this strategy using three different oligopeptide systems: (i) the MMP-9 prodomain and Regasepin1, (ii) APP-IP and its derivative, and (iii) the natural substrate and its derivative.
The oligopeptide inhibitor Regasepin1 is specific to MMP-9 (IC50 = 1 μM); however, its interaction mechanism has not been characterized. Following up on the original work presented by Hu et al.,5 we found that its sequence is similar to that of the prodomain of MMP-9. By using the X-ray structure of full-length MMP-9, we constructed a complex made up of the MMP-9 catalytic domain and the part of the prodomain formed after the proteolytic activation reactions. We found that the MMP-9–Regasepin1 complex is characterized by a stable zinc finger motif composed of two cysteines from the inhibitor and two histidines from the enzyme. This explains the dramatic differences31 in the binding properties of Inhpro, which spontaneously dissociates from MMP-9 and Regasepin1. The main structural drawback of Regasepin1 is that it has a twisted loop structure, and therefore, it interacts modestly with the binding sites of the catalytic domain.
According to the X-ray data, the latter problem can be resolved for another oligopeptide inhibitor, APP-IP. It binds tightly to the MMP-2 catalytic domain and is located in the channel formed by the amino acids of the binding sites, which explains its low IC50 value of 0.03 μM. Armed with the knowledge of the Regasepin1 binding mechanism, we proposed a mutated variant of APP-IP capable of forming a Cys2His2 zinc finger motif upon complexation with MMP-2. The suggested point mutations do not affect binding properties of the peptidomimetic, but provide additional attractive interactions with Zn2+: APP-IP coordinates the Zn2+ion via the carboxylate group, whereas APP-IPZnF does this via two thiolate groups.
It should be noted that both APP-IP and Regasepin1 are oriented in the opposite N-to-C direction compared with the natural substrates. Such an arrangement disturbs the reactive configuration of the active site, displacing a catalytic water molecule in particular. To the best of our knowledge, neither structural data nor other evidence of oligopeptide-based inhibitors that bind to MMP in a direction similar to the native substrate has been reported. We suppose that the main reason for this is that MMP binding sites are constructed in such a manner that a substrate binds in a specific way. Namely, the side chain of the amino acid immediately preceding the scissile bond is oriented to the solution, whereas the next one is oriented toward the enzyme. This provides the free volume in the active site that is occupied by the catalytic water molecule. Correspondingly, we suggested introducing d-Cys before the scissile fragment in the substrate. Its side chain orients toward the active site of MMP, opposite to the l-form’s orientation, pushing out a catalytic water molecule and coordinating the Zn2+ instead. Another Cys was introduced to the sequence of the natural substrate to help create the zinc finger motif. The presence of a d-amino acid in the oligopeptide has an additional advantage in that this inhibitor should be more resistant to other proteases in in vivo applications.
We observed similar behavior by the side chains of key amino acids in all structures of MMP with inhibitors capable of forming the zinc finger motif. Upon binding, one of the introduced cysteines substitutes the histidine from the coordination sphere. Figures 1–3 show that it can be either His1 or His5, depending on the direction of the cysteine attack. Namely, in the MMP–Regasepin1 complex, Cys101 substitutes His1 from the coordination sphere to form a new Zn2+ tetrahedral coordination. For APP-IPZnF and Inhd-Cys, Cys3 and Cys, respectively, push out His5.
Conclusion
Encouraged by research showing that complexation of MMP-9 with its inhibitor Regasepin1 is accompanied by formation of a structural pattern known as the zinc finger motif, Zn2+(Cys2His2), in place of the initial Zn2+(His3) construct in the enzyme active site, we suggested a novel strategy to assemble efficient MMP inhibitors on the basis of oligopeptide ligands. The approach was to introduce cysteine residues into appropriate places in natural substrates while keeping their native outermost amino acid residues to guarantee specific binding to the enzyme. Exploring this hypothesis, we constructed, in silico, a more promising variant of the MMP-2 inhibitor APP-IP by considering two replacements in the substrate: Tyr3Cys and Asp6Cys. Molecular modeling shows that this peptide variant blocks an enzyme active site via the Zn2+(Cys2His2) zinc finger construct. A natural substrate of MMP-2, Ace-Gln-Gly ∼ Ile-Ala-Gly-Nme, can also be converted to an inhibiting compound by two replacements, Ile by Cys and Gly, by the d-isomer of Cys, thus favoring formation of the zinc finger motif.
Acknowledgments
A.N. and M.K. thank the Russian Science Foundation (Project No. 14-03-00124) for support of this study. M.K. acknowledges stipend support from the Dynasty Foundation Fellowship. We acknowledge the use of supercomputer resources of the Lomonosov Moscow State University39 and of the Joint Supercomputer Center of the Russian Academy of Sciences. I.T. thanks the staff and administration of the Advanced Biomedical Computing Center for their partial support of the project with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Supporting Information Available
Calculated partial charges on atoms in the active site of the MMP-9-Regasepin1 complex (Table S1) and comparison of the results of different MD protocols for the MMP-9-Regasepin1 complex (Figures S1–S3, Table S2). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gupta S. P., Ed.; Matrix Metalloproteinase Inhibitors. Specificity of Binding and Structure-Activity Relationships; Experientia Supplementum; Springer: Basel, 2012; p 103. [PubMed] [Google Scholar]
- Dormán G.; Cseh S.; Hajdú I.; Barna L.; Kónya D.; Kupai K.; Kovács L.; Ferdinandy P. Matrix Metalloproteinase Inhibitors. A Critical Appraisal of Design Principles and Proposed Therapeutic Utility. Drugs 2010, 70, 949–964. [DOI] [PubMed] [Google Scholar]
- Jacobsen J. A.; Jourden J. L. M.; Miller M. T.; Cohen S. M. To Bind Zinc or Not To Bind Zinc: An Examination of Innovative Approaches to Improved Metalloproteinase Inhibition. Biochim. Biophys. Acta 2010, 1803, 72–94. [DOI] [PubMed] [Google Scholar]
- Ndinguri M. W.; Bhowmick M.; Tokmina-Roszyk D.; Robichaud T. K.; Fields G. B. Peptide-Based Selective Inhibitors of Matrix Metalloproteinase-Mediated Activities. Molecules 2012, 17, 14230–14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.; Fiten P.; Van den Steen P. E.; Chaltin P.; Opdenakker G. Simulation of Evolution-Selected Propeptide by High-Throughput Selection of a Peptidomimetic Inhibitor on a Capillary DNA Sequencer Platform. Anal. Chem. 2005, 77, 2116–2124. [DOI] [PubMed] [Google Scholar]
- Laitaoja M.; Valjakka J.; Jänis J. Zinc Coordination Spheres in Protein Structures. Inorg. Chem. 2013, 52, 10983–10991. [DOI] [PubMed] [Google Scholar]
- Higashi S.; Miyazaki K. Identification of a Region of β-Amyloid Precursor Protein Essential for its Gelatinase A Inhibitory Activity. J. Biol. Chem. 2003, 278, 14020–14028. [DOI] [PubMed] [Google Scholar]
- Blomberg M. R. A.; Borowski T.; Himo F.; Liao R.-Z.; Siegbahn P. E. M. Quantum Chemical Studies of Mechanisms for Metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [DOI] [PubMed] [Google Scholar]
- Topol I. A.; Casas-Finet J. R.; Gussio R.; Burt S. K.; Erickson J. W. A Quantum-Mechanical Study of Metal Binding Sites in Zinc Finger Structures. J. Mol. Struct. (Theochem) 1998, 423, 13–28. [Google Scholar]
- Topol I. A.; Nemukhin A. V.; Chao M.; Iyer L. K.; Tawa G. J.; Burt S. K. Quantum Chemical Studies of Reactions of the Cyclic Disulfides with the Zinc Finger Domains in the HIV-1 Nucleocapsid Protein (NCp7). J. Am. Chem. Soc. 2000, 122, 7087–7094. [Google Scholar]
- Topol I. A.; Nemukhin A. V.; Dobrogorskaya Y. I.; Burt S. K. Interactions of Azodicarbonamide (ADA) Species with the Model Zinc Finger Site: Theoretical Support of the Zinc Finger Domain Destruction in the HIV-1 Nucleocapsid Protein (NCp7) by ADA. J. Phys. Chem. B 2001, 105, 11341–11350. [Google Scholar]
- Antony S.; Baysem C. A. Density Functional Theory Study of the Attack of Ebselen on a Zinc-Finger Model. Inorg. Chem. 2013, 52, 13803–13805. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Levitt M. Theoretical Studies of Enzymic Reactions: Dielectric, Electrostatic and Steric Stabilization of the Carbonium Ion in the Reaction of Lysozyme. J. Mol. Biol. 1976, 103, 227–249. [DOI] [PubMed] [Google Scholar]
- Senn H. M.; Thiel W. QM/MM Methods for Biomolecular Systems. Angew. Chem., Int. Ed. 2009, 48, 1198–1229. [DOI] [PubMed] [Google Scholar]
- Li X.; Hayik S. A.; Merz K. M. Jr. QM/MM X-Ray Refinement of Zinc Metalloenzymes. J. Inorg. Biochem. 2010, 104, 512–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelmenschikov V.; Siegbahn E. M. Catalytic Mechanism of Matrix Metalloproteinases: Two-Layered ONIOM Study. Inorg. Chem. 2002, 41, 5659–5666. [DOI] [PubMed] [Google Scholar]
- Diaz N.; Suarez D. Peptide Hydrolysis Catalyzed by Matrix Metalloproteinase 2: A Computational Study. J. Phys. Chem. B 2008, 112, 8412–8424. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Tao P.; Fisher J. F.; Shi Q.; Mobashery S.; Schlegel H. B. QM/MM Studies of the Matrix Metalloproteinase 2 (MMP-2) Inhibition Mechanism of (S)-SB-3CT and its Oxirane Analogue. J. Chem. Theory Comput. 2010, 6, 3580–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khrenova M. G.; Nemukhin A. V.; Savitsky A. P. Computational Characterization of Ketone–Ketal Transformations at the Active Site of Matrix Metalloproteinases. J. Phys. Chem. B 2014, 118, 4345–4350. [DOI] [PubMed] [Google Scholar]
- Valiev M.; Bylaska E. J.; Govind N.; Kowalski K.; Straatsma T. P.; van Dam H. J. J.; Wang D.; Nieplocha J.; Apra E.; Windus T. L.; et al. NWChem: A Comprehensive and Scalable Open-Source Solution for Large Scale Molecular Simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar]
- Zhao Y.; Lynch B. J.; Truhlar D. G. Development and Assessment of a New Hybrid Density Functional Model for Thermochemical Kinetics. J. Phys. Chem. A 2004, 108, 2715–2719. [Google Scholar]
- Kästner J.; Thiel S.; Senn H. M.; Sherwood P.; Thiel W. Exploiting QM/MM Capabilities in Geometry Optimization: A Microiterative Approach Using Electrostatic Embedding. J. Chem. Theory Comp. 2007, 3, 1064–1072. [DOI] [PubMed] [Google Scholar]
- VandeVondele J.; Krack M.; Mohamed F.; Parrinello M.; Chassaing T.; Hutter J. Quickstep: Fast and Accurate Density Functional Calculations Using a Mixed Gaussian and Plane Waves Approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar]
- Laino T.; Mohamed F.; Laio A.; Parrinello M. An Efficient Real Space Multigrid QM/MM Electrostatic Coupling. J. Chem. Theory Comput. 2005, 1, 1176–1184. [DOI] [PubMed] [Google Scholar]
- Lippert G.; Hutter J.; Parrinello M. A Hybrid Gaussian and Plane Wave Density Functional Scheme. Mol. Phys. 1997, 92, 477–488. [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- Goedecker S.; Teter M.; Hutter J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [DOI] [PubMed] [Google Scholar]
- Søndergaard C. R.; Olsson M. H. M.; Rostkowski M.; Jensen J. H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- Sang Q.-X.; Birkedal-Hansen H.; Van Wart H. E. Proteolytic and Non-Proteolytic Activation of Human Neutrophil Progelatinase B. Biochim. Biophys. Acta 1995, 1251, 99–108. [DOI] [PubMed] [Google Scholar]
- Elkins P. A.; Ho Y. S.; Smith W. W.; Janson C. A.; D’Alessio K. J.; McQueney M. S.; Cummings M. D.; Romanic A. M. Structure of the C-Terminally Truncated Human ProMMP9, a Gelatin-Binding Matrix Metalloproteinase. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002, 58, 1182–1192. [DOI] [PubMed] [Google Scholar]
- Hashimoto H.; Takeuchi T.; Komatsu K.; Miyazaki K.; Sato M.; Higashi S. Structural Basis for Matrix Metalloproteinase-2 (MMP-2)-Selective Inhibitory Action of β-Amyloid Precursor Protein-Derived Inhibitor. J. Biol. Chem. 2011, 286, 33236–33243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom L. M.; Brinkmeyer M. K.; Ha Y.; Raetz A. G.; Hedman B.; Hodgson K. O.; Solomon E. I.; David S. S. A Zinc Linchpin Motif in the MUTYH Glycosylase Interdomain Connector Is Required for Efficient Repair of DNA Damage. J. Am. Chem. Soc. 2014, 136, 7829–7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K. L.; Bakman I.; Marts A. R.; Batir Y.; Dowd T. L.; Tierney D. L.; Gibney B. R. Characterization of the Zn(II) Binding Properties of the Human Wilms’ Tumor Suppressor Protein C-terminal Zinc Finger Peptide. Inorg. Chem. 2014, 53, 6309–6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghimire-Rijal S.; Maynard E. L. Jr. Comparative Thermodynamic Analysis of Zinc Binding to the His/Cys Motif in Virion Infectivity Factor. Inorg. Chem. 2014, 53, 4295–4302. [DOI] [PubMed] [Google Scholar]
- Min D.; Chen M.; Zheng L.; Jin Y.; Schwartz M. A.; Sang Q. X.; Yang W. Enhancing QM/MM Molecular Dynamics Sampling in Explicit Environments via an Orthogonal-Space-Random-Walk-Based Strategy. J. Phys. Chem. B 2011, 115, 3924–3935. [DOI] [PubMed] [Google Scholar]
- Yoshida T.; Hitaoka S.; Mashima A.; Sugimoto T.; Matoba H.; Chuman H. Combined QM/MM (ONIOM) and QSAR Approach to the Study of Complex Formation of Matrix Metalloproteinase-9 with a Series of Biphenylsulfonamides–LERE-QSAR Analysis (V). J. Phys. Chem. B 2012, 116, 10283–10289. [DOI] [PubMed] [Google Scholar]
- Voevodin Vl.V.; Zhumatiy S. A.; Sobolev S. I.; Antonov A. S.; Bryzgalov P. A.; Nikitenko D. A.; Stefanov K. S.; Voevodin Vad.V. Practice of “Lomonosov” Supercomputer. Open Syst. J. - Moscow 2012, 36–39. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.