

Abstract

Extensive all-atom molecular dynamics (∼24 μs total) allowed exploration of configurational space and calculation of lateral diffusion coefficients of the components of a protein-embedded, cholesterol-containing model bilayer. The three model membranes are composed of an ∼50/50 (by mole) dipalmitoylphosphatidylcholine (DPPC)/cholesterol bilayer and contained an α-helical transmembrane protein (HIV-1 gp41 TM). Despite the high concentration of cholesterol, normal Brownian motion was observed and the calculated diffusion coefficients (on the order of 10–9 cm2/s) are consistent with experiments. Diffusion is sensitive to a variety of parameters, and a temperature difference of ∼4 K from thermostat artifacts resulted in 2–10-fold differences in diffusion coefficients and significant differences in lipid order, membrane thickness, and unit cell area. Also, the specific peptide sequence likely underlies the consistently observed faster diffusion in one leaflet. Although the simulations here present molecular dynamics (MD) an order of magnitude longer than those from previous studies, the three systems did not approach ergodicity. The distributions of cholesterol and DPPC around the peptides changed on the microsecond time scale, but not significantly enough to thoroughly explore configurational space. These simulations support conclusions of other recent microsecond MD in that even longer time scales are needed for equilibration of model membranes and simulations of more realistic cellular or viral bilayers.
Introduction
The dynamics of lipids and cholesterol in model membranes have been studied both experimentally and computationally as cholesterol is an important cellular and viral membrane component. A large amount of cholesterol is characteristic of cellular microdomains and some viral membranes such as that of HIV-1, which require cholesterol for infection.1,2 Although it has been demonstrated that lateral diffusivities in model mixed bilayers depend on temperature and composition, the measurement time scale also partially determines these diffusivities. In fact, variations in diffusion coefficients of ≥2 orders of magnitude are a result of the different time scales measured by experiments.3,4 Quasi-elastic neutron scattering measures diffusion on the picosecond time scale, while, for instance, fluorescence correlation spectroscopy (FCS) and fluorescence recovery after photobleaching (FRAP) measure diffusion on longer time scales, showing signatures of Brownian motion. However, a detailed picture of the lateral motion of molecules in a mixed bilayer with cholesterol is still lacking.
Molecular simulation approaches, in particular all-atom molecular dynamics (MD), are suitable for use on model membranes to study diffusion on time scales from picoseconds to microseconds and to observe concerted diffusion of lipids with atomic resolution.5,6 Previously, lateral diffusion coefficients of lipids, cholesterol, and/or proteins in model membranes have been calculated for time scales of up to 150 ns in cholesterol-free membranes and up to 500 ns in cholesterol-containing membranes with all-atom MD.5−10 Bilayers with a high cholesterol content (up to 50%) have been difficult to study with MD as such large amounts of cholesterol slow molecular diffusion, making it difficult to generate sufficient statistics on typical simulation time scales.10,11 Therefore, the distributions of membrane components in most MD simulations with cholesterol to date are strongly dependent on initial positions; it is unclear whether any mixed bilayer systems studied with MD have achieved ergodicity. Careful attention is needed for protein-embedded bilayers in which the initial arrangement of lipids and cholesterol around a protein depends on where a user has chosen to insert the protein during the setup of the system. It is therefore also important to understand the requirements for removing initial condition bias when using MD simulations of protein-embedded, cholesterol-containing, mixed-bilayer systems. The computational power necessary to reach beyond microsecond time scales in all-atom MD has only recently become available, giving the opportunity to explore ergodicity in such cholesterol-containing bilayers.
In this paper, three systems of a model membrane composed of ∼50% cholesterol, ∼50% DPPC, and a single α-helical transmembrane protein [the HIV-1 gp41 transmembrane (TM) domain] were simulated using all-atom molecular dynamics on the microsecond time scale using the Anton supercomputer.12,13 Importantly, Brownian motion was observed for up to 10 μs for a single trajectory, an order of magnitude longer than that previously examined using all-atom MD, and sufficient statistics allowed calculation of all diffusion coefficients. Slower processes were also observed, such as undulations, cholesterol flip-flops, and changes in membrane area, order, and thickness. Despite the length of the trajectories, the membranes did not achieve complete ergodicity; time scales longer than 10 μs seem to be required for complete configurational sampling of bilayers with ∼50% cholesterol and proteins, at least at temperatures between 300 and 310 K. Discrepancies in self-diffusion coefficients are partially attributed to sequence-specific effects and to small thermostating artifacts that cooled systems by a few degrees relative to their set points, highlighting the need for more careful temperature control in microsecond MD, especially in membrane systems.
Computational Methods
Setup and Equilibration
System setup details were published previously.14,15 Briefly, the system designated WT1 was generated by pulling the α-helical peptide with 27 residues (HIV-1 gp41 TM, 681-KLFIMIVGGLVGLRIVFAVLSIVNRVR-707) into a model bilayer patch of an ∼50/50 (by mole) dipalmitoylphosphatidylcholine (DPPC)/cholesterol mixture generated using the CHARMM-GUI membrane builder.16 The membrane was cropped into a smaller patch, resulting in a bilayer comprised of 151 DPPC molecules (45.2% by mole) and 183 cholesterol molecules (54.8% by mole). Another initial condition, WT2, was generated by pulling the peptide into a location in the final bilayer different from that of WT1, resulting in a different local lipid composition near the peptide. A mutant system, R694L, was generated from the WT1 system by mutating residue 694 from arginine to leucine. As part of the previous study,15 all systems were then simulated in the NPT ensemble for 300 ns using NAMD.17 Each of the three systems has ∼58000 atoms. The systems were neutralized and ionized to 0.1 M NaCl. Periodic boundary conditions were used. These systems have dimensions of roughly 80 Å × 80 Å × 80 Å.
Microsecond MD on Anton
The WT1, WT2, and R694L systems after 300 ns MD were then run for 9.98, 6.45, and 8.06 μs NPT MD, respectively, on the Anton supercomputer, a highly specialized machine specifically built for MD.12,13 The CHARMM force field with recent lipid-based corrections and explicit TIP3P water were used.18−21 Verlet integration was applied with a 2 fs time step. Long-range electrostatics were handled with the Gaussian Split Ewald method.22 The Nosé–Hoover thermostat was set to 310 K because the systems here are models of the HIV-1 viral membrane at body temperature. The Martyna–Tobias–Klein barostat was set to a semi-isotropic pressure of 1 atm.23,24 Trajectories were visualized with VMD.25 The complete set of Anton parameters for WT1 (identical to WT2 and R694L) is given in the Supporting Information.
The choice of thermostat is important for accurate dynamics and ensemble sampling, because all thermostats change simulation dynamics to some degree. Thermostats that reinitialize velocities, such as Andersen and Langevin dynamics, generally do not accurately replicate dynamics. Thermostats that rescale velocities, such as Nosé–Hoover and Berendsen thermostats, are usually more accurate at replicating transport properties (although the Berendsen thermostat does not correctly sample the ensemble), but their accuracy also depends on other parameters, like coupling constants, etc.26 The only thermostat available with the version of Anton software utilized in this paper was the Nosé–Hoover thermostat, which rescales velocities. System center-of-mass (COM) motion is normally removed on the fly to avoid the “flying ice cube” phenomenon, although this does not prevent dumping of energy into rotational motion or large velocities of the individual leaflet COMs. To minimize postprocessing errors and to avoid fictitious forces between monolayers, we did not remove system COM motion during the simulation. This set of conditions led to slight cooling of the WT1, WT2, and R694L systems to 306.7, 302.6, and 302.2 K, respectively [average of the last microsecond (see Figure S1 of the Supporting Information)]; this cooling is similar to the flying ice cube effect,26−28 except that the temperatures stabilized instead of continuously decreasing. This artifact can also be seen by comparing system COM versus time (Figure S1 of the Supporting Information); the systems with larger temperature drops (WT2 and R694L) have the greatest COM velocity.
Observables Computed
Unique water molecules per residue and helix tilt were computed as previously described.15 Atomic mass density distributions, ρi(x,y,z), were computed by histogramming mass into 1 Å3 bins from trajectories in which coordinates were shifted to bring the protein x,y-COM to the x,y-origin and the membrane z-COM to the z-origin. All components were then rewrapped into the primary cell. We report integrated forms of this mass density, including atomic density profiles along z for molecules within 8 Å of the protein, and lateral density maps of cholesterol hydroxyl oxygen or DPPC phosphorus atoms in the lower leaflets in 4 Å × 4 Å patches. Membrane thickness as a function of lateral position was measured by determining the difference between z-COMs of lipid headgroups in upper and lower leaflets in 4 Å × 4 Å patches, averaged over the trajectory for each patch. The tilt angle of cholesterol molecules was measured by the angle between the vector connecting C3 and C17 carbon atoms and membrane normal. The DPPC order parameters (SCD) were calculated for each tail according to
| 1 |
where θ is the angle between the carbon–hydrogen vector in a lipid acyl chain and bilayer normal.29 The radial distribution functions g(r) were calculated using VMD, with a δ of 0.1 Å.30 Chosen for pair selections were cholesterol hydroxyl oxygen with respect to itself, DPPC phosphate oxygens with respect to cholesterol hydroxyl hydrogen, and DPPC carbonyl oxygens with respect to cholesterol hydroxyl hydrogen.
The lateral self-diffusion coefficient of species i, Di,lat, is calculated from the Einstein relation:
| 2 |
Here, the sum of mean-square displacements (MSD) runs over all molecules of species i, and rj(t) refers to a particular molecule’s center of mass at time t. The angle brackets refer to an average over all time origins resulting in intervals that fit within the trajectory. Measurements were taken for intervals between 1 and 2–5 μs, when the MSD was observed to be linear in time, and with either the bilayer or each monolayer rewrapped to the origin. (For the protein, the intervals were usually 0.1–2.5 μs.) The upper and lower leaflets (UL and LL, respectively) were considered separately. An analogous Einstein relation was used to convert mean-square angular displacements (MSAD) versus time to measurements of species-specific one-dimensional rotational diffusion coefficients, Di,rot.
Results and Discussion
Peptide Properties on Microsecond Time Scales Consistent with Nanosecond Time Scales
All-atom models of the transmembrane domain of HIV-1 gp41 in a bilayer with a high cholesterol content were used to explore the dynamics of cholesterol, lipids, and peptides on 6–10 μs MD time scales. Representative system configurations are shown in Figure 1. We showed previously that the TM peptide remains α-helical and membrane-spanning for 300 ns, despite solvation of its midspan arginine.15 Here the systems achieve run times that are >20 times longer and show that the TM peptide still remains helical and membrane-spanning, as seen in plots of the root-mean-square deviation (rmsd) of backbone atoms from initial structures (Figure 2). The wild-type peptides in systems WT1 and WT2 explored up to 3 Å from their initial structures, with intermittent unfolding of the C-terminal residues. The system with a mutant sequence, R694L, had less conformational flexibility than WT1 and WT2 and explored up to 2 Å from its initial structure. The WT1 and WT2 systems solvate their midspan arginines (R694) with water defects that also remain stable on microsecond time scales, and the average number of unique water molecules per residue was consistent with measurements made on the 300 ns time scale (Figure 3A). To facilitate this solvation, both WT1 and WT2 peptides tilt to allow R694 to snorkel to water and lipid headgroups, as illustrated by the tilt angle traces shown in Figure 3B. The R694L system does not need to solvate its midspan leucine and consequently has a smaller tilt. Overall, the properties of TM and its mutant are stable during the trajectories and are consistent with previous, shorter simulations (300 ns).15
Figure 1.
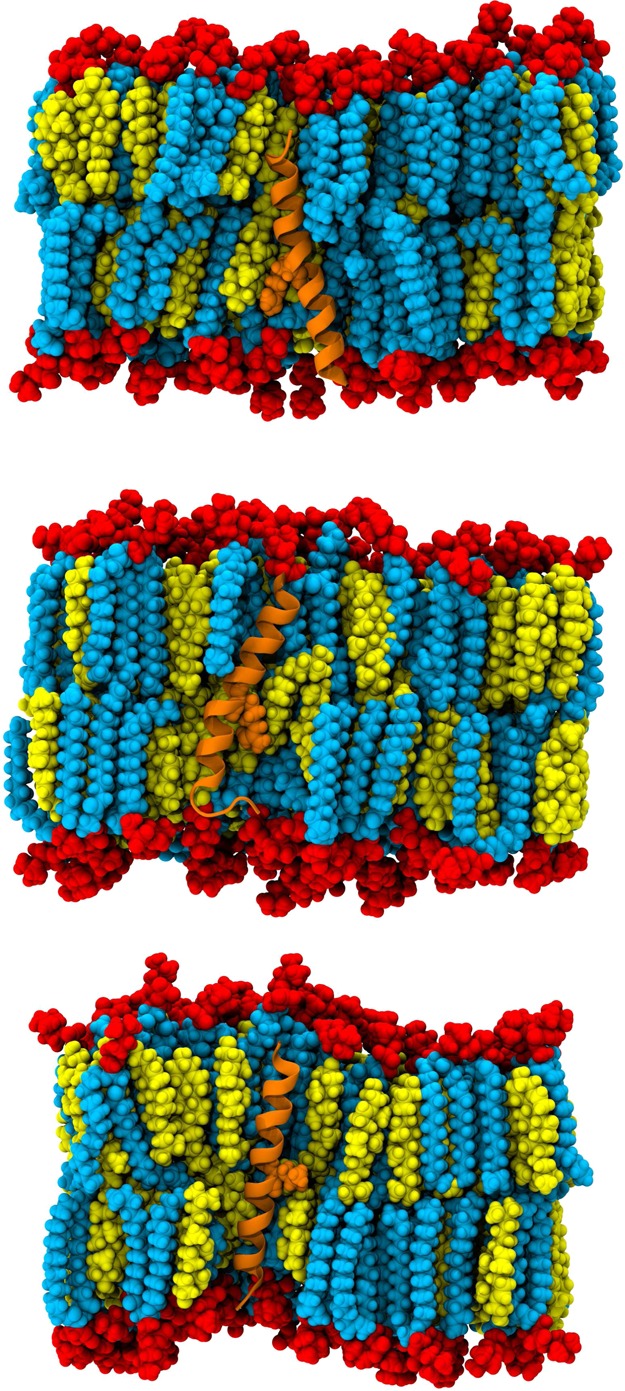
Representative system configurations at 9.98, 6.45, and 8.06 μs of WT1, WT2, and R694L systems, respectively, rendered in VMD. Lipid headgroups are shown in red vdW and lipid tails in cyan vdW. Cholesterol is shown in yellow vdW, the peptide in orange new cartoon, and residue 694 in orange vdW. For the sake of clarity, lipids and cholesterol molecules in the foreground and all water molecules have been omitted.
Figure 2.

rmsd of backbone atoms compared to frame 0 in angstroms vs simulation time in microseconds for the (A) WT1, (B) WT2, and (C) R694L systems.
Figure 3.
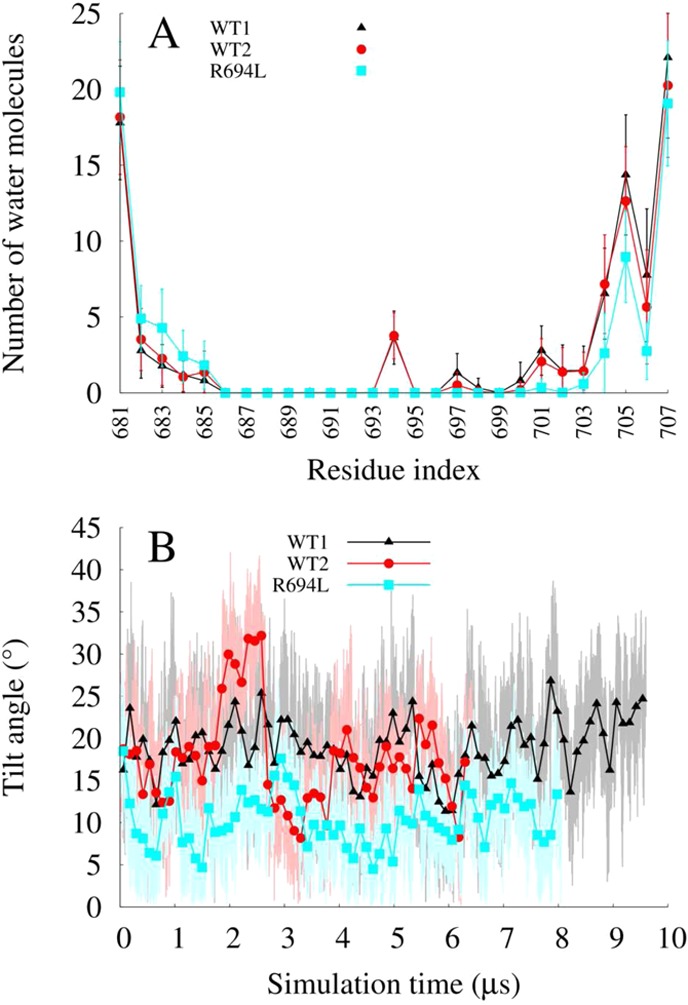
(A) Number of unique water molecules within 4 Å of protein vs amino acid averaged over entire MD trajectories. Error bars represent the standard deviation. (B) Helix tilt angle from membrane normal in degrees vs simulation time in microseconds.
The Membrane Distribution Local to Peptide Changes Slowly
These simulations sought to address the extent to which initial condition bias is removed and the extent to which ergodicity is achieved in multiple-microsecond MD simulations of cholesterol-containing bilayers. Figure 4 shows initial and final system snapshots of the lower leaflets viewed along membrane normal in which a selection of DPPC molecules initially within 6 Å of the peptide are colored uniquely, for each of the WT1, WT2, and R694L systems. (These images are of the systems in which the coordinates are centered on the peptide center of mass and rewrapped into the periodic box.) Figure 5 shows analogous snapshots of cholesterol molecules. These images illustrate that some of the molecules initially surrounding the peptide do indeed leave its immediate vicinity on the microsecond time scale. However, a few lipid and cholesterol molecules that interact with the protein throughout the trajectories remain. This is more evident for WT2 and R694L in the same figures, as the lipids and cholesterol do not diffuse away from the protein as much as they do in WT1. Maps of average cholesterol hydroxyl oxygen mass density in the lower leaflet for protein-centered WT1, WT2, and R694L systems are shown in the top row of Figure 6, while the bottom row shows analogous plots for the DPPC phosphorus atom. The WT1 system has an average uniform distribution of cholesterol. However, WT2 and R694L systems show increased cholesterol density near the proteins. WT1 seems to have increased density representing two DPPC molecules, and WT2 has one DPPC molecule near the protein. The surprising implication of these data is that lipids and cholesterol may effectively form long-lived complexes with peptides that only slowly convert between bound and unbound states.
Figure 4.
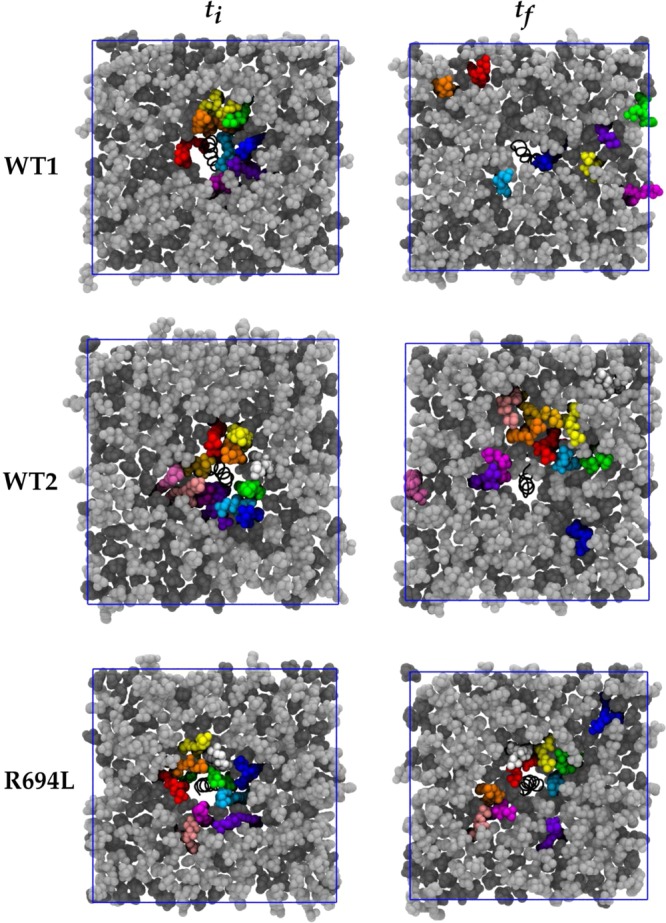
DPPC lipid molecules within 6 Å of protein in the lower leaflet in bright colors at the first simulation frame (ti) and the same molecules at the final simulation frame (tf). Lipid molecules are colored light gray and cholesterol molecules dark gray. The protein is represented as a black spiral. The blue square represents the x and y periodic boundary conditions.
Figure 5.

Cholesterol molecules within 6 Å of protein in the lower leaflet in bright colors at the first simulation frame (ti) and the same molecules at the final simulation frame (tf). Cholesterol molecules are colored dark gray, and the protein is represented as a black spiral. Lipid molecules are not shown for the sake of clarity. The blue square represents the x and y periodic boundary conditions.
Figure 6.

Maps of average mass (amu) per frame 20 Å from protein in the lower leaflet for WT1, WT2, and R694L. The top row shows cholesterol hydroxyl oxygen mass and the second row DPPC phosphorus mass. The peptides are centered in x and y, and the azimuthal orientation of the midspan residue (arginine for WT1 and WT2 and leucine for R694L) is aligned along the x-axis in each map, as shown by the white arrow in the bottom right panel.
To more quantitatively assess ergodicity in the systems, the local density of each species, ρi(x,y,z), in a protein-centered coordinate system was computed from the MD trajectories. MD simulations that significantly explore configurational space will have ρi’s different from those of the starting configurations. This condition is only partially met when considering local density profiles for atoms <8 Å from the peptide along the membrane normal coordinate. In Figure 7 are shown local density profiles from the initial and final two microseconds of each simulation. Differences among the initial profiles reflect the purposeful choice to begin the simulations with different initial configurations. All three systems have more similar final distributions, with more cholesterol than DPPC density near the peptide in the lower leaflet. Similar graphs corresponding to shorter time intervals show a gradual change from initial to final density profiles for each system. However, the systems do not exhibit the full range of possible local density profiles during the multiple-microsecond trajectories, and it seems this time scale is not sufficiently long for these systems to approach ergodicity.
Figure 7.

Mass density within 8 Å of the protein along membrane normal for cholesterol (red squares), lipid headgroups (cyan circles), and lipid tails (black diamonds) for (A–C) the first 2 μs MD and (D–F) the last 2 μs MD for WT1, WT2, and R694L systems. The orientation of the upper and lower leaflets is noted.
Although the membrane components changed their distribution near the peptide, the lipid and cholesterol molecules remain randomly distributed in the bilayer and do not cluster, as determined by radial distribution functions g(r) over time (data not shown). Figure S2 of the Supporting Information shows the average g(r) for various atomic pairs for the three systems; they are essentially identical between systems for all atomic sets and are consistent with those from similar, previous studies.31
Diffusion Coefficients from Microsecond Simulations Are Comparable to Those from Experiments
Sufficient statistics of 6.45–9.98 μs MD allowed analysis of the lateral diffusion of the components of protein-embedded membranes with a high cholesterol content. The mean-square displacements of the lipids (by leaflet), cholesterol (by leaflet), and protein were measured over time on the unwrapped trajectories and are shown in Figure 8A–C for leaflet-centered and Figure 8D–F for bilayer-centered unwrapping, respectively. At short times, ballistic motion is present and diffusive motion occurs between 100 ns and 1 μs. Using Einstein’s relation, the lateral self-diffusion coefficients (Di,lat) are listed in Table 1 and are on the order of 10–9 cm2/s. The fit to linear regimes is shown in Figure S3 of the Supporting Information. Even though WT1 and WT2 are replicas, WT1 exhibits 3-fold faster diffusion in the upper leaflet for both DPPC and cholesterol and 5-fold faster diffusion in the lower leaflet for DPPC and cholesterol, compared to that of WT2. In all three systems, the upper leaflet has diffusion coefficients higher than those of the lower leaflet. The diffusion coefficients were calculated two different ways based on the postprocessing of the trajectories. It is suggested measurements of lipid displacements should be taken after bilayer COM has been removed, because measurements after removal of leaflet COM tend to underestimate diffusion coefficients.8,32 Figure 8 and Table 1 show data from both methods, to determine the extent of system size effects. Previous MD has shown that the difference between these two methods decreases as system size increases.8,32 Diffusion coefficients measured here based on bilayer COM removal are 1.1–1.8 times larger than those measured after monolayer COM removal, indicating some system size dependence. Also, the diffusion coefficients measured after bilayer COM removal have slightly smaller differences between leaflets. Even though the number of lipids and cholesterol in these simulations (334 total) is larger than the number in the largest systems with insignificant system size dependence in the cited studies, the high concentration of cholesterol here may increase the correlation length of the lipids.
Figure 8.

Log–log plots of mean-square displacements vs time for DPPC, cholesterol (excluding flip-flops), and the gp41 TM α-helix. The displacements measured after monolayer COM motion was removed are graphed in panels A–C and after bilayer COM motion was removed in panels D–F. In panels A, B, D, and E, “UL” and “LL” refer to upper and lower leaflets, respectively. Panels C and F both show a sudden drop in diffusivity from 103 to 104 caused by the lack of statistics for the peptide at long time scales.
Table 1. Lateral Diffusion Coefficients (×10–9 cm2/s).
| |
monolayer
COM removed |
bilayer
COM removed |
|||||
|---|---|---|---|---|---|---|---|
| WT1 | WT2 | R694L | WT1 | WT2 | R694L | ||
| protein | LL | 1.50 | 0.87 | 0.30 | 1.83 | 0.63 | 0.20 |
| DPPC | UL | 7.49 | 2.71 | 0.95 | 9.03 | 3.03 | 1.31 |
| LL | 4.98 | 1.01 | 0.41 | 6.30 | 1.32 | 0.74 | |
| cholesterol | UL | 8.90 | 2.90 | 1.20 | 9.98 | 3.20 | 1.54 |
| LL | 6.50 | 1.34 | 0.47 | 7.62 | 1.60 | 0.77 | |
| temperature (K) | 306.7 ± 1.22 | 302.6 ± 1.22 | 302.2 ± 1.24 | 306.7 ± 1.22 | 302.6 ± 1.22 | 302.2 ± 1.24 | |
The lateral self-diffusion coefficients measured here compare reasonably well with those from experiments, which can also be highly variable. Experimental measurements of lateral diffusion of components in model bilayers depend on the temperature, lipid phase, lipid composition, cholesterol content, protein content, ion concentration, hydration level, and time scale of measurement. Although the time scale of NMR might not be appropriate for comparison here, Scheidt et al. used PFG MAS (1H pulsed field gradient magic angle spinning) NMR spectroscopy on 50% DPPC/50% cholesterol bilayers at 309 K and measured values of 33 × 10–9 cm2/s for DPPC and 37 × 10–9 cm2/s for cholesterol.33 Our results are within 1 order of magnitude of those of Scheidt et al. and agree qualitatively in that DPPC has a diffusion coefficient lower than that of cholesterol. Filippov et al. measured diffusion using NMR on bilayers composed of 58% DPPC and 42% cholesterol, resulting in Di,lat values for DPPC of 25 and 75 × 10–9 cm2/s at 308 and 313 K, respectively.34 Diffusion of 50% DPPC and 50% cholesterol at room temperature measured using FCS by Scherfeld et al. yielded a diffusion coefficient of 4.5 × 10–9 cm2/s for the dye.35 In addition to the conditions listed above, computational measurements of lateral diffusion also depend on force field, system size, MD parameters, simulation length, and area per lipid. Falck et al. simulated 100 ns MD of 50% DPPC and 50% cholesterol at 323 K, which yielded a Di,lat of 2 × 10–9 cm2/s for both DPPC and cholesterol, although the authors cautioned about the accuracy of diffusion measurements for systems with 50% cholesterol on the 100 ns time scale.11 The diffusion coefficients measured here are comparable to both experimental and computational measurements of diffusion, although it is difficult to directly compare them.
The mean-square angular displacements are shown in Figure S4 of the Supporting Information. From these, we extract species rotational diffusion constants Di,rot, which are also reported in Table S1 of the Supporting Information. Di,rot shows system-to-system and leaflet-to-leaflet trends identical to those of Di,lat; for instance, WT1 has orientational diffusivity for all components faster than those in the WT2 and R694L systems.
Examination of Interdependent Variables That Manifest the Intersystem Discrepancies in Di,lat
As mentioned in Computational Methods, use of specific MD parameters (Nosé–Hoover thermostat and no removal of system COM velocity) resulted in cooling of the three systems from the 310 K set point to 306.7, 302.6, and 302.2 K [WT1, WT2, and R694L, respectively (Figure S1 of the Supporting Information)]. The system-to-system trends in Di,lat and Di,rot directly correlate to the observed temperature in each system, with the coolest systems showing the lowest diffusivities and with the differences between WT1 and WT2 being generally larger than those between WT2 and R694L. The temperature differences during the simulation result in changes in unit cell area, lipid order parameters, membrane thickness, and, as discussed, diffusion. The other interdependent variables are now examined.
The WT1 system has a stable total unit cell area for the entire trajectory, as shown in Figure S5 of the Supporting Information, of ∼69 nm2. (Because the membranes contain two components and a protein, determining the area per lipid is not trivial. The total unit cell area, from periodic boundary conditions employed in MD, is useful for looking at changes in membrane area.) This correlates with the stable system temperature of WT1 (Figure S1 of the Supporting Information). Both the WT2 and R694L systems take ∼3 μs to relax into a smaller unit cell area of ∼64 nm2. This is a result of the systems adjusting to lower simulation temperatures. Also evident in Figure S5 of the Supporting Information are instances in which the R694L system exhibits a much smaller area, corresponding to trajectory segments where the membrane experiences undulatory motions. These undulations have been seen in other microsecond simulations, and the undulations increase the difference between the “true” area and the unit cell area.36 Interestingly, mean-square displacement measurements taken during and after undulatory motions of R694L show the same spread of MSDs for the undulations and undulation-free sections. No pattern was discernible, even when the curvature of the undulations was taken into account. Apparently, these undulations do not significantly affect diffusion in the R694L system.
Lipid order parameters (SCD) of DPPC also reflect the temperature change and resultant unit cell area shrinkage. SCD was averaged for each leaflet over the whole trajectory for each system and is shown in Figure 9. The SCD data here are similar to those from other studies with shorter durations with similar lipid compositions.31 Both leaflets of WT1 have lipid order parameters lower than those of the other systems’ leaflets over the entire trajectories. Also, the WT1 upper leaflet has an SCD lower than that of the WT1 lower leaflet. These trends correspond inversely with the trends seen in the diffusion coefficients. The WT1 upper leaflet has the lowest SCD and also has the fastest diffusion. This trend also occurs for the WT2 and R694L leaflets. R694L has the highest SCD (is the most ordered) and has the slowest dynamics. The systems also show changes in lipid order on the microsecond time scale. For example, the DPPC lipids in the upper leaflet of the R694L system are more ordered from 2 to 8.02 μs, than during the first 2 μs, as seen in Figure S6 of the Supporting Information. This correlates with the change in the R694L bilayer to a different configuration at a lower temperature and a smaller total unit cell area, as discussed above. Figure S7 of the Supporting Information shows that the levels of order of both leaflets in WT2 also increase compared to the first 2 μs. The level of order of the upper leaflet of WT1 increases from 4 to 6 μs but then returns to its initial value, as seen in Figure S8 of the Supporting Information.
Figure 9.
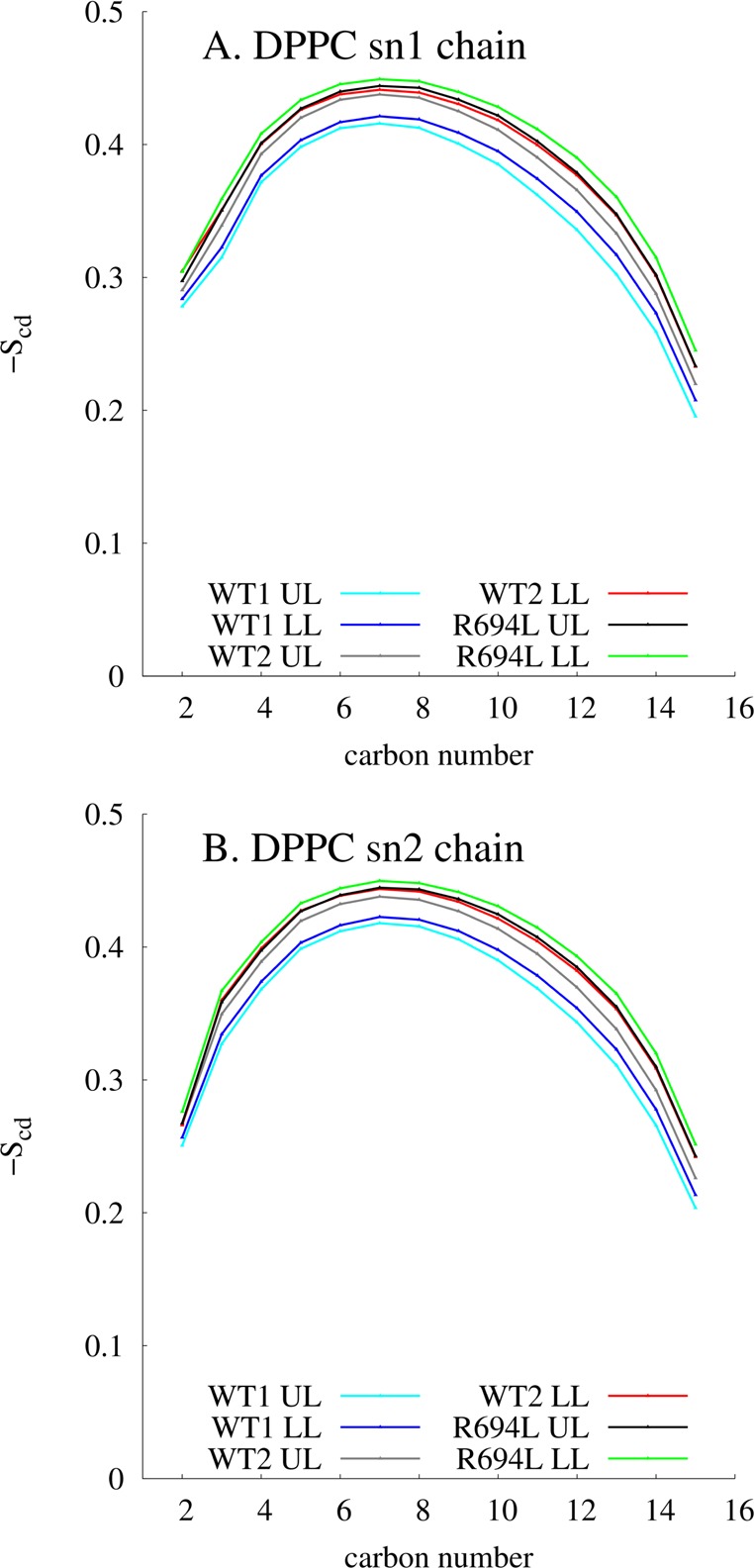
DPPC order parameters for each chain, averaged over the entire trajectory. “UL” and “LL” refer to upper and lower leaflets, respectively.
As expected, the membrane thickness also correlates with the lipid order parameters and unit cell area. The average distance between the headgroups of DPPC in the lower and upper leaflets in 4 Å × 4 Å lateral squares was histogrammed for each system, as shown in Figure S9 of the Supporting Information. WT1, the more disordered system, has a smaller median membrane thickness. The more ordered systems, WT2 and R694L, have larger membrane thicknesses.
The ∼4 K difference in simulation temperatures between the WT1 system and the WT2 and R694L systems resulted in differences in both diffusion and membrane area. The cooler systems (WT2 and R694L) have ∼10-fold slower diffusion and 5% smaller membrane areas. The lower temperature results in more ordered systems. These trends among diffusion, membrane order, area, and thickness have been seen in other systems, as well, and our simulations support those observations.37 Here, they were the result of choosing to operate the thermostat in such a way as to determine the true displacements and reduce the likelihood of fictitious interleaflet motion. These small temperature decreases also emphasize the extreme sensitivity of diffusion measurements in nearly similar systems.
Speculation about the Interleaflet Discrepancies in Self-Diffusion Coefficients
All systems consistently exhibited higher diffusivities in the upper leaflet (which houses the N-terminus of the peptide) than in the lower leaflet. Systems WT1 and WT2 displayed a stable water defect on microsecond time scales because of solvation of the midspan arginine and the consequently larger tilt angle of the WT peptide compared to that of the R694L peptide. Slower self-diffusion in the lower leaflets relative to the upper leaflets cannot necessarily be attributed to the water defect because the R694L system also displayed the interleaflet discrepancy and did not contain a water defect. The two terminal arginines, via specific interactions with lipids and cholesterol, may have effectively lowered the self-diffusion coefficients for lipids and cholesterol in the lower leaflets relative to those in the upper leaflets. We have not performed multiple-microsecond simulations to address this hypothesis; however, previous 300 ns simulations of the systems add support. Previously,15 300 ns NPT MD of WT1, WT2, and R694L in an ∼50/50 DPPC/cholesterol bilayer were compared to 300 ns of a protein-free bilayer control of the same composition. SCD values by leaflet for the protein-containing systems show interleaflet discrepancies over the trajectories, indicating their presence before simulation for many microseconds on Anton (Figure S10 of the Supporting Information). However, the protein-free control has similar SCD values for both its upper and lower leaflets over 300 ns (Figure S10 of the Supporting Information). The specific sequence of the peptide seems to manifest differences between the leaflets. Computational resources for microsecond MD are limited, so we have not yet been able to investigate specific residues. The sequences of the 13 C-terminal residues of the three peptides are the same and contain two C-terminal arginines, which may help order the bilayer.
Examination of Rare Cholesterol Flip-Flop Events
Three cholesterols attempted to “flip-flop” from one leaflet to the other in the simulations. (There were no lipid molecules that attempted to flip-flop.) The z-position over the trajectory of these three cholesterols is shown in Figure 10A–C. WT1 has one cholesterol that quickly switches leaflets in ∼30 ns (panel A) and one cholesterol that reaches the membrane interface but becomes “stuck” for more than 2 μs before returning to its original leaflet (panel B). The cholesterols in WT1 are able to enter a leaflet when their tilt angle becomes sufficiently large. For the cholesterol in panels A and D, it tilts 0.5 ns before a change in z-position; the tilt increases until it reaches a maximum 15 ns before it reaches the other leaflet. This qualitatively agrees with the PMF of cholesterol flip-flop in DPPC calculated by Jo et al. in that the cholesterol prefers to tilt first before moving to the membrane interface.38 The cholesterol in panels B and E does not tilt first but moves to the membrane interface. It remains there for more than 2 μs and then tilts 75 ns before it reaches its original leaflet from the membrane interface. None of the cholesterols in WT2 attempt to flip-flop. In R694L, one cholesterol is stuck at the membrane interface for the majority of the simulation, perhaps because it does not achieve a sufficiently large tilt at first. Eventually, it returns to its original leaflet after changes in tilt (panels C and F).
Figure 10.

Z-Positions of oxygen atoms vs simulation time of two cholesterol molecules from WT1 (A and B) and one cholesterol molecule from R694L (C) that flip-flop during the trajectories. (D–F) Tilts of the cholesterol ring with respect to membrane normal vs simulation time, corresponding to cholesterol molecules in panels A–C, respectively.
These rare events suggest that if a cholesterol does not immediately increase its tilt by >150° and flip-flop to the other leaflet, it will remain in the membrane interface for a significant amount of time. The nanosecond to microsecond flip-flop events here do not agree with the faster and more numerous events during a 15 μs simulation by Choubey et al.,39 although we have a much higher percentage of cholesterol, which may increase the free energy barrier to flip-flop.40 However, both the nanosecond and microsecond time scales are considered fast for cholesterol flip-flop compared to those in experiments, and disagreement between experiments and simulations of cholesterol flip-flop times, like with membrane diffusion, is not too surprising because there may not always be an overlap in time scales between them. The force field used here, CHARMM36, may not adequately be able to handle parallel orientations of cholesterol in the membrane interface.38 An update to the cholesterol force field parameters improved accuracy in this regard;41 however, we were not able to use this updated force field on Anton.
The three cholesterol molecules identified as flip-floppers were excluded from the intraleaflet mean-square displacement calculations, and the lateral self-diffusion coefficients listed in Table 1 reflect these exclusions. Excluding the two flip-flops in WT1 decreased the coefficient from 6.53 to 6.50 × 10–9 cm2/s in cholesterol diffusion in the lower leaflet. The diffusion coefficient in the lower leaflet of R694L decreased by more than half (from 1.02 to 0.47 × 10–9 cm2/s) as the quick movement of the trapped cholesterol (as shown in Figure 10C) was removed from the MSD measurement. Clearly, differences in the flip-flops between the systems did not significantly contribute to the interleaflet discrepancies of the self-diffusion coefficients discussed previously.
Conclusion
Microsecond-long simulations of peptide-containing membranes with ∼50% cholesterol and ∼50% DPPC allowed observation of diffusion of lipids, cholesterol, and protein at an order of magnitude longer than that of previous all-atom MD. The diffusion coefficients measured (on the order of 10–9 cm2/s) were consistent with experiments; however, there was variation among the three systems because of simulation temperature differences of 0.4–4.5 K. The lower leaflets in the three systems were more ordered and were slower than the corresponding upper leaflets, possibly because of the specific sequence of the peptide, which contains two C-terminal arginines.
Other trends observed agree with those from previous experiments. For example, we also observed a correlation between an increased level of DPPC tail order and decreased diffusivities. Also, we observed that the diffusivity of cholesterol is greater than that of DPPC because of its smaller size, even in ∼50% composition bilayers. The systems here have the same membrane compositions and show for the first time, to the best of our knowledge, that small temperature differences during microsecond simulations can result in significant changes in area, thickness, DPPC order, and diffusivity, at least around temperatures between 300 and 310 K. Therefore, temperature control for microsecond simulations of membranes is extremely important. The lipid composition near the protein did change from their initial condition but did not explore the full range of profiles. Therefore, the simulations here also suggest that membranes with a high cholesterol content require longer than microsecond simulations to approach ergodicity. For instance, a recent umbrella sampling calculation of the binding of a small peptide to a bilayer required windows of 4 μs.42 It seems more research on model membranes on longer time scales is needed before equilibrium properties of model bilayers can be related to cellular and viral membranes.
Acknowledgments
Financial support from the National Institutes of Health (Grant R01 AI-084117-01) is acknowledged. Anton computer time was provided under allocations PSCA10042P by the Pittsburgh Supercomputing Center (PSC) and the National Center for Multiscale Modeling of Biological Systems (MMBioS) through Grant P41GM103712-S1 from the National Institutes of Health. The Anton machine at NRBSC/PSC was generously made available by D. E. Shaw Research. This research was supported in part by the National Science Foundation through TeraGrid resources under Grant TG-MCB070073N.
Supporting Information Available
Rotational diffusion coefficients (Table S1), simulation temperature versus time (A) and system COM velocity versus time (B) (Figure S1), g(r) values for several atomic pairs (Figure S2), plot of MSD versus time showing linear fits (Figure S3), plot of MSAD versus time (Figure S4), plot of unit cell area versus time and a snapshot of R694L undulation (Figure S5), SCD versus carbon number at 2 μs intervals for R694L (Figure S6), SCD versus carbon number at 2 μs intervals for WT2 (Figure S7), SCD versus carbon number at 2 μs intervals for WT1 (Figure S8), membrane thickness histograms (Figure S9), SCD for shorter simulations of WT1, WT2, R694L, and a protein-free control (Figure S10), and Anton configuration parameters. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Guyader M.; Kiyokawa E.; Abrami L.; Turelli P.; Trono D. Role for Human Immunodeficiency Virus Type 1 Membrane Cholesterol in Viral Internalization. J. Virol. 2002, 76, 10356–10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed A. A.; Freed E. O. Lipids and Membrane Microdomains in HIV-1 Replication. Virus Res. 2009, 143, 162–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlert J.; Edholm O. Dynamics in Atomistic Simulations of Phospholipid Membranes: Nuclear Magnetic Resonance Relaxation Rates and Lateral Diffusion. J. Chem. Phys. 2006, 125, 204703-1–204703-10. [DOI] [PubMed] [Google Scholar]
- Sanderson J. M. Resolving the Kinetics of Lipid, Protein and Peptide Diffusion in Membranes. Mol. Membr. Biol. 2012, 29, 118–143. [DOI] [PubMed] [Google Scholar]
- Falck E.; Róg T.; Karttunen M.; Vattulainen I. Lateral Diffusion in Lipid Membranes through Collective Flows. J. Am. Chem. Soc. 2008, 130, 44–45. [DOI] [PubMed] [Google Scholar]
- Niemelä P. S.; Miettinen M. S.; Monticelli L.; Hammaren H.; Bjelkmar P.; Murtola T.; Lindahl E.; Vattulainen I. Membrane Proteins Diffuse as Dynamic Complexes with Lipids. J. Am. Chem. Soc. 2010, 132, 7574–7575. [DOI] [PubMed] [Google Scholar]
- Lindahl E.; Edholm O. Molecular Dynamics Simulation of NMR Relaxation Rates and Slow Dynamics in Lipid Bilayers. J. Chem. Phys. 2001, 115, 4938–4950. [Google Scholar]
- Roark M.; Feller S. E. Molecular Dynamics Simulation Study of Correlated Motions in Phospholipid Bilayer Membranes. J. Phys. Chem. B 2009, 113, 13229–13234. [DOI] [PubMed] [Google Scholar]
- Andoh Y.; Okazaki S.; Ueoka R. Molecular Dynamics Study of Lipid Bilayers Modeling the Plasma Membranes of Normal Murine Thymocytes and Leukemic GRSL Cells. Biochim. Biophys. Acta 2013, 1828, 1259–1270. [DOI] [PubMed] [Google Scholar]
- Jeon J.-H.; Monne H. M.-S.; Javanainen M.; Metzler R. Anomalous Diffusion of Phospholipids and Cholesterols in a Lipid Bilayer and Its Origins. Phys. Rev. Lett. 2012, 109, 188103-1–188103-5. [DOI] [PubMed] [Google Scholar]
- Falck E.; Patra M.; Karttunen M.; Hyvönen M. T.; Vattulainen I. Lessons of Slicing Membranes: Interplay of Packing, Free Area, and Lateral Diffusion in Phospholipid/Cholesterol Bilayers. Biophys. J. 2004, 87, 1076–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw D. E.; Deneroff M. M.; Dror R.; Kuskin J. S.; Larson R. H.; Salmon J. K.; Young C.; Batson B.; Bowers K. J.; Chao J. C.; et al. Anton, a Special-Purpose Machine for Molecular Dynamics Simulation. Commun. ACM 2008, 91–97. [Google Scholar]
- Klepeis J. L.; Lindorff-Larsen K.; Dror R. O.; Shaw D. E. Long-Timescale Molecular Dynamics Simulations of Protein Structure and Function. Curr. Opin. Struct. Biol. 2009, 19, 120–127. [DOI] [PubMed] [Google Scholar]
- Gangupomu V. K.; Abrams C. F. All-Atom Models of the Membrane-Spanning Domain of HIV-1 gp41 from Metadynamics. Biophys. J. 2010, 99, 3438–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M. K.; Gangupomu V. K.; Abrams C. F. Characterization of the Water Defect at the HIV-1 gp41 Membrane Spanning Domain in Bilayers With and Without Cholesterol using Molecular Simulations. Biochim. Biophys. Acta 2014, 694, 26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Lim J. B.; Klauda J. B.; Im W. CHARMM-GUI Membrane Builder for Mixed Bilayers and its Application to Yeast Membranes. Biophys. J. 2009, 97, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Feller S. E.; MacKerell A. D. An Improved Empirical Potential Energy Function for Molecular Simulations of Phospholipids. J. Phys. Chem. B 2000, 104, 7510–7515. [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor R. W.; MacKerell A. D. Development of the CHARMM Force Field for Lipids. J. Phys. Chem. Lett. 2011, 2, 1526–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y.; Klepeis J. L.; Eastwood M. P.; Dror R. O.; Shaw D. E. Gaussian Split Ewald: A Fast Ewald Mesh Method for Molecular Simulation. J. Chem. Phys. 2005, 122, 054101-1–054101-13. [DOI] [PubMed] [Google Scholar]
- Martyna G. J.; Tobias D. J.; Klein M. L. Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar]
- Lippert R. A.; Predescu C.; Ierardi D. J.; Mackenzie K. M.; Eastwood M. P.; Dror R. O.; Shaw D. E. Accurate and Efficient Integration for Molecular Dynamics Simulations at Constant Temperature and Pressure. J. Chem. Phys. 2013, 139, 164106-1–164106-11. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 27–28, 33–38. [DOI] [PubMed] [Google Scholar]
- Basconi J. E.; Shirts M. R. Effects of Temperature Control Algorithms on Transport Properties and Kinetics in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2013, 9, 2887–2899. [DOI] [PubMed] [Google Scholar]
- Harvey S. C.; Tan R. K.-Z.; Cheatham T. E. The Flying Ice Cube: Velocity Rescaling in Molecular Dynamics Leads to Violation of Energy Equipartition. J. Comput. Chem. 1998, 19, 726–740. [Google Scholar]
- Chiu S.-W.; Clark M.; Subramaniam S.; Jakobsson E. Collective Motion Artifacts Arising in Long-Duration Molecular Dynamics Simulations. J. Comput. Chem. 2000, 21, 121–131. [Google Scholar]
- Vermeer L. S.; de Groot B. L.; Réat V.; Milon A.; Czaplicki J. Acyl Chain Order Parameter Profiles in Phospholipid Bilayers: Computation from Molecular Dynamics Simulations and Comparison with 2H NMR Experiments. Eur. Biophys. J. 2007, 36, 919–931. [DOI] [PubMed] [Google Scholar]
- Levine B. G.; Stone J. E.; Kohlmeyer A. Fast Analysis of Molecular Dynamics Trajectories with Graphics Processing Units: Radial Distribution Function Histogramming. J. Comput. Phys. 2011, 230, 3556–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cournia Z.; Ullmann G. M.; Smith J. C. Differential Effects of Cholesterol, Ergosterol and Lanosterol on a Dipalmitoyl Phosphatidylcholine Membrane: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2007, 111, 1786–1801. [DOI] [PubMed] [Google Scholar]
- Klauda J. B.; Brooks B. R.; Pastor R. W. Dynamical Motions of Lipids and a Finite Size Effect in Simulations of Bilayers. J. Chem. Phys. 2006, 125, 144710-1–144710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidt H. A.; Huster D.; Gawrisch K. Diffusion of Cholesterol and its Precursors in Lipid Membranes Studied by 1H Pulsed Field Gradient Magic Angle Spinning NMR. Biophys. J. 2005, 89, 2504–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov A. V.; Rudakova M. A.; Oradd G.; Lindblom G. Lateral Diffusion of Saturated Phosphatidylcholines in Cholesterol-Containing Bilayers. Biophyzika 2007, 52, 476–485. [PubMed] [Google Scholar]
- Scherfeld D.; Kahya N.; Schwille P. Lipid Dynamics and Domain Formation in Model Membranes Composed of Ternary Mixtures of Unsaturated and Saturated Phosphatidylcholines and Cholesterol. Biophys. J. 2003, 85, 3758–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A. R.; Brandt E. G.; Edholm O.; Nagle J. F.; Sachs J. N. Determination of Electron Density Profiles and Area From Simulations of Undulating Membranes. Biophys. J. 2011, 100, 2112–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apajalahti T.; Niemelä P.; Govindan P. N.; Miettinen M. S.; Salonen E.; Marrink S.-J.; Vattulainen I. Concerted Diffusion of Lipids in Raft-Like Membranes. Faraday Discuss. 2010, 144, 411–430. [DOI] [PubMed] [Google Scholar]
- Jo S.; Rui H.; Lim J. B.; Klauda J. B.; Im W. Cholesterol Flip-Flop: Insights From Free Energy Simulation Studies. J. Phys. Chem. B 2010, 114, 13342–13348. [DOI] [PubMed] [Google Scholar]
- Choubey A.; Kalia R.; Malmstadt N.; Nakano A.; Vashishta P. Cholesterol Translocation in a Phospholipid Membrane. Biophys. J. 2013, 104, 2429–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett W. F. D.; MacCallum J. L.; Hinner M. J.; Marrink S. J.; Tieleman D. P. Molecular View of Cholesterol Flip-Flop and Chemical Potential in Different Membrane Environments. J. Am. Chem. Soc. 2009, 131, 12714–12720. [DOI] [PubMed] [Google Scholar]
- Lim J. B.; Rogaski B.; Klauda J. B. Update of the Cholesterol Force Field Parameters in CHARMM. J. Phys. Chem. B 2012, 116, 203–210. [DOI] [PubMed] [Google Scholar]
- Neale C.; Hsu J. C. Y.; Yip C. M.; Pomès R. Indolicidin Binding Induces Thinning of a Lipid Bilayer. Biophys. J. 2014, 106, L29–L31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.