

Abstract
Objectives
The objective of this study was to identify novel survival-associated biomarkers in oral rinse samples collected from patients with oral squamous cell carcinoma (OSCC).
Materials & Methods
We screened for putative survival-associated markers using publicly available methylation array data from 88 OSCC tumors. Cox models were then fit to methylation array data restricted to these putative loci in oral rinse samples of 82 OSCC patients from greater Boston. Pyrosequencing assays were designed for each locus that replicated in the oral rinse samples and applied to a validation set of oral rinse samples from another 61 OSCC patients.
Results
We identified 7 survival-associated methylation markers in oral rinse samples from OSCC patients, and have validated one, located in the body of GABBR1, by pyrosequencing.
Conclusion
The 7 CpG loci identified through this study represent novel prognostic biomarkers for patients with OSCC that can be detected using a non-invasive oral rinse collection technique.
Keywords: Oral cancer, prognosis, mouthwash, epigenomics, GABBR1, OPCML, ADCK4, ZFYVE26, POLR3E, KIF11
INTRODUCTION
It is projected that 27,450 new cases of oral cancer will be diagnosed in the United States in 2013 [1], and more than 90% of these cancers will be oral squamous cell carcinomas (OSCC) [2]. The prognosis for OSCC remains relatively poor, with an overall 5-year survival rate around 60%, which is a value that has remained virtually unchanged over the past three decades [3], and sharply declines with increasing stage at diagnosis [4]. However, the only prognostic biomarkers currently in routine clinical use relate to HPV16 positivity. Thus, there is a need to identify novel molecular markers that could aid in outcome prediction, helping to mitigate the clinical uncertainty for the patients and providers.
Development of OSCC is a multistep process involving an accumulation of genetic and epigenetic alterations resulting in cellular dysregulation and uncontrolled growth. It has long been appreciated that increased exposure of the oral epithelium to tobacco and alcohol carcinogens can give rise to fields of altered cells [5]. The observation that second primary tumors are often clonally related to the primary tumor [6] has led to the formulation of the expanding fields model, which proposes that a single stem cell in the basal layer of the epithelium undergoes a genetic or epigenetic transformation that confers a growth advantage, clonally expands, and gradually replaces the normal epithelium giving rise to a field defect. As cells within the expanding field acquire new alterations, various subclones develop within the field, which can eventually propagate into distinct but related tumors. These models help to explain, in part, the high rate of local recurrences and development of second primaries associated with OSCC, which are a major reason for treatment failure and adversely impact long-term survival [7, 8]. Thus measurement of field defects may provide valuable insight into overall patient prognosis.
Oral rinse (i.e. mouthwash) is a non-invasive sample collection technique that can be used to obtain DNA from the oral epithelium. The broad collection of exfoliated epithelial cells across the oral cavity may be advantageous in the assessment of epigenetic alterations in the tumor cells and/or surrounding epithelium. While a number of studies have reported on the diagnostic potential of DNA methylation markers in oral rinse [9-19], few have evaluated the prognostic value [18, 20-22], and those that did were focused solely on promoter methylation of a limited subset of candidate tumor suppressor genes. Such a candidate approach has clear limitations, particularly given our incomplete understanding of genes involved in the biology of head and neck cancer and the crucial involvement of CpG methylation outside of the context of promoter regions in transcriptional regulation and carcinogenesis. This includes recent evidence of the important role of CpG island shores (defined as sequences within 2kb distance of a CpG island) in transcriptional control and differentiation that has begun to emerge in the recent literature [23, 24]. Further, methylation of CpG motifs located outside of CpG islands or shores, including cis or trans enhancer regions, also play a role in gene regulation [25], tissue differentiation [26] and genomic stability [27].
The primary objective of this study was to apply a comprehensive epigenome-wide strategy to oral rinse samples collected from OSCC patients for identification of novel survival-associated biomarkers. To this end, we have employed a three-stage study design to a prospective cohort of OSCC patients to identify and validate epigenetic markers of OSCC prognosis using an array-based approach to interrogate nearly 400,000 CpG loci spanning 99% of annotated autosomal human genes.
MATERIALS AND METHODS
Study design
We applied a three-stage epigenome-wide approach to prospectively collected survival data from two independent cohorts of OSCC patients for identification of novel prognostic DNA methylation markers in oral rinse samples. This entailed an initial (1) Discovery stage using publicly available Infinium HumanMethylation450 BeadArray data for OSCC tumor tissue from The Tumor Genome Atlas (TCGA) to identify putative methylation targets for the non-invasive oral rinse samples with biological-relevance with regard to tumor behavior, followed by (2) Replication in HumanMethylation450 BeadArray data for oral rinse samples from OSCC patients, and then (3) Validation of significant loci that replicated in a second set of oral rinse samples using custom pyrosequencing assays.
The Cancer Genome Atlas OSCC Tumor Data
Infinium HumanMethylation450 BeadArray data for tumor tissue from 88 OSCC patients (Discovery), along with corresponding clinical data, were obtained from TCGA (http://cancergenome.nih.gov/). Level 1 methylation data (raw signal intensities) were requested and downloaded on March 7, 2013, and were pre-processed using GenomeStudio v. 2011.1 (Illumina, San Diego, CA).
Collaborative Study of Head and Neck Diseases (CoHANDS)
The Replication set was comprised of oral rinse samples (n = 82) that were collected from patients with incident initial primary squamous cell carcinoma arising in the oral cavity (OSCC; ICD-9 codes 141.1-141.5, 141.8, 141.9, 143-145.2, 145.5-145.9, 149.8, 149.9) diagnosed between October 2006 and June 2011 at major teaching hospitals located in Boston, Massachusetts (Brigham and Women’s Hospital, Beth Israel Deaconess Medical Center, Boston Medical Center, Dana-Farber Cancer Institute, Massachusetts Eye and Ear Infirmary, Massachusetts General Hospital, and New England Medical Center; which together see the vast majority of cases in the region) as part of a population-based study of head and neck cancer in the greater-Boston area [28, 29] (Collaborative Study of Head and Neck Diseases: CoHANDS). For inclusion in the study, cases were required to reside in Boston or any of 162 contiguous cities and towns within approximately one hour drive from Boston at the time of diagnosis; the case participation rate was 78%. Cases with a prior history of malignancy other than non-melanoma skin cancer were excluded from the analyses.
An additional 61 oral rinse samples were available from a second set of CoHANDS OSCC patients for use as the Validation set (pyrosequencing). These cases included patients diagnosed between October 2006 and June 2011 but with scant amount of total DNA, limiting the application toward the Infinium HumanMethylation450 BeadArray due to the input requirements, and study subjects enrolled as part of an earlier CoHANDS enrollment period (diagnosed between December 1999 and December 2003).
All patients included in the analyses provided written informed consent prior to enrollment in the study, as approved by the Institutional Review Boards of the participating institutions.
Oral Rinse Collection
Study subjects were asked to vigorously swish with approximately 30 ml of commercial alcohol-free mouthwash (Act™) for 30 seconds. Samples were then centrifuged into cell pellets and stored at -80°C in cryovials until DNA extraction.
DNA Extraction and Bisulfite Modification
DNA was extracted using the QIAamp Blood Kit (Qiagen, Valencia, CA), modified to accommodate oral rinse samples. Extracted DNA was bisulfite modified using the EZ-96 DNA Methylation-Direct Kit (Zymo Research, Irvine, CA) according to the manufacturer’s recommendations.
Infinium HumanMethylation450 BeadArray
Methylation analysis was performed on bisulfite modified DNA obtained from oral rinse samples from 82 CoHANDS OSCC patients using the HumanMethylation450 BeadArray (Illumina, San Diego, CA), which interrogates > 450,000 CpG loci spanning 99% of RefSeq genes, at the University of California San Francisco Institute for Human Genomics Core Facility. All array data points are represented by fluorescent signals from both methylated (Cy5) and unmethylated (Cy3) alleles, and average methylation level (β) is derived from the ~18 replicate methylation measurements, β= (max(Cy5, 0))/(|Cy3| + |Cy5| + 100). Beta (β) = 1 indicates complete methylation; β = 0 represents no methylation. Outliers were assessed using array control probes to diagnose problems such as poor bisulfite conversion, batch or BeadChip effect, or color-specific problems. Mahalanobis distances were determined based on fitted mean vector and variance-covariance matrix, and arrays with large distances inconsistent with multivariate normality [30] were discarded. Logit-transformed beta values were adjusted for potential batch effect prior to analysis using a linear mixed effects model approach with an indicator for BeadChip included as the random effect variable.
Pyrosequencing Assays
Custom pyrosequencing assays were designed for all CpG loci associated with survival in the array-based data from oral rinse samples during the Replication stage (n = 82) and were applied to oral rinse samples from OSCC patients in the Validation set (n = 61). Primers were designed using the PSQ Assay Design software (Qiagen, Valencia, CA) to amplify the CpG of interest using a bisulfite modified DNA template. This allowed for re-sequencing of the CpG site targeted by the array, as well as any surrounding CpG sites. A dilution curve of methylated:unmethylated referent DNA was established for each pyrosequencing assay to detect for PCR bias. All samples were run in triplicate using the highly sensitive PyroMark Q96 MD system (Qiagen, Valencia, CA), yielding quantitative methylation for the targeted CpG (0-100%), quantitative methylation for each additional CpG captured, and average percent methylation across all measured CpGs. Primer sequences and PCR conditions for the pyrosequencing assays are provided in Supplementary Table S1.
HPV16 Serology
Serologic HPV16 testing for E6 and E7 viral protein antibodies was performed on CoHANDS study subjects as a biomarker of HPV16-transformed invasive tumors [31]. HPV16 E6 and E7 antibodies were detected by using a glutathione S-transferase capture enzyme-linked immunosorbent assay in combination with fluorescent bead technology [32]. Subjects were considered to be HPV16-positive if serology was positive for either HPV16 E6 or E7 antibodies.
Statistical Analysis
We first fit a series of Cox proportional hazards models, adjusted for age, gender, smoking status (never, former, current), and stage at diagnosis, across all 384,475 autosomal CpGs (after exclusion of loci with probe sequences containing one or more genetic variants) to the TCGA OSCC tumor Infinium HumanMethylation450 BeadArray dataset (n = 88) to screen for loci for which methylation was associated with overall survival (Discovery); HPV16 serology was not available for TCGA subjects (14 had HPV immunohistochemistry results available, all of which were negative), although it should be noted that the expected frequency of HPV16-positive oral cavity tumors is much lower compared to tumors arising in the oropharynx (not included in this study) [33]. Significance was considered at an unadjusted p-value ≤ 0.05 (to minimize false-negative results during the initial screen for putative markers). A second series of Cox proportional hazards models (adjusted for age, gender, smoking, HPV16 E6/E7 serology and stage at diagnosis) were then fit to the oral rinse methylation array data (Replication; n = 82) for all loci that were significantly associated with survival in the TCGA tumor samples. To account for multiple comparisons, we controlled for false-discovery rate (FDR) using the methods of Benjamini and Hochberg [34], with significance considered where Q ≤ 0.10. For the purpose of computational efficiency for each of the aforementioned series of Cox models, missing covariate data were coded with a dummy indicator.
To assist in visualization of the data, Kaplan-Meier plots for 5-year survival were generated for the Replication and Validation sets for methylation above and below the median for each respective study for each CpG identified during the Replication stage. Differences in the curves by methylation status were assessed using the log-rank test, considered significant at p ≤ 0.05.
Separate multivariable Cox proportional hazards models were refit for each of the significant loci identified in the prior step (Replication) and fit to the average methylation data from each of the pyrosequencing assays (Validation), adjusted for age, gender, smoking pack-years, HPV16 serology, and stage at diagnosis. Multiple imputation by chained equations [35] (MICE) was applied to account for missing covariate data, using 20 iterations and logit prediction for ever-smoking, HPV16 serology, and stage at diagnosis; predictive mean matching (PMM) for pack-years (conditional on ever-smoking); and multivariate logit prediction for alcohol consumption; age and gender were also included as independent predictors. HPV16 serology and stage at diagnosis were not included as predictors for the other missing covariates. The proportional hazards assumption was tested for each model using an approach based on the slope of scaled Schoenfeld’s residuals as a function of time [36].
Statistical analyses were conducted using Stata 13 (Stata Corp, College Station, TX) and R version 3.0.1 (http://www.r-project.org). All statistical tests were two-sided.
RESULTS
A schematic overview of the three-stage workflow is provided in Figure 1. Specific details for the study populations and results by stage are presented below.
Figure 1.
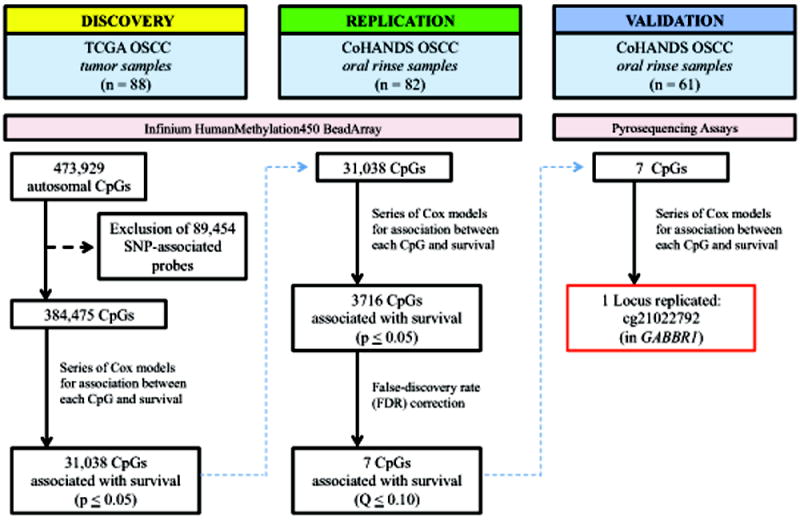
Schematic of study workflow for identification and validation of novel epigenetic predictors of overall survival in oral rinse samples from OSCC patients.
Description of Study Populations
The study populations for each of the three datasets were similar in terms of age (with median ages of 61, 58, and 61 years for the Discovery, Replication, and Validation populations respectively) and gender but differed by race (p = 0.02), smoking (p = 0.01), and stage at diagnosis (p < 0.001). The Validation study population (CoHANDS) included a higher proportion of non-white subjects relative to the Discovery (TCGA) and Replication (CoHANDS) sets. The patients included as part of the TCGA Discovery population had a much higher frequency of advanced stage tumors (AJCC Stage III or IV) and more oft self-reported as a current smoker and less as a never-smoker. A complete description of each of the study populations is provided in Table 1.
Table 1.
Description of the three study populations used for each of the three respective stages of the analysis.
| TCGA (tumor)
|
CoHANDs (oral rinse)
|
pdifference | ||
|---|---|---|---|---|
| Discovery (n = 88) | Replication (n = 82) | Validation (n = 61) | ||
| Age, median years (range) | 61.0 (19-90) | 58.0 (23-85) | 61.0 (23-88) | 0.17‡ |
| Gender, n (%) | ||||
| Female | 28 (31.8%) | 36 (43.9%) | 24 (39.3%) | 0.26§ |
| Male | 60 (68.2%) | 46 (56.1%) | 37 (60.7%) | |
| Race, n (%) | ||||
| White | 85 (96.6%) | 79 (96.3%) | 52 (85.3%) | 0.02§ |
| Other | 3 (3.4%) | 3 (3.7%) | 9 (14.8%) | |
| Smoking status, n (%) | ||||
| Never | 19 (21.6%) | 30 (36.6%) | 20 (32.8%) | 0.01§ |
| Former | 42 (47.7%) | 36 (43.9%) | 28 (45.9%) | |
| Current | 25 (28.4%) | 8 (9.8%) | 5 (8.2%) | |
| Missing | 2 (2.3%) | 8 (9.8%) | 8 (13.1%) | |
| Smoking pack-years*, median (range) | 40.0 (1.0-300.0) | 21.9 (0.9-102.5) | 31.5 (0.5-129.0) | 0.03‡ |
| Alcohol consumption, n (%) | ||||
| None | 3 (3.4%) | 7 (8.5%) | 7 (11.5%) | 0.22§ |
| ≤ 2 drinks per day | 11 (12.5%) | 46 (56.1%) | 27 (44.3%) | |
| > 2 drinks per day | 14 (15.9%) | 20 (24.4%) | 19 (31.2%) | |
| Missing | 60 (68.2%) | 9 (11.0%) | 8 (13.1%) | |
| HPV16 serology†, n (%) | ||||
| Negative | --- | 64 (78.0%) | 32 (52.5%) | 0.50§ |
| Positive | --- | 9 (11.0%) | 2 (3.3%) | |
| Missing | --- | 9 (11.0%) | 27 (44.3%) | |
| Stage at diagnosis, n (%) | ||||
| Local (stage I or II) | 15 (17.0%) | 48 (58.5%) | 29 (47.5%) | < 0.001§ |
| Advanced (III or IV) | 69 (78.4%) | 34 (41.5%) | 29 (47.5%) | |
| Missing | 4 (4.5%) | 0 (0.0%) | 3 (4.9%) | |
Ever-smokers only (i.e. former or current). Pack-year data is missing for 24 Discovery subjects, 8 Replication subjects, and 8 Validation subjects
Based on HPV16 E6 or E7 serology; no HPV serology was available for the TCGA samples (14 had HPV immunohistochemistry results available, all of which were negative)
Kruskal-Wallis equality-of-populations rank test
Fisher’s exact test for non-missing values
The three study sets also varied by follow-up time and observed events (deaths) with respect to overall survival. The OSCC patients from the TCGA Discovery dataset had a median survival time of 13.3 months (interquartile range: 7.0-30.5 months) with 42 deaths observed; the CoHANDS Replication set had a median survival time of 42.2 months (interquartile range: 31.9-54.7 months) with 23 deaths observed; and the CoHANDS Validation set had 76.1 months (interquartile range: 56.8-97.5 months) with 21 deaths observed.
Discovery of putative survival-associated CpGs in OSCC tumor tissue
The first step in the three-stage process was to screen for putative survival-associated CpG loci using the Infinium HumanMethylation450 BeadArray tumor data for OSCC patients in the publicly available TCGA database (Discovery). A total of 88 OSCC samples with methylation array data from tumor tissue were available through TCGA. After exclusion of 89,454 loci with probes containing one or more single-nucleotide polymorphism (SNP), 384,475 autosomal CpG loci remained, of which 31,038 were found to be significantly associated with overall survival (p ≤ 0.05), adjusted for age, gender, smoking and stage at diagnosis.
Replication of survival-associated CpGs in oral rinse samples
We next attempted to replicate the putative survival-associated CpG loci identified in tumor tissue in an independent set of oral rinse samples from OSCC patients enrolled in CoHANDS (Replication; n = 82) by fitting a series of multivariable Cox proportional hazards models across each of the 31,038 loci, adjusted for age, gender, smoking, HPV16 serology, and stage at diagnosis. This resulted in 3,716 survival-associated CpGs at a nominal p-value ≤ 0.05 (Supplementary Figure S1). After adjusting for false-discovery rate, seven of those loci remained significantly associated with survival in the oral rinse samples (Q ≤ 0.10). A description of each of those seven CpGs and associated genes (where applicable) is presented in Table 2.
Table 2.
Description of the seven significant survival-associated loci identified in oral rinse samples during the Replication stage and their associated genes if applicable.
| Locus ID | Chromosome | Located in a CpG Island | Located in an enhancer region | UCSC RefGene Group | Associated Gene | Associated Gene Descriptionb |
|---|---|---|---|---|---|---|
| cg02319972 | 15 | No | Yes | [intergenic] | ||
| cg03784083 | 11 | No | Yes | Body | OPCML | Opioid binding-cell adhesion molecule; highly conserved member of the IgLON subfamily of immunoglobulins; two transcript variants have been identified |
| cg15740054 | 19 | Yes | No | 5’UTR; TSS200 | ADCK4 | Uncharacterized AarF Domain Containing Kinase 4; unknown function, although it contains kinase domain; multiple transcript variants have been identified |
| cg18928362 | 14 | No | No | TSS1500 | ZFYVE26 | Encodes a protein which contains a FYVE zinc finger binding domain; involved in cytokinesis; may have involvement in homologous recombination DNA double-strand break repair |
| cg21022792 | 6 | No | No | Body | GABBR1 | Gamma-Aminobutyric Acid B Receptor 1; receptor for a major inhibitory neurotransmitter (GABA); multiple splice variants have been identified |
| cg21702497 | 16 | Yes | No | 5’UTR | POLR3E | RNA polymerase III; catalyzes the transcription of DNA into RNA; involved with innate immune response |
| cg25914931 | 10 | Yes | No | TSS200 | KIF11 | Kinase Family Member 11; involvement in spindle dynamics |
Abbreviations: 5’UTR = 5’-untranslated region; TSS200 = within 200 bases of the transcription start site; TSS1500 = within 1500 bases of the transcription start site
Validation of oral rinse markers by pyrosequencing
In an attempt to further validate the survival-associated CpG loci identified in oral rinse samples, we developed individual pyrosequencing assays designed to interrogate each of the seven CpGs described in Table 2 and applied them to oral rinse samples from a separate set of CoHANDS OSCC patients that were not used in the Infinium HumanMethylation450 BeadArray analysis (Validation; n = 61).
The distribution of methylation for the seven loci in oral rinse samples from the Replication (Infinium HumanMethylation450 BeadArray) and Validation (pyrosequencing) study sets is presented in Figure 2. It was anticipated that measured signals would be greatly attenuated in oral rinse samples since the samples contain a heterogeneous mixture of normal cells and field defects or residual tumor cells. While the methylation patterns for each of the loci are similar between study sets in a relative sense, it is important to note the lack of methylation levels for three of the seven loci in the pyrosequencing data with essentially no variability (cg18928362, cg21702497, and cg25914931); control DNA yielded expected levels of methylation for these assays. This lack of observed methylation in the validation set adversely impacts our ability to validate any survival association in these loci.
Figure 2.
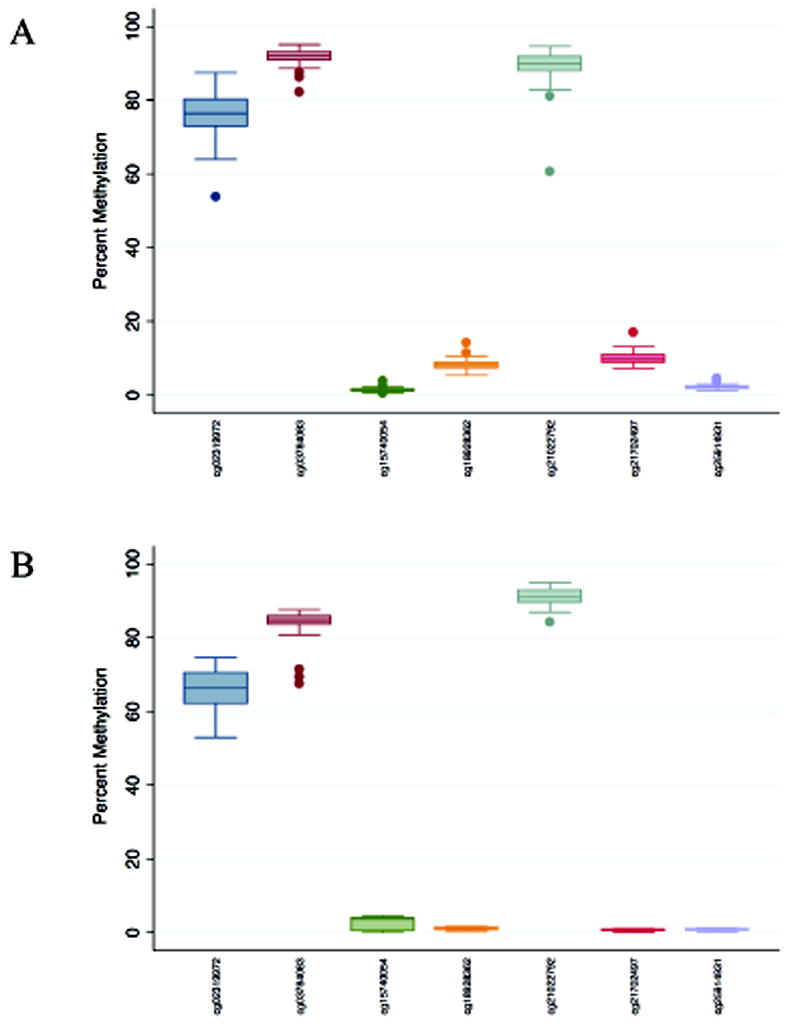
Distribution of percent methylation for each of seven survival-associated CpG loci (labeled on the x-axis by Infinium Array locus identification number) in oral rinse samples from oral squamous cell carcinoma (OSCC) patients for the (A) Replication set (n = 82) using the Infinium HumanMethylation450 BeadArray; and (B) Validation set (n = 61) using pyrosequencing assays.
A side-by-side contrast of the Kaplan-Meier 5-year survival functions (comparing methylation levels above and below the median) for the Replication and Validation study sets is presented in Figure 3. No univariate differences in survival were observed for any of the seven loci using the pyrosequencing (Validation) data, although it must be considered that division at the median is an arbitrary assignment. However, a marginal association was observed between each additional 1% of the average methylation measured by the cg21022792 pyrosequencing assay (in GABBR1) and overall survival (HR = 0.78, 95% CI: 0.61,1.01) after adjusting for age, gender, smoking pack-years, HPV16 serology, and stage at diagnosis in the Cox proportional hazards model (Table 3). The magnitude and direction of effect is comparable to what was found in the Infinium HumanMethylation450 BeadArray data in the Replication set (HR = 0.84, 95% CI: 0.77, 0.91).
Figure 3.
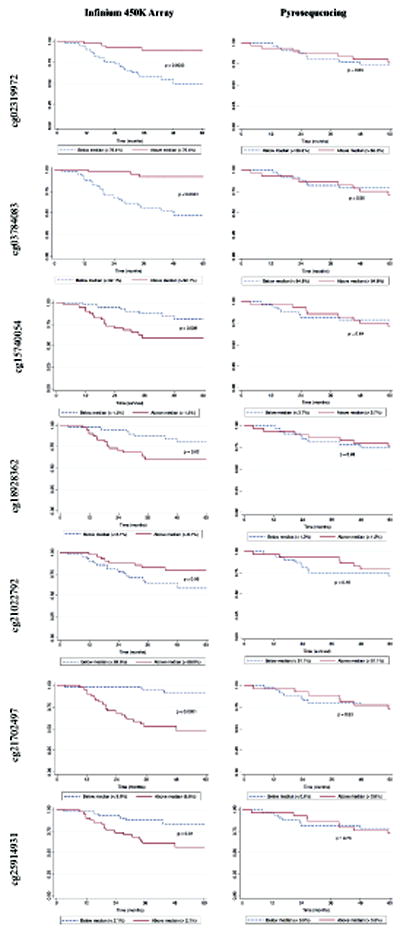
Kaplan-Meier survival functions for the seven significant CpG loci identified in the oral rinse array Replication dataset (n = 82) compared to those from the pyrosequencing assay Validation set (n = 61).
Table 3.
Comparison of survival estimates per 1% methylation of the 7 CpG loci identified from the oral rinse Infinium HumanMethylation450 BeadArray to the corresponding estimates from the pyrosequencing data set.
| Locus ID | Infinium HumanMethyaltion450 BeadArray
|
Pyrosequencing
|
||
|---|---|---|---|---|
| Crude HR (95% CI) | Adjusted HR* (95% CI) | Crude HR (95% CI) | Adjusted HR* (95% CI) | |
| cg02319972 | 0.87 (0.82, 0.92) | 0.85 (0.80, 0.91) | 1.00 (0.92, 1.09) | 1.01 (0.91, 1.11) |
|
| ||||
| cg03784083 | 0.74 (0.65, 0.85) | 0.70 (0.60, 0.82) | 0.95 (0.87, 1.04) | 0.96 (0.82, 1.12) |
|
| ||||
| cg15740054 | 3.65 (2.09, 6.32) | 6.31 (3.05, 13.05) | 1.00 (0.76, 1.33) | 0.91 (0.66, 1.24) |
|
| ||||
| cg18928362 | 1.72 (1.32, 2.25) | 1.60 (1.22, 2.12) | 0.84 (0.21, 3.40) | 0.36 (0.07, 1.77) |
|
| ||||
| cg21022792 | 0.85 (0.79, 0.92) | 0.84 (0.77, 0.91) | 0.94 (0.76, 1.15) | 0.78 (0.61, 1.01) |
|
| ||||
| cg21702497 | 1.48 (1.23, 1.79) | 1.54 (1.20, 1.98) | 1.00 (0.17, 5.79) | 0.79 (0.09, 7.21) |
|
| ||||
| cg25914931 | 3.24 (1.86, 5.66) | 4.45 (2.17, 9.15) | 1.03 (0.17, 6.23) | 0.53 (0.08, 3.51) |
Adjusted for age, gender, smoking pack-years, HPV16 and stage at diagnosis
DISCUSSION
We have identified 7 novel candidate prognostic DNA methylation markers in oral rinse samples from OSCC patients, one of which we were able to validate using a custom pyrosequencing assay in a discrete study set. Despite the marginal association in the validation study, given the rigor of our multistage study algorithm, including stringent control for false-discovery rate in the Replication stage, and the similar magnitude and direction of the survival effects, we accept this as strong evidence of the observed survival association. The validated locus is situated in the gene body of Gamma-Aminobutyric Acid B Receptor 1 (GABBR1), a gene encoding a receptor for a major inhibitory neurotransmitter for which multiple transcript variants have been identified [37]. We did not, however, observe a significant relationship with GABBR1 expression using the mRNA-sequencing data for 85 OSCC tumors available through TCGA (data not shown), although we cannot rule out a trans effect on the transcription of other genes. It is also of note that several studies have identified polymorphisms in GABBR1 that are associated with nasopharyngeal carcinoma [38-40].
The lack of validation of the remaining 6 loci identified in the Replication stage is not necessarily indicative of a null effect, particularly given the limited study power in the Validation stage and virtual absence of a methylation signal (and accordingly with no variability) in the respective CpGs associated with ZFYVE26, POLR3E, and KIF11. There are several factors that can influence this outcome, including differences in the study populations. The Validation study subjects tended to have better overall survival compared to the Replication subjects, despite deriving from the same study (CoHANDS). While this could be due to random chance, it is also conceivable that the lower DNA yield for many of the Validation subjects relates to the tumor characteristics and behavior. Further, as there is a somewhat higher frequency of non-White subjects in the Validation population, we cannot rule out with absolute certainty the possibility that our findings do not generalize to patients from other racial backgrounds, although we consider this to be an unlikely scenario.
Of the remaining six loci that did not validate, all were either located in a CpG island and/or promoter or enhancer region with two of the associated genes (OPCML and ADCK4) reported as having multiple transcript variants [41, 42]. KIF11 is a protein kinase with involvement in spindle dynamics [43] that was recently identified as essential for head and neck squamous cell carcinoma cell survival in a functional genetic screen [44]. The survival-associated locus that we identified is situated in a CpG island within 200 bases of the transcriptional start site of KIF11 and the methylation pattern observed in oral rinse would be consistent with aberrant promoter methylation. Additionally, OPCML has been widely reported to undergo down-regulation in ovarian adenocarcinomas [45] along with several other solid tumor types [41, 46-50]; ADCK4 has been reported to be associated with resistance to cisplatin [42] (a common front-line treatment for OSCC); and functional mutations [51] and overexpression [52] of ZFYVE26 has been reported for breast cancer cells.
A major strength of this study was the rigorous three-stage analytic strategy with stringent control for false-discovery rate in the Replication stage in which the seven loci were identified. Further, our application of an epigenome-wide array-based approach for discovery allowed us to interrogate a broad sampling of CpGs situated in varying genomic contexts without introducing bias that arises from a candidate approach, which is based on our limited understanding of cancer genomics/epigenomics. In other words, the array-based approach allows for identification of novel markers in an unbiased way. Additional strengths of this study include the prospective collection of survival data, our use of TCGA data to screen for putative survival-associated loci with biological relevance to the tumor, and availability of clinical and demographic data in all three data sets enabling us to account for potential confounding by clinical and personal/ behavioral characteristics in the statistical analyses.
However, there are also some limitations to this study. Our statistical power to detect an association with survival may have been limited by the modest size of our study sets. This could potentially lead to both false-negative results in the Discovery and Replication phases, resulting in oversight of some potentially clinically relevant CpGs, as well as false-negatives in the Validation stage, as previously discussed. The latter concern is exacerbated by the relatively low mortality and enhanced survival observed in the Validation cohort. Thus it must be stressed that lack of validation should not preclude future consideration of the six non-validated loci identified in the Replication stage, particularly given that these loci were observed to be significantly associated with survival in two other independent data sets (Discovery and Replication). Additionally, our survival data were passively collected and so we have no available data regarding the impact of these loci (or possibly others) on risk for recurrence or development of second primary tumors. The influence of epigenetics on tumor recurrence and origination of second primaries should be the target of future research in an effort to address the mechanism behind the observed association with survival.
We have identified and validated a novel survival-associated DNA methylation marker in DNA collected at diagnosis by oral rinse from OSCC patients, located in the body of GABBR1 (cg21022792). We have additionally identified six other CpG loci that were not validated in our limited study set but should be considered in future studies using better-powered prospective cohorts to assess clinical impact. Continued efforts to identify and develop innovative clinical tools that enhance prediction of oral cancer outcomes will help to drive forward patient care and continue to advance our comprehension of the underlying biology of this devastating disease.
Supplementary Material
We sought to identify survival-associated methylation loci in oral cancer patients
This research entails a comprehensive 3-stage epigenomic approach
Used non-invasive oral rinse samples
We have identified 7 novel DNA methylation loci
One of the 7 loci was validated using a custom pyrosequencing assay
Acknowledgments
Funding Sources
This work was supported by the National Cancer Institute (K22CA172358 to S.M.L. and R01CA100679 to K.T.K.). The funding sources were not involved in the collection, analysis and interpretation of data; in the writing of the report; or in the decision to submit the article for publication.
Footnotes
DISCLOSURE STATEMENT
We have no conflicts to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Carvalho AL, Nishimoto IN, Califano JA, Kowalski LP. Trends in incidence and prognosis for head and neck cancer in the United States: a site-specific analysis of the SEER database. Int J Cancer. 2005;114:806–16. doi: 10.1002/ijc.20740. [DOI] [PubMed] [Google Scholar]
- 3.Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet. 2008;371:1695–709. doi: 10.1016/S0140-6736(08)60728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altekruse SF, Kosary CL, Krapcho M, Neyman N, Aminou R, Waldron W, et al. SEER Cancer Statistics Review, 1975-2007. Bethesda, MD: National Cancer Institute; 2010. [Google Scholar]
- 5.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–8. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 6.Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727–30. [PubMed] [Google Scholar]
- 7.Lippman SM, Hong WK. Second malignant tumors in head and neck squamous cell carcinoma: the overshadowing threat for patients with early-stage disease. Int J Radiat Oncol Biol Phys. 1989;17:691–4. doi: 10.1016/0360-3016(89)90126-0. [DOI] [PubMed] [Google Scholar]
- 8.Tabor MP, Brakenhoff RH, Ruijter-Schippers HJ, Van Der Wal JE, Snow GB, Leemans CR, et al. Multiple head and neck tumors frequently originate from a single preneoplastic lesion. Am J Pathol. 2002;161:1051–60. doi: 10.1016/S0002-9440(10)64266-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carvalho AL, Jeronimo C, Kim MM, Henrique R, Zhang Z, Hoque MO, et al. Evaluation of promoter hypermethylation detection in body fluids as a screening/diagnosis tool for head and neck squamous cell carcinoma. Clin Cancer Res. 2008;14:97–107. doi: 10.1158/1078-0432.CCR-07-0722. [DOI] [PubMed] [Google Scholar]
- 10.Demokan S, Chang X, Chuang A, Mydlarz WK, Kaur J, Huang P, et al. KIF1A and EDNRB are differentially methylated in primary HNSCC and salivary rinses. International journal of cancer Journal international du cancer. 2010;127:2351–9. doi: 10.1002/ijc.25248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guerrero-Preston R, Soudry E, Acero J, Orera M, Moreno-Lopez L, Macia-Colon G, et al. NID2 and HOXA9 promoter hypermethylation as biomarkers for prevention and early detection in oral cavity squamous cell carcinoma tissues and saliva. Cancer Prev Res (Phila) 2011;4:1061–72. doi: 10.1158/1940-6207.CAPR-11-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kusumoto T, Hamada T, Yamada N, Nagata S, Kanmura Y, Houjou I, et al. Comprehensive epigenetic analysis using oral rinse samples: a pilot study. J Oral Maxillofac Surg. 2012;70:1486–94. doi: 10.1016/j.joms.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 13.Langevin SM, Stone RA, Bunker CH, Grandis JR, Sobol RW, Taioli E. MicroRNA-137 promoter methylation in oral rinses from patients with squamous cell carcinoma of the head and neck is associated with gender and body mass index. Carcinogenesis. 2010;31:864–70. doi: 10.1093/carcin/bgq051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Zhou ZT, He QB, Jiang WW. DAPK promoter hypermethylation in tissues and body fluids of oral precancer patients. Med Oncol. 2012;29:729–33. doi: 10.1007/s12032-011-9953-5. [DOI] [PubMed] [Google Scholar]
- 15.Lopez M, Aguirre JM, Cuevas N, Anzola M, Videgain J, Aguirregaviria J, et al. Gene promoter hypermethylation in oral rinses of leukoplakia patients--a diagnostic and/or prognostic tool? European journal of cancer. 2003;39:2306–9. doi: 10.1016/s0959-8049(03)00550-1. [DOI] [PubMed] [Google Scholar]
- 16.Nagata S, Hamada T, Yamada N, Yokoyama S, Kitamoto S, Kanmura Y, et al. Aberrant DNA methylation of tumor-related genes in oral rinse: a noninvasive method for detection of oral squamous cell carcinoma. Cancer. 2012;118:4298–308. doi: 10.1002/cncr.27417. [DOI] [PubMed] [Google Scholar]
- 17.Pattani KM, Zhang Z, Demokan S, Glazer C, Loyo M, Goodman S, et al. Endothelin receptor type B gene promoter hypermethylation in salivary rinses is independently associated with risk of oral cavity cancer and premalignancy. Cancer Prev Res (Phila) 2010;3:1093–103. doi: 10.1158/1940-6207.CAPR-10-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Righini CA, de Fraipont F, Timsit JF, Faure C, Brambilla E, Reyt E, et al. Tumor-specific methylation in saliva: a promising biomarker for early detection of head and neck cancer recurrence. Clin Cancer Res. 2007;13:1179–85. doi: 10.1158/1078-0432.CCR-06-2027. [DOI] [PubMed] [Google Scholar]
- 19.Schussel J, Zhou XC, Zhang Z, Pattani K, Bermudez F, Jean-Charles G, et al. EDNRB and DCC salivary rinse hypermethylation has a similar performance as expert clinical examination in discrimination of oral cancer/dysplasia versus benign lesions. Clin Cancer Res. 2013;19:3268–75. doi: 10.1158/1078-0432.CCR-12-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun W, Zaboli D, Wang H, Liu Y, Arnaoutakis D, Khan T, et al. Detection of TIMP3 promoter hypermethylation in salivary rinse as an independent predictor of local recurrence-free survival in head and neck cancer. Clin Cancer Res. 2012 doi: 10.1158/1078-0432.CCR-11-2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carvalho AL, Henrique R, Jeronimo C, Nayak CS, Reddy AN, Hoque MO, et al. Detection of promoter hypermethylation in salivary rinses as a biomarker for head and neck squamous cell carcinoma surveillance. Clin Cancer Res. 2011;17:4782–9. doi: 10.1158/1078-0432.CCR-11-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rettori MM, de Carvalho AC, Bomfim Longo AL, de Oliveira CZ, Kowalski LP, Carvalho AL, et al. Prognostic significance of TIMP3 hypermethylation in post-treatment salivary rinse from head and neck squamous cell carcinoma patients. Carcinogenesis. 2013;34:20–7. doi: 10.1093/carcin/bgs311. [DOI] [PubMed] [Google Scholar]
- 23.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–3. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harmston N, Lenhard B. Chromatin and epigenetic features of long-range gene regulation. Nucleic acids research. 2013;41:7185–99. doi: 10.1093/nar/gkt499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slieker RC, Bos SD, Goeman JJ, Bovee JV, Talens RP, van der Breggen R, et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 2013;6:26. doi: 10.1186/1756-8935-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 28.Applebaum KM, McClean MD, Nelson HH, Marsit CJ, Christensen BC, Kelsey KT. Smoking modifies the relationship between XRCC1 haplotypes and HPV16-negative head and neck squamous cell carcinoma. International journal of cancer Journal international du cancer. 2009;124:2690–6. doi: 10.1002/ijc.24256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang C, McClean MD, Marsit C, Christensen B, Peters E, Nelson HH, et al. A population-based case-control study of marijuana use and head and neck squamous cell carcinoma. Cancer Prev Res (Phila) 2009;2:759–68. doi: 10.1158/1940-6207.CAPR-09-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Houseman E, Coull B, Ryan L. A functional-based distribution diagnostic for a linear model with correlated outcomes. Biometrika. 2006;93:911–26. [Google Scholar]
- 31.Liang C, Marsit CJ, McClean MD, Nelson HH, Christensen BC, Haddad RI, et al. Biomarkers of HPV in head and neck squamous cell carcinoma. Cancer Res. 2012;72:5004–13. doi: 10.1158/0008-5472.CAN-11-3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waterboer T, Sehr P, Michael KM, Franceschi S, Nieland JD, Joos TO, et al. Multiplex human papillomavirus serology based on in situ-purified glutathione s-transferase fusion proteins. Clin Chem. 2005;51:1845–53. doi: 10.1373/clinchem.2005.052381. [DOI] [PubMed] [Google Scholar]
- 33.Michaud DS, Langevin SM, Eliot M, Nelson HH, Pawlita M, McClean MD, et al. High-risk HPV types and head and neck cancer. International journal of cancer Journal international du cancer. 2014 doi: 10.1002/ijc.28811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995;29:1165–88. [Google Scholar]
- 35.White IR, Royston P, Wood AM. Multiple imputation using chained equations: Issues and guidance for practice. Stat Med. 2011;30:377–99. doi: 10.1002/sim.4067. [DOI] [PubMed] [Google Scholar]
- 36.Grambsch PM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika. 1994;81:515–26. [Google Scholar]
- 37.Martin SC, Russek SJ, Farb DH. Human GABA(B)R genomic structure: evidence for splice variants in GABA(B)R1 but not GABA(B)R2. Gene. 2001;278:63–79. doi: 10.1016/s0378-1119(01)00678-3. [DOI] [PubMed] [Google Scholar]
- 38.Hsu WL, Tse KP, Liang S, Chien YC, Su WH, Yu KJ, et al. Evaluation of human leukocyte antigen-A (HLA-A), other non-HLA markers on chromosome 6p21 and risk of nasopharyngeal carcinoma. PloS one. 2012;7:e42767. doi: 10.1371/journal.pone.0042767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Fu L, Wong AM, Fan YH, Li MX, Bei JX, et al. Identification of genes with allelic imbalance on 6p associated with nasopharyngeal carcinoma in southern Chinese. PloS one. 2011;6:e14562. doi: 10.1371/journal.pone.0014562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tse KP, Su WH, Chang KP, Tsang NM, Yu CJ, Tang P, et al. Genome-wide association study reveals multiple nasopharyngeal carcinoma-associated loci within the HLA region at chromosome 6p21.3. Am J Hum Genet. 2009;85:194–203. doi: 10.1016/j.ajhg.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reed JE, Dunn JR, du Plessis DG, Shaw EJ, Reeves P, Gee AL, et al. Expression of cellular adhesion molecule ‘OPCML’ is down-regulated in gliomas and other brain tumours. Neuropathology and applied neurobiology. 2007;33:77–85. doi: 10.1111/j.1365-2990.2006.00786.x. [DOI] [PubMed] [Google Scholar]
- 42.Zhou J, Wei YH, Liao MY, Xiong Y, Li JL, Cai HB. Identification of Cisplatin-Resistance Associated Genes through Proteomic Analysis of Human Ovarian Cancer Cells and a Cisplatin-resistant Subline. Asian Pac J Cancer Prev. 2012;13:6435–9. doi: 10.7314/apjcp.2012.13.12.6435. [DOI] [PubMed] [Google Scholar]
- 43.Rath O, Kozielski F. Kinesins and cancer. Nat Rev Cancer. 2012;12:527–39. doi: 10.1038/nrc3310. [DOI] [PubMed] [Google Scholar]
- 44.Martens-de Kemp SR, Nagel R, Stigter-van Walsum M, van der Meulen IH, van Beusechem VW, Braakhuis BJ, et al. Functional genetic screens identify genes essential for tumor cell survival in head and neck and lung cancer. Clin Cancer Res. 2013;19:1994–2003. doi: 10.1158/1078-0432.CCR-12-2539. [DOI] [PubMed] [Google Scholar]
- 45.Wu SY, Sood AK. New roles opined for OPCML. Cancer Discov. 2012;2:115–6. doi: 10.1158/2159-8290.CD-11-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Selamat SA, Galler JS, Joshi AD, Fyfe MN, Campan M, Siegmund KD, et al. DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PloS one. 2011;6:e21443. doi: 10.1371/journal.pone.0021443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sriraksa R, Zeller C, El-Bahrawy MA, Dai W, Daduang J, Jearanaikoon P, et al. CpG-island methylation study of liver fluke-related cholangiocarcinoma. British journal of cancer. 2011;104:1313–8. doi: 10.1038/bjc.2011.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duarte-Pereira S, Paiva F, Costa VL, Ramalho-Carvalho J, Savva-Bordalo J, Rodrigues A, et al. Prognostic value of opioid binding protein/cell adhesion molecule-like promoter methylation in bladder carcinoma. European journal of cancer. 2011;47:1106–14. doi: 10.1016/j.ejca.2010.12.025. [DOI] [PubMed] [Google Scholar]
- 49.Cui Y, Ying Y, van Hasselt A, Ng KM, Yu J, Zhang Q, et al. OPCML is a broad tumor suppressor for multiple carcinomas and lymphomas with frequently epigenetic inactivation. PloS one. 2008;3:e2990. doi: 10.1371/journal.pone.0002990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ye F, Zhang SF, Xie X, Lu WG. OPCML gene promoter methylation and gene expression in tumor and stroma cells of invasive cervical carcinoma. Cancer investigation. 2008;26:569–74. doi: 10.1080/07357900701837044. [DOI] [PubMed] [Google Scholar]
- 51.Sagona AP, Nezis IP, Bache KG, Haglund K, Bakken AC, Skotheim RI, et al. A tumor-associated mutation of FYVE-CENT prevents its interaction with Beclin 1 and interferes with cytokinesis. PloS one. 2011;6:e17086. doi: 10.1371/journal.pone.0017086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guillaud-Bataille M, Brison O, Danglot G, Lavialle C, Raynal B, Lazar V, et al. Two populations of double minute chromosomes harbor distinct amplicons, the MYC locus at 8q24.2 and a 0.43-Mb region at 14q24.1, in the SW613-S human carcinoma cell line. Cytogenet Genome Res. 2009;124:1–11. doi: 10.1159/000200082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.