

Abstract
Allosteric proteins have great potential in synthetic biology, but our limited understanding of the molecular underpinnings of allostery has hindered the development of designer molecules, including transcription factors with new DNA-binding or ligand-binding specificities that respond appropriately to inducers. Such allosteric proteins could function as novel switches in complex circuits, metabolite sensors, or orthogonal regulators for independent, inducible control of multiple genes. Advances in DNA synthesis and next-generation sequencing technologies have enabled the assessment of millions of mutants in a single experiment, providing new opportunities to study allostery. Using the classic LacI protein as an example, we describe a genetic selection system using a bidirectional reporter to capture mutants in both allosteric states, allowing the positions most critical for allostery to be identified. This approach is not limited to bacterial transcription factors, and could reveal new mechanistic insights and facilitate engineering of other major classes of allosteric proteins such as nuclear receptors, two-component systems, G-protein coupled receptors and protein kinases.
Unlocking the power of allostery in synthetic biology
Allosteric regulation mediates virtually every biological process, including transcription, signal transduction, enzyme activity and transport. Allostery can be broadly defined as activity at one site in a protein regulating function at a spatially distant site. Allosteric regulation occurs through an allosteric effector, generally a small molecule, which binds at one active site and triggers a conformational change that affects function at the distant site. Because of their ability to respond to small molecules by a change of state, allosteric proteins play an important role in synthetic biology. But our ability to engineer allosteric proteins is highly constrained by our limited understanding of the molecular details of allostery., and thus, we have barely scratched the surface of how allosteric proteins can be applied in this emerging field.
Allosteric proteins are used as switches in synthetic circuits. Although synthetic biologists would like to build more complex circuits, a major limitation is the lack of orthogonal switches (allosteric proteins that bind to different inducers and different DNA sequences with little crosstalk). A suite of well-characterized orthogonal switches would vastly enhance our ability to build higher-order synthetic circuits with real-world applicability [1]. For example, such switches could serve as analog-to-digital converters that convert a continuous chemical gradient into a digital output. Bacteria possessing synthetic circuits combining many such analog-to-digital converters could then be used as whole-cell biosensors of the gut [1].
Allosteric proteins can also be used as in vivo metabolite sensors for engineering biosynthetic pathways [2]. These sensors detect and respond to the level of some sought-after metabolite, enabling genetic selections in which the best producers are identified from a large number of variant organisms. Despite an increasing demand for allosteric sensors detecting industrially useful chemicals, we are limited to the ligand-binding domains of known transcription factors; however, this bottleneck could be removed by designing new allosteric proteins. For instance, new small molecule sensors could be generated from chimeras of a well-characterized DNA-binding domain with ligand-binding domains identified in the sequences of metagenomic samples. Alternatively, we might be able to mutate binding site residues in an existing sensor to create new ligand specificities without affecting allosteric communication [3].
Besides their biotechnological applications, designer allosteric proteins can provide independent, temporal regulation of multiple genes, a useful tool for developmental biology. The Tet-On/Off activator system, based on the E. coli Tet repressor, is widely used for mammalian gene regulation, but it does not allow the independent control of multiple genes. With multiple orthogonal regulators similar to the Tet-On/Off elements, we could, for instance, gain exquisite control over stem cell differentiation pathways by modulating each differentiation factor independently.
Finally, redesigning allosteric proteins to respond to molecules that cross the blood-brain barrier would enable the activation of specific neural circuits in the brains of live animals simply by incorporating the inducers in the diet. In order to engineer allosteric proteins, however, we need to take a closer look at how allostery works at the molecular level.
Efforts to understand allostery have largely focused on biophysical models to explain the conformational transition between two states, corresponding to the presence and absence of the effector [4]. Protein dynamics shows that allosteric transitions occur as a consequence of local conformational changes such as disorder or unfolding that are propagated to distal regions, shifting the overall conformational equilibrium [1, 5–7]. Whereas these models focus on the thermodynamic drivers of allostery, little is understood about the underlying molecular basis. Alternatively, a genetics approach to understanding allostery could be taken by making large numbers of single mutations and combinations of double mutations in an allosteric protein and then determining which mutant proteins maintain allosteric coupling. This approach, in conjunction with biophysical measurements derived from NMR or molecular dynamics simulations, may tell us how nature designs allosteric control, and might let us develop rules for engineering allosteric proteins to use in synthetic biology applications. The grand goal would be to engineer de novo an allosteric transition in a protein that is not normally allosteric. For instance, could we convert an immunoglobulin domain to actuate an allosteric response when it binds to an antigen? While this may appear a daunting challenge, a near-term achievable goal might combine domains from two different allosteric proteins to result in a functional chimera.
Our ability to engineer proteins to bind DNA, small molecules or other proteins draws from a wealth of research studying these interactions at molecular resolution. However, allostery has proven to be recalcitrant to engineering because we do not fully understand the molecular connectivity involved in allosteric communication. Previous studies indicate that a network of structurally contiguous residues act in concert to transmit the allosteric signal [1, 8–11]. Here, we review a classic allosteric regulator and describe how new technologies – based on protein-wide mutational perturbation – could build a molecular “wiring diagram” of allostery by deconstructing the role of each amino acid in transmission of the allosteric signal. Knowledge of the wiring diagram should allow us to preserve allosteric connections as we design new functions into an allosteric protein. We conclude with how similar strategies might be applied to other broad classes of allosteric proteins.
LESSONS FROM LACI
Allosteric transcription factors in bacteria, one of the largest annotated families of proteins, regulate adaptive responses to environmental cues. The best- and longest-studied allosteric protein is the E. coli lac repressor, LacI, which regulates the lactose operon [2, 12]. LacI is composed of ligand-binding and DNA-binding domains. In the absence of the ligand, LacI has high affinity for DNA; when bound to the inducer, the protein undergoes a conformational change that causes the DNA-binding domain to lose affinity for DNA, thereby coming off the operator site and unblocking the path of RNA polymerase to transcribe downstream genes.
The structure of LacI [3, 12, 13] can be divided into three sections: the N-terminal 60 residues form a helix-turn-helix motif that binds DNA; the core of the protein (residues 61–330) is made up of N- and C-terminal subdomains where the ligand binds; and the C-terminal 30 residues are involved in tetramerization (Fig. 1). LacI is a functional dimer that makes extensive monomer-monomer contacts across the N- and C-terminal ligand-binding subdomains [4, 12, 13]. The ligand binds in the cleft between the core subdomains, inducing a Venus flytrap-like allosteric motion. Upon induction, the allosteric signal is communicated by the relative motion between the N- and C-terminal ligand-binding subdomains, causing the DNA-binding domain to undergo a helix-to-coil structural transition, and hence lose its DNA-binding activity.
Figure 1. Engineering novel allosteric proteins.

(A) Designing novel ligand specificities: a known allosteric protein can be made to recognize different small molecule inducers by mutating the amino acids in the ligand binding domain. (B) Designing novel DNA specificities: allosteric transcription factors can be targeted to new DNA sequences by altering the DNA-binding domain. (C) Novel allosteric chimeras: allosteric protein domains that are not capable of binding DNA, such as periplasmic binding proteins, can be attached to a DNA-binding domain to create novel, chimeric transcription factors.
The molecular basis of allosteric communication in LacI has been the focus of extensive genetic and biochemical studies. Jeffrey Miller’s laboratory made 4000 point mutants of LacI [14–16] and used a sensitive genetic screen to classify each mutant into one of three phenotypes: wild type-like activity (WT); unable to bind to DNA (I−); or unable to bind to inducer or broken allostery (Is). These studies showed how structural integrity, dimerization, allosteric signal transduction, ligand-binding and DNA-binding are tightly interlinked and act in concert for protein function.
The I− phenotype was generally caused by mutations in the DNA-binding domain or in the C-terminal ligand-binding subdomain, which acts as a scaffold for the dimerization required for DNA-binding. Long-range allosteric communication was evident when over 20 second site mutations scattered across the structure reverted the I− phenotype of Y282D, a change that disrupted dimerization [17, 18] (Fig. 1). These second site mutations were clustered in the core-pivot region.
The Is phenotype resulted from mutations that affected the extensive interactions between the DNA-binding domain and the N-terminal ligand-binding subdomain. Mutation at a single position, A110, in the N-terminal ligand-binding subdomain dimerization interface caused opposite phenotypes depending on the substitution: higher inducer affinity and lower DNA affinity, or lower inducer affinity and higher DNA affinity, highlighting the key role of the interface in allostery [19].
Co-evolution is a molecular signature of functionally coupled residues undergoing coordinated sequence changes across multiple members of a protein family. In order to preserve protein function, co-evolving pairs undergo mutually compensatory changes during evolution, which provides insight into residue connectivity [20]. The LacI family of bacterial transcription factors share the same protein architecture yet have a sequence similarity less than 30% [21]. Although residues in the ligand-binding pocket and at the DNA-binding interface vary depending on inducer and operator sequence, residues involved in allostery have lower sequence entropy. Indeed, most co-evolving residues involved in allostery within a subfamily were found in spatially disconnected regions, showing that complex epistatic networks participate in allostery [22, 23]. In LacI, these positions are scattered across the N- and C-terminal dimerization interfaces, core-pivot and hinge regions, and DNA-binding domain, consistent with genetic and biochemical studies of the mutants [21].
Within a close subfamily, structural residues in the ligand-binding pocket are more conserved and ligand specificity is achieved through mutations at less conserved residues. Comparison across more distant families shows that approximately 19% of the residues are conserved, suggesting that these positions may be indispensable or “hardwired” for structural integrity and function for that protein fold family [21]. As the scaffold evolved to acquire specific functions, it may have adapted to a particular niche through minor alterations in mechanism. For instance, although LacI and PurR share highly similar structures and sequences, their allosteric wiring is opposite: upon ligand binding, LacI dislodges from DNA but PurR binds to DNA.
DEEP MUTATIONAL SCANNING USING A TOGGLED SELECTION SYSTEM FOR ALLOSTERY
Biochemical and evolutionary studies of LacI suggest that amino acids involved in allostery are intricately coupled with amino acids recognizing ligand and DNA. Thus, a first step toward unraveling this allosteric network would be to determine the role of all the amino acids in LacI function, classifying each as participating in one of the following categories: binds to DNA or to ligand, required for structural stability, involved in allostery or none of the above. As with conventional protein biochemistry, the functional importance of each amino acid can be determined by mutational analysis. Because allostery is a systemic effect, however, we need to scale up mutagenesis by orders of magnitude in order to interrogate the effect of every single mutation and every combination of double mutations. The double mutations are particularly important because they can reveal long-range allosteric interactions, as seen with the Y282D mutation in LacI. Though allostery most likely depends on a higher-order network interactions beyond pair-wise couplings, sufficient pairs of long-range double mutants should identify linchpin positions in this network.
How can we obtain the phenotype associated with every single and double mutation in an allosteric protein? An approach called “deep mutational scanning” [24] can generate such a comprehensive mutation map, which shows the effect of all neutral and loss-of-function, and any gain-of-function or hyper-activating changes, as well as double mutations that restore function lost in a single mutant or that exacerbate the combined effect of single mutations more than predicted. Deep mutational scanning has succesfully been used to analyze antibody affinity maturation [25, 26], protein-peptide interactions [27, 28], protein-small molecule affinity [29], ubiquitination [30], protein stability [31], splicing [32] and antibiotic resistance [33] among other protein activities.
Deep mutational scanning leverages two major advances: first, the generation of targeted DNA libraries with over a million unique sequences through doped oligonucleotide assembly or pre-specified microarray-generated oligonucleotides [34]; and second, enormous read numbers from next-generation sequencing that allow thousands of clones to be assayed simultaneously by linking activity to read frequency of each unique sequence [31]. Genotype and phenotype are linked through an in vitro or in vivo selection that amplifies mutants with the desired function. Because the total sequence read count is orders of magnitude greater (e.g. 400 million reads with the Illumina HiSeq) than the number of selected genotypes, the activity of a protein variant, to a first approximation, is reflected in the relative sequence abundance of its encoding DNA. Enthalpies derived from sequencing statistics has been shown to linearly correlate with experimental measurements and computational structure-based ΔΔG predictions [26, 35]. For LacI, the library of all single mutations is just over 6000, and even the library of all double mutations, at nearly 2.5 × 107, is well within the transformation efficiency (109) of commercially competent cells.
Once the mutants are constructed, they need to be assayed in a suitable selection system. Selection systems typically enrich for only one function; for instance, with protein-peptide interactions, phage display enriches for stronger binders. However, a selection system for allosteric proteins should enrich for either of two states: the induced or the uninduced. Such selections can be accomplished with a dual selectable marker as the reporter gene. For example, the E. coli tolC gene [36, 37] or the Saccharomyces cerevisiae URA3 gene allows enrichment of cells expressing (positive selection) or not expressing (negative selection) the reporter using different selection agents. Dual selection can thus identify mutants with the three phenotypes found in Miller’s study: WT, I− and Is.
First, subjecting the library to positive selection in the absence of the inducer reveals mutants that are not capable of binding DNA (I− phenotype), because they constitutively express the reporter and are enriched (Fig. 2, left arrow). Second, subjecting the library to negative selection in the absence of the inducer identifies mutants that bind to DNA; these repress the reporter and are insensitive to negative selection (Fig. 2, center arrow). Subjecting this DNA-bound population to positive selection in the presence of the inducer enriches for mutants with WT phenotype, because they respond to the inducer to activate the reporter (Fig. 2, center arrow). Finally, exposing the DNA-bound population to negative selection in the presence of the inducer enriches for mutants that do not respond to the inducer (Is phenotype), while the ones exhibiting WT-like activation perish (Fig. 2, right arrow).
Figure 2. LacI protein architecture and long-range allosteric connections.

Structure of a LacI dimer bound to DNA is shown on the left side, key regions highlighted in different colors. The dotted lines on the right side show long-range connection between four mutations (M42, A133, D149 and S151) that rescue allostery in Y282D.
The next step is to identify specific positions from the I− and Is set that play a role in allostery. To delineate this subset, we need to reclassify I− and Is into subgroups. The I− set comprises mutants that do not recognize DNA, structurally unstable mutants and mutants that have broken allostery. The Is set comprises mutants that do not recognize the inducer and those that fail to transmit the allosteric signal despite inducer binding. To narrow down the allosteric subset from the I- and Is sets, we can carry out computational structure-based ΔΔG calculations of folding, ligand-binding and DNA-binding of each mutant. Because the allosteric subset is deduced by the process of elimination, we need to prune this set further to isolate a minimal subset of positions that maximally contribute to allostery. NMR chemical shift perturbation measurement detects the local change in chemical environment for each amino acid upon conformational change. This approach has been used to identify the allosteric connectivity in protein kinase A [11]. Because amino acids in the vicinity of the allosteric positions are likely to undergo conformational change, chemical shift perturbations alone cannot identify the allosteric network. However, via the intersection of mutational data from genetic screens, structure-based enthalpy calculations and biophysical measurements of local conformational change by NMR, we can pinpoint the minimal set of amino acids that constitute the allosteric network. Molecular simulations can independently validate the allosteric network by computing the conformational change of individual amino acids across the two allosteric states [8, 38].
Thus, in one multiplexed assay, millions of LacI variants can be functionally characterized in a manner that is facile and might be extensible to other proteins. Extending this selection system for other members of the LacI family might be accomplished by swapping the promoter regulating the reporter with a promoter that binds a different allosteric protein.
ENGINEERING ALLOSTERIC PROTEINS
New ligand and DNA specificities engineered into existing proteins
To engineer an orthogonal transcription factor, we can begin by redesigning the ligand- and DNA-binding specificities of natural transcription factors. Once the allosteric connections have been identified, and residues responsible for allostery have been distinguished from those involved in binding ligand or DNA, we can incorporate this information into the design protocol. For example, in the case of LacI, mutating residues that cause an Is phenotype is more likely to result in altered specificity because a subset of Is mutants retain allostery despite losing IPTG binding. On the other hand, to change the DNA specificity, we target I− mutants in order to restore allostery and to achieve new specificity because both are intimately linked in the DNA-binding domain of LacI.
Despite different ligand specificities, the overall binding pocket architecture of LacI-like proteins is similar. This similarity suggests that an altered specificity for a new ligand can be achieved while still preserving allostery. The binding pocket of each allosteric protein family can, in principle, be redesigned to access chemical diversity around its cognate ligand. For instance, the LacI family of proteins may be able to accommodate many derivatives of sugars. With respect to DNA recognition, the helix-turn-helix domain is commonly used across several bacterial transcription factor families. Thus, by analyzing the binding site preferences across helix-turn-helix family members, we might gain the knowledge to engineer new DNA specificity without compromising allostery.
Chimeric allosteric proteins
An alternative to redesigning existing proteins is to engineer new allosteric proteins by mixing and matching protein domains. The LacI family is thought to have evolutionarily diverged from the structurally similar periplasmic-binding protein family by acquiring a DNA-binding domain [39]. Periplasmic-binding proteins respond to a large repertoire of small molecules by regulating ABC transporters. A chimera of a periplasmic-binding protein and a DNA-binding domain could be engineered such that an allosteric change in the periplasmic-binding protein is communicated to the DNA-binding domain to change DNA affinity. Another DNA-binding domain family that might be amenable to engineering is the zinc finger protein class, which is comparable in size to the helix-turn-helix domain class. Because the specificity of these proteins can be programmed, we can build completely orthogonal switches. Yet another approach is to engineer a chimeric protein that upon ligand binding exposes a binding surface that recruits a sigma factor, resulting in the activation of transcription. Engineering functional chimeras will involve extensive optimization of the linker between the domains, the inter-domain interface and the dimerization interface.
Although thousands of bacterial allosteric transcription factors have been annotated in the sequences of metagenomic samples, their ligand and DNA specificities remain unknown. However, a clue to the type of ligand recognized might come from the characterization of the nearby operon that is regulated by a transcription factor. This set of transcription factors is a treasure trove for the design of biosensors for many industrially valuable molecules, allowing their biosynthetic pathways to be optimized for higher production. If the rules of allostery are known, we can build a functional chimera with a known DNA-binding domain and identify its inducer using a reporter-gene assay against a panel of ligands.
In general, knowledge of allosteric connections can help favorably bias the search for new function by avoiding changes that break or weaken allostery. Computational and directed evolution design protocols could incorporate deep mutational scanning data when choosing residues that can be mutated.
APPLICATION TO THE WIDER WORLD OF ALLOSTERY
As DNA synthesis and sequencing become increasingly cheaper and higher in throughput, the rate-limiting step for the analysis of other classes of allosteric proteins is the development of functional assays. The bacterial transcription factor family allows one of the easiest functional assays because allostery is directly coupled to transcription. In this section, we briefly describe functional assays for other major classes of allosteric proteins.
Direct transcriptional readout (nuclear receptors)
The toggled selection scheme for LacI can be adapted for other allosteric transcription factors, like nuclear receptors (Fig. 3A). Binding of the ligand to the nuclear receptor induces a reorientation of the ligand-binding domain, and in the case of steroid receptors, directly leads to transcriptional activation. Other nuclear receptors, such as the receptors for retinoic acid and vitamin D, may also be successfully interrogated with this strategy, though upon ligand binding they have a more complex set of binding interactions involving additional transcriptional activators and repressors [40, 41].
Figure 3. Toggled selection scheme for high-throughput functional evaluation of LacI mutants.

LacI mutants are shown in yellow; red circle represents a mutation. Positive selection in the absence of the inducer enriches for I- mutants (left arrow). Negative selection without inducer followed by positive selection with inducer enriches for WT-like mutants (center arrow). Negative selection with inducer enriches for Is mutants (right arrow).
Indirect transcriptional readout (two-component system)
Many allosteric proteins alter gene expression indirectly by regulating downstream transcription factors. A transcriptional readout may be employed to evaluate such allosteric proteins as long as target gene expression robustly reflects protein activity (Fig. 3B). An example of such pathways is the bacterial two-component system, containing a membrane-bound histidine kinase, serving as an environmental sensor, and a cytoplasmic response regulator, which is phosphorylated by the activated kinase and often acts directly as a transcription factor [42]. Systematic mutagenesis could identify the amino acids needed for the sensor histidine kinase to change conformation upon ligand activation [43], with a toggled selection marker at the locus bound by the response regulator as a readout of pathway activation.
Split reporter assay (G-protein coupled receptors and receptor tyrosine kinases)
For allosteric receptors far upstream of the transcription factors in their pathway, a different readout is needed. Membrane-bound receptors such as GPCRs and RTKs, upon allosteric activation, co-localize with a partner: GPCRs associate with beta-arrestin [44] and EGFR family RTKs can form heterodimers [45]. Activity of these proteins can be measured with a split reporter assay in which the receptor is fused to one domain of a reporter protein and the recruited partner is fused to the other domain; when brought into close proximity the two domains can combine into a functional whole (Fig. 3C) [46–49]. This assay can be performed using a divided GFP and FACS to sort cells based on fluorescence intensity.
Environment-sensitive fluorophores (protein kinases)
Allosteric protein kinases can be difficult to study due to the transitory nature of their interactions with their substrates. Kinases that do not directly regulate transcription factors may be interrogated using fluorescent sensors dependent on target phosphorylation. For tyrosine kinases whose phosphorylated targets are recognized by SH2 domains, the kinase target can be fused to SH2, along with two FRET-compatible fluorophores at either end of the construct, such that SH2 binding to the phosphotyrosine brings the fluorophores together. Alternatively, as a more general tyrosine/serine phosphorylation sensor, a peptide substrate of the kinase is attached to a fluorophore with a separation of only several angstroms; when the peptide is phosphorylated, the change in the microenvironment activates the fluorophore (Fig 3D) [50–52]. FACS can then be used to sort cells based on fluorescence intensity.
Domain-inserted reporter
As a general methodology for cytoplasmic allosteric proteins, various domain-inserted reporter systems have been developed. The allosteric protein is inserted into a reporter protein in such a way that reporter activity is dependent on the allosteric protein’s conformation (Fig. 3E). β-lactamase has been used as such a reporter with maltose binding protein, leading to maltose-inducible ampicillin resistance [53]; similarly, a calcium-sensitive GFP reporter was made through insertion of calmodulin [54]. In this way, a wide array of allosteric proteins, such as enzymes and ion sensors that do not regulate transcription, can be tied directly to fluorescent and antibiotic resistance readouts.
Concluding remarks
The ability to engineer the above allosteric protein classes paves the way for new synthetic biology applications: designer GPCRs that can respond to a drug overdose, two-component system proteins that enable bacterial chemotaxis towards a specific molecule, dynamic rewiring of kinase signaling, and controlling the composition and function of engineered microbiota with quorum-sensing switches.
The approach outlined here shows how the power of deep sequencing can be harnessed to address a longstanding question in biology: how protein sequence affects allostery. We envision that this rich mutational data set will motivate new studies in kinetics of allostery through molecular dynamics and NMR experiments.
Figure 4. Functional assays for other allosteric protein classes.
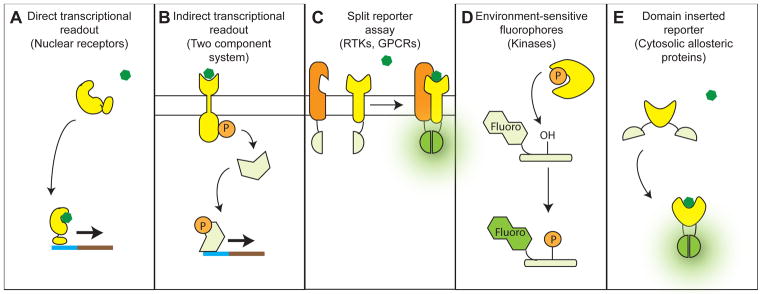
(A) Direct transcriptional readout: applicable to allosteric transcription factors like nuclear receptors. Presence of the receptor ligand is necessary to activate expression of a selectable marker. (B) Indirect transcriptional readout: applicable to two-component systems. Allosteric activation of a histidine kinase causes phosphorylation of a response regulator, leading to transcription of a selectable marker. (C) Split reporter assay: applicable to GPCRs and heterodimeric receptor tyrosine kinases. Complementary halves of GFP are attached to the receptor and the factor recruited upon allosteric activation. A functional GFP is formed only when the activated allosteric protein binds its partner and brings the two GFP halves into sufficient proximity. (D) Environment-sensitive fluorophores: applicable to protein kinases. A fluorophore is attached to a peptide substrate of the kinase in close proximity to the phosphorylated residue, such that the fluorophore will fluoresce only when the phosphate group is present. (E) Domain-inserted reporter: applicable to cytosolic allosteric proteins. The allosteric protein is inserted into GFP, such that a functional GFP is formed only when the allosteric protein changes to a ligand-bound conformation.
Acknowledgments
We thank Lea Starita and James Carothers for comments on the manuscript. This work has been supported by the Wyss Technology Development Fellowship to S.R, US DOE (DE-FG02-02ER63445 to G.M.C.), and P41 GM103533 (to S.F.). S.F. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lu TK, et al. Next-generation synthetic gene networks. Nat Biotechnol. 2009;27:1139–1150. doi: 10.1038/nbt.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dietrich JA, et al. Transcription factor-based screens and synthetic selections for microbial small-molecule biosynthesis. ACS Synth Biol. 2013;2:47–58. doi: 10.1021/sb300091d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodriguez GJ, et al. Evolution-guided discovery and recoding of allosteric pathway specificity determinants in psychoactive bioamine receptors. Proc Natl Acad Sci USA. 2010;107:7787–7792. doi: 10.1073/pnas.0914877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hilser VJ, et al. Structural and Energetic Basis of Allostery. Annu Rev Biophys. 2012;41:585–609. doi: 10.1146/annurev-biophys-050511-102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai F, et al. Conformational spread as a mechanism for cooperativity in the bacterial flagellar switch. Science. 2010;327:685–689. doi: 10.1126/science.1182105. [DOI] [PubMed] [Google Scholar]
- 6.Hilser VJ. An Ensemble View of Allostery. Science. 2010;327:653–654. doi: 10.1126/science.1186121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 8.Amaro RE, et al. A network of conserved interactions regulates the allosteric signal in a glutamine amidotransferase. Biochemistry. 2007;46:2156–2173. doi: 10.1021/bi061708e. [DOI] [PubMed] [Google Scholar]
- 9.Gandhi PS, et al. Structural identification of the pathway of long-range communication in an allosteric enzyme. Proc Natl Acad Sci USA. 2008;105:1832–1837. doi: 10.1073/pnas.0710894105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ricketson D, et al. A conformational switch in the ligand-binding domain regulates the dependence of the glucocorticoid receptor on Hsp90. J Mol Biol. 2007;368:729–741. doi: 10.1016/j.jmb.2007.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masterson LR, et al. Allosteric cooperativity in protein kinase A. Proc Natl Acad Sci USA. 2008;105:506–511. doi: 10.1073/pnas.0709214104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis M. The lac repressor. Comptes Rendus Biologies. 2005;328:521–548. doi: 10.1016/j.crvi.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Bell CE, Lewis M. A closer view of the conformation of the Lac repressor bound to operator. Nat Struct Biol. 2000;7:209–214. doi: 10.1038/73317. [DOI] [PubMed] [Google Scholar]
- 14.Kleina LG, Miller JH. Genetic studies of the lac repressor. XIII. Extensive amino acid replacements generated by the use of natural and synthetic nonsense suppressors. J Mol Biol. 1990;212:295–318. doi: 10.1016/0022-2836(90)90126-7. [DOI] [PubMed] [Google Scholar]
- 15.Markiewicz P, et al. Genetic studies of the lac repressor. XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as “spacers” which do not require a specific sequence. J Mol Biol. 1994;240:421–433. doi: 10.1006/jmbi.1994.1458. [DOI] [PubMed] [Google Scholar]
- 16.Suckow J, et al. Genetic studies of the Lac repressor. XV: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J Mol Biol. 1996;261:509–523. doi: 10.1006/jmbi.1996.0479. [DOI] [PubMed] [Google Scholar]
- 17.Xu J, Matthews KS. Flexibility in the Inducer Binding Region Is Crucial for Allostery in the Escherichia coli Lactose Repressor. Biochemistry. 2009;48:4988–4998. doi: 10.1021/bi9002343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swint-Kruse L, et al. Perturbation from a Distance: Mutations that Alter LacI Function through Long-Range Effects †. Biochemistry. 2003;42:14004–14016. doi: 10.1021/bi035116x. [DOI] [PubMed] [Google Scholar]
- 19.Müller-Hartmann H, Müller-Hill B. The side-chain of the amino acid residue in position 110 of the Lac repressor influences its allosteric equilibrium. J Mol Biol. 1996;257:473–478. doi: 10.1006/jmbi.1996.0176. [DOI] [PubMed] [Google Scholar]
- 20.de Juan D, et al. Emerging methods in protein co-evolution. Nature Publishing Group. 2013;14:249–261. doi: 10.1038/nrg3414. [DOI] [PubMed] [Google Scholar]
- 21.Parente DJ, Swint-Kruse L. Multiple Co-Evolutionary Networks Are Supported by the Common Tertiary Scaffold of the LacI/GalR Proteins. PLoS ONE. 2013;8:e84398. doi: 10.1371/journal.pone.0084398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Süel GM, et al. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol. 2002;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 23.Shulman AI, et al. Structural determinants of allosteric ligand activation in RXR heterodimers. Cell. 2004;116:417–429. doi: 10.1016/s0092-8674(04)00119-9. [DOI] [PubMed] [Google Scholar]
- 24.Fowler DM, Fields S. Deep mutational scanning: a new style of protein science. Nat Methods. 2014;11:801–807. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujino Y, et al. Robust in vitro affinity maturation strategy based on interface-focused high-throughput mutational scanning. Biochem Biophys Res Commun. 2012;428:395–400. doi: 10.1016/j.bbrc.2012.10.066. [DOI] [PubMed] [Google Scholar]
- 26.Whitehead TA, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol. 2012;30:543–548. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ernst A, et al. Coevolution of PDZ domain–ligand interactions analyzed by high-throughput phage display and deep sequencing. Mol Bio Syst. 2010;6:1782. doi: 10.1039/c0mb00061b. [DOI] [PubMed] [Google Scholar]
- 28.Araya CL, et al. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc Natl Acad Sci USA. 2012;109:16858–16863. doi: 10.1073/pnas.1209751109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tinberg CE, et al. Computational design of ligand-binding proteins with high affinity and selectivity. Nature. 2013;501:212–216. doi: 10.1038/nature12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Starita LM, et al. Activity-enhancing mutations in an E3 ubiquitin ligase identified by high-throughput mutagenesis. Proc Natl Acad Sci USA. 2013;110:E1263–72. doi: 10.1073/pnas.1303309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fowler DM, et al. high-resolution mapping of protein sequence- function relationships. Nat Methods. 2010;7:741–746. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Findlay GM, et al. Saturation editing of genomic regions by multiplex homology-directed repair. Nature. 2014 doi: 10.1038/nature13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng AA, et al. Enhanced killing of antibiotic-resistant bacteria enabled by massively parallel combinatorial genetics. Proc Natl Acad Sci USA. 2014;111:12462–12467. doi: 10.1073/pnas.1400093111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kosuri S, et al. Scalable gene synthesis by selective amplification of DNA pools from high-fidelity microchips. Nat Biotechnol. 2010;28:1295–1299. doi: 10.1038/nbt.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLaughlin RN, Jr, et al. The spatial architecture of protein function and adaptation. Nature. 2013;490:138–142. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeVito JA. Recombineering with tolC as a selectable/counter-selectable marker: remodeling the rRNA operons of Escherichia coli. Nucleic Acids Res. 2008;36:e4. doi: 10.1093/nar/gkm1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Isaacs FJ, et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science. 2011;333:348–353. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daily MD, et al. Contact rearrangements form coupled networks from local motions in allosteric proteins. Proteins. 2008;71:455–466. doi: 10.1002/prot.21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fukami-Kobayashi K, et al. Parallel evolution of ligand specificity between LacI/GalR family repressors and periplasmic sugar-binding proteins. Mol Biol Evol. 2003;20:267–277. doi: 10.1093/molbev/msg038. [DOI] [PubMed] [Google Scholar]
- 40.Gronemeyer H, et al. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 41.Wurtz JM, et al. A canonical structure for the ligand-binding domain of nuclear receptors. Nat Struct Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- 42.Galperin MY. Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J Bacteriol. 2006;188:4169–4182. doi: 10.1128/JB.01887-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Capra EJ, et al. Systematic dissection and trajectory-scanning mutagenesis of the molecular interface that ensures specificity of two-component signaling pathways. PLoS Genet. 2010;6:e1001220. doi: 10.1371/journal.pgen.1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferguson SS, et al. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- 45.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 46.Stynen B, et al. Diversity in genetic in vivo methods for protein-protein interaction studies: from the yeast two-hybrid system to the mammalian split-luciferase system. Microbiol Mol Biol Rev. 2012;76:331–382. doi: 10.1128/MMBR.05021-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petschnigg J, et al. The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat Methods. 2014 doi: 10.1038/nmeth.2895. [DOI] [PubMed] [Google Scholar]
- 48.Kittanakom S, et al. CHIP-MYTH: A novel interactive proteomics method for the assessment of agonist-dependent interactions of the human β2-adrenergic receptor. Biochem Biophys Res Commun. 2014;445:746–756. doi: 10.1016/j.bbrc.2014.02.033. [DOI] [PubMed] [Google Scholar]
- 49.Yan YX, et al. Cell-based high-throughput screening assay system for monitoring G protein-coupled receptor activation using beta-galactosidase enzyme complementation technology. J Biomol Screen. 2002;7:451–459. doi: 10.1177/108705702237677. [DOI] [PubMed] [Google Scholar]
- 50.González-Vera JA. Probing the kinome in real time with fluorescent peptides. Chem Soc Rev. 2012;41:1652–1664. doi: 10.1039/c1cs15198c. [DOI] [PubMed] [Google Scholar]
- 51.Morris MC. Fluorescent biosensors - probing protein kinase function in cancer and drug discovery. Biochim Biophys Acta. 2013;1834:1387–1395. doi: 10.1016/j.bbapap.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 52.Chen CA, et al. Biosensors of protein kinase action: from in vitro assays to living cells. Biochim Biophys Acta. 2004;1697:39–51. doi: 10.1016/j.bbapap.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 53.Guntas G, et al. Directed evolution of protein switches and their application to the creation of ligand-binding proteins. Proc Natl Acad Sci USA. 2005;102:11224–11229. doi: 10.1073/pnas.0502673102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baird GS, et al. Circular permutation and receptor insertion within green fluorescent proteins. Proc Natl Acad Sci USA. 1999;96:11241–11246. doi: 10.1073/pnas.96.20.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]