

Abstract
Osteoclast (OC) progenitors (OCP) have been defined in the bone marrow (BM) as CD3−CD45R(B220)−GR1−CD11blo/−CD115+ (dOCP) and more recently in the peripheral blood (PB) as Lym−Ly6G−CD11b+Ly6C+. These progenitors respond to stimuli, including LPS from periopathogenic Aggregatibacter actinomycetemcomitans, activating MAPK signaling, resulting in cytokine/chemokine-mediated osteoclastogenesis. Intracellular negative signaling pathways, including MAPK phosphatase-1 (MKP-1, gene Dusp1) deactivate MAPK pathways (p-p38 and p-JNK) and reduce inflammatory cytokines/chemokines.
Objective
To delineate the role of MKP-1 in chemokine-mediated OC formation using defined OC progenitor populations. Given its role in innate immune inflammatory signaling, we hypothesize that MKP-1 regulates LPS-induced OC formation from BM OCP through deregulated chemokines.
Methods
BM and PB from WT and Dusp1−/− female mice (8–12wks) was obtained and sorted into defined progenitor populations. BM sorted dOCP were primed with MCSF and RANKL (48hrs), blocked with vehicle or chemokine blocking antibodies and stimulated with LPS (48–96hrs). TRAP assay and OC activity were measured for OC formation and activity following treatments. Nanostring Array and qPCR were utilized for gene expression analysis.
Results
Dusp1−/− dOCPs formed more and larger osteoclasts from CD11bhi and dOCP compared to matched WT (P<0.05 each). PB-derived dOCP produced larger and more functional osteoclasts from Dusp1−/− mice compared to WT controls. Nanostring array data revealed significant deregulation in chemokine expression from Dusp1−/− vs. WT cells. qPCR validation of target genes revealed that Dusp1 deficient CD11b+ populations display 1.5–3.5-fold greater expression of CXCL1 and 2–3-fold greater expression of CXCL2 compared to WT in CD11bhi and dOCP (P<0.05 each). Antibody blocking studies using anti-CXCL1 and CXCL2 antibodies blunted osteoclastogenesis in Dusp1−/− cells.
Conclusion
MKP-1 negatively regulates chemokine-driven OC formation and subsequent bone resorption in response to LPS stimulation. Collectively, these data provide useful insight into mechanisms potentially leading to the development of therapeutic treatment of periodontal disease.
Keywords: Osteoclasts, chemokines, MKP-1, lipopolysaccharide, periodontal diseases
1. Introduction
Periodontal diseases are characterized by gingival inflammation, cellular infiltration and oral bone loss in response to pathogens within the oral microbiome [1]. Osteoclasts (OC) are specialized multinucleated cells capable of bone resorption arising from hematopoietic progenitor stem cells (HSCs) within the bone marrow (BM). Defined OC progenitor (dOCPs) populations from BM have been defined as CD3e−CD45R−Gr1−CD11blo/−CD115+ or from peripheral blood (PB) as Lymph−CD11blo/−Ly6CloCD115+ [2, 3]. Numerous studies have shown that varying levels of macrophage-1 antigen (Mac-1)/CD11b can be used to determine the potential to differentiate into a distinct monocyte lineage. Based on this concept, high levels of CD11b are considered macrophage-committed monocytes, while CD11blo cells have the highest osteoclast potential [4] [5]. Consistent with these observations, we and others have shown that BM dOCP enrichment of the CD11blo population yields large, highly functional OC in response to physiological stimuli, macrophage colony stimulating factor (M-CSF) and receptor activator of NF-κB ligand (RANKL) [6].
Within the cell, RANKL signaling activates a multitude of transcription factors through the MAPK and NF-κB signaling cascades [7]. These signals in turn lead to nuclear translocation of NFATc1, thus initiating the process of osteoclastogenesis (OCgen) through transcriptional initiation of genes crucial to dOCP fusion, including DCSTAMP [8, 9]. Under pathogenic conditions, stimuli such as lipopolysaccharide (LPS), a major virulence factor on gram-negative bacteria including the periodontal pathogen Aggregatibacter actinomycetemcomitans, commandeer the process of OCgen through by production of inflammatory cytokines/chemokines induced by activated MAPK (p38, JNK and ERK) and NF-kB pathways [10]. In response to LPS stimulation, these pathways are able to modify and transduce signals in order to affect downstream innate immune responses to microbial antigens [11]. Specifically, JNK and p38 MAPK regulate AP-1, a transcription factor dually involved in NFATc1 promoter binding and multiple cytokine genes transcriptional activation [12].
Once the innate immune signaling has been initiated in response to pathogenic stimuli, a number of intracellular signaling mechanisms exist to help return to cellular homeostasis. MAPK phosphatases (MKP) are a family of phosphatases that deactivate MAPKs at threonine and tyrosine residues. MKP-1 (gene Dusp1), the family archetype, preferentially dephosphorylates phospho (p)-p38 and p-JNK [13]. With regard to oral bone loss, our group has demonstrated that alveolar bone loss is more pronounced in LPS-induced in mice lacking MKP-1 and that overexpression of MKP-1 protects from LPS-induced bone loss [14, 15]. Given the ability of MKP-1 to negatively regulate MAPK induced inflammatory cytokines in response to LPS, we hypothesized that bone loss was due to an exacerbated cytokine response within the periodontal microenvironment. While the mechanisms of inflammatory bone loss were not explored, our data supports the concept that excessive cytokines as a result of non-attenuated LPS-induced inflammation in Dusp1 deficient mice. With regard to intracellular signaling mechanisms, our group has shown that excessive p38 MAPK and subsequent cytokine expression correlated with human periodontal disease severity [16]. While inflammatory-driven bone loss is the hallmark of periodontitis, the mechanisms of inflammatory-induced OC dysregulation are not well appreciated.
Chemokines and their receptors belong to a superfamily of signaling molecules whose expression is driven and regulated by cells that regulate inflammation. Chemokine receptors are primarily located on leukocytes and have been characterized as seven-transmembrane domain spanning G protein-coupled receptors [17]. Chemokine ligands are specific heterotrimeric proteins secreted by activated leukocytes which bind to their cognate receptors G-protein, engaging Gα or Gβγ subunits, thereby activating signal transduction cascades. These second messengers function to control a number of critical responses to inflammation including control of migration, survival and adhesion of leukocytes. It is by this engagement that naïve cells become localized to the site of infection and can engage in pathogenic clearance and the necessary inflammatory response needed for injury to subside. While necessary, this process needs to be highly regulated as persistent influx of innate immune cells may result in excess inflammation. With regard to OCs, it has been shown that numerous chemokines and their receptors can stimulate pre-OC migration as well as formation [18–21]
CXCL2 was first identified as a major chemokine produced by endotoxin-treated macrophages and functions as an inducer of inflammation. CXCL2 binds to its cognate receptor, CXCR2 to induce cellular adherence or cell-cell fusion. While CXCL2 has been shown to be involved in LPS-driven OCgen, the signaling mechanisms governing chemokines in OCgen remain to be defined [19]. In this study, we evaluated the role of MKP-1 in controlling OC-directed bone turnover in response to the periodontal pathogen A. actinomycetemcomitans LPS[15]. As a negative regulator of inflammatory MAPK pathways and subsequent cytokines/chemokines, we hypothesized that MKP-1 may control inflammatory driven OC formation through activity of the chemokine CXCL2. Interestingly, our data indicates that in addition to CXCL2, an isomer, CXCL1 participate in LPS-induced OC formation. Moreover, it was shown that MKP-1 regulates these chemokines and their ability to modify OC formation in response to LPS. These data strongly implicate MKP-1 is a key negative signaling molecule of chemokine-induced OCgen. The results of this study provide insight into pathogenic mechanisms that may allow for development of targeted therapeutic interventions.
2. Materials and Methods
2.1 Isolation of Hematopoietic Progenitors
WT and Dusp1−/− mice on a mixed C57/129 background were used to obtain primary bone marrow cells by flushing tibiae, femurs and humerus bones from age matched female mice (8–12 weeks) with α-modified MEM media (Invitrogen) containing 10% heat-inactivated Hyclone FBS (Thermo-Fisher) and 1% Pen-Strep. Using the AutoMACS sorter (Miltenyi Biotec), bone marrow HSCs were able to be sorted into three distinct groups based on expression of CD11b: CD11bhi (macrophage lineage), CD11blo (dOCPlo) and CD11bneg (dOCP−). Cells were evaluated for enrichment using flow cytometry using the following antibodies: GR-1-Vioblue, CD115-PE, CD11b-APC, lymph-FITC. All experiments were performed using cells from the first passage following cell sorting.
2.2 Osteoclast Differentiation and TRAP Assay
CD11bhi, dOCPlo and dOCP− populations were plated in 96-well or 6-well plates at a density between 5×104–2×106 cells/well and primed with 10ng/ml M-CSF supplemented media overnight. Following initial priming, cells were treated with media containing M-CSF (25ng/ml) and RANKL (50ng/ml). After 48 hours, TRAP-positive, pre-OC cells appeared. Primed pre-OC (herein referred to as Day 0) were harvested for OC formation and RNA. Remaining pre-OC were then stimulated with 50ng/ml of A. actinomycetemcomitans derived LPS (strain Y4) for 48 hours and harvested for TRAP and RNA isolation. Finally, remaining cells were stimulated an additional 48 hours and harvested. For blocking experiments, cells were primed exactly as above but were treated in presence of CXCL1 (Monoclonal Rat IgG2A Clone #48415) and CXCL2 (Monoclonal Rat IgG2B Clone#40605) blocking antibodies and matching IgG controls (R&D Systems Inc. Minneapolis, MN) each at a concentration of 2ug/ml thirty minutes prior to stimulation with M-CSF and LPS. For conditioned media (CM), cells, n=3 WT mice were sacrificed and sorted for monocyte/macrophage/osteoclast progenitors and primed as described above. Media from primed, LPS stimulated cells was collected on day 2 and day 4 from WT and Dusp1−/− cell cultures and used to treat sorted and primed WT cells. Additionally, for LPS bypass studies, primed WT and Dusp1−/− cells were treated with 1ug/ml of recombinant CXCL1 or 0.3ug/ml recombinant CXCL1 (R&D Systems Inc.) For all treatments, M-CSF and appropriate IgGs or vehicles were used as a negative controls, M-CSF and RANKL were used as a positive control. Following treatments cells were fixed or harvested for downstream enumeration or analytical applications. Treated cells were fixed with 10% glutaraldehyde and TRAP stained as previously described [22].
2.3 OC Enumeration and Pit Assay
Differences in OC between WT and Dusp1−/− were determined using the following criteria; 1) number of OC per field of view, 2) number of nuclei per OC (≥3) and 3) size of OC. To measure OC activity, sorted, primed OCP were seeded onto bovine cortical bone slices and treated with LPS as described previously[23]. Bone slices were harvested eight-days following initial priming and pits were enumerated based on field of view at 20x magnification under an inverted microscope.
2.4 Protien Isolation and Western Blot Analysis
Isolated WT and Dusp1−/− dOCP were plated at 1×106 cells per well and stimulated with MCSF and RANKL for 48-hours as described above. Primed cells were then treated with LPS (50ng/ml) for 30, 60 and 120-min. Following stimulation, cells were washed with PBS and total cells lysates were harvested using RIPA buffer (Sigma). Protein content from lysates was determined using a BCA protein assay kit (ThermoScientific), Based on content from each lysate, protein was loaded onto 10% acrylamide gels and electrophoresed to separate proteins by molecular weight. The separated protein was then blotted onto Nitrocellulose membranes using the turbo-blot apparatus (Bio-Rad, Hercules, CA). Blots were blocked with 5% milk protein and probed for total amd phosphorylated MAPK family members including p-p38, p38, pJNK, pERK and ERK (Cell signaling, Beverly, MA ). Additionally, total JNK was obtained from Santa Cruz Biotech (Santa Cruz, CA). All blots were probed and 1:1000 and developed using anti-rabbit-HRP conjugated secondary along with West Pice ECL (ThermoScientific).
2.5 RT-qPCR and Nanostring
mRNA was isolated from Day 0 and from cells stimulated with LPS for 48- and 96-hours using the TriZOL reagent (Invitrogen) according to the manufacturer’s specifications. Amplified cDNA was then probed using amplicon primers obtained from Applied Biosystems including Cxcl1, Cxcl2, Cxcr2, Tnfrsf1 (RANK), Tnfsf11 (RANKL) and Tnfsf11b (OPG). GAPDH was used as endogenous loading control for normalization. Normalized amplicon values were expressed as fold change compared to the M-CSF only controls. For Nanostring analysis, samples isolated above were assessed for RNA purity using a BioAgilent analyzer. Pure samples with purity (RIN) score greater than 8 were used from representative WT and Dusp1−/− samples from each time point and sent to the Nanostring laboratories (Seattle, WA) for analysis using a pre-assembled mouse inflammatory code-set with 179 genes relevant to innate and adaptive immune regulation.
2.6 Statistical Analysis
Data were analyzed with GraphPad Prism software, version 5.0. To compare values between two groups, an unpaired t-test and subsequent Mann-Whitney test were used. Comparison of values between multiple groups was measured using two-way ANOVA analysis followed by Bonferroni’s multiple comparison test. Results were expressed as mean ± SD of at least three (n=3) biological replicates for each experiment. A P value of less than 0.05 was considered to be statistically significant.
3. Results
3.1 MKP-1 Regulates the Distribution of Defined OC Progenitors
Previous research published by our group and others have shown that isolation of defined OC progenitors (dOCP, defined as CD45R−CD3e−GR1−CD11blo/−CD115+) generated robust OC in response to M-CSF and RANKL stimulation. To further identify OC progenitors, we defined the distribution of dOCP in whole BM cells by depleting cells positive for lymphocyte markers (CD45R and CD3e) and the pan-granulocyte marker Gr1. The remaining cells depicted in Figure 1A, were analyzed for expression of CD11b versus CD115 and enumerated based on lineage expression. Results in Figure 1B show a number of lineage combinations result from dOCP isolation. Interestingly, our analysis shows that lymph−Gr1−CD11b−CD115+ population (non-macrophage/monocyte dOCP) was significantly more abundant in the Dusp1−/− population (P<0.05). Conversely, the true dOCP (lymph−Gr1−CD11bloCD115+) had significantly less OC as a result of Dusp1 deficiency. While these results of interest to the animal as a whole, their bearing on OC formation in cell culture was controlled through seeding density normalization in downstream experiments.
Figure 1. MKP-1 Regulates Pre-Osteoclast Monocyte Distribution in Bone Marrow Hematopoietic Progenitor Cells.
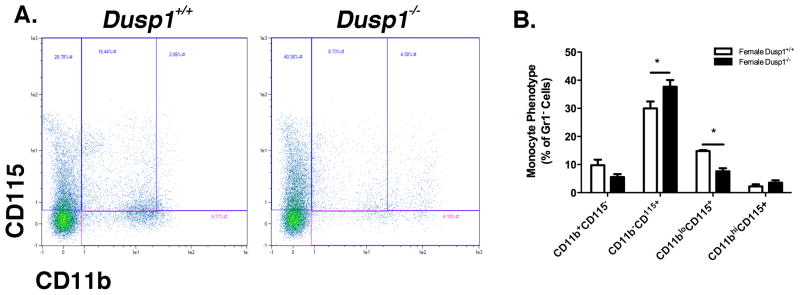
BM HSCs from WT and Dusp1−/− were depleted of Lymphocytes (CD45R, CD3e) and granulocytes (Gr1). Remaining cells were analyzed for expression of CD11b versus CD115 using flow cytometry. (A) Representative dot-plot gated on varying expression of CD11b versus CD115. (B) Enumeration of monocyte and defined OC progenitor populations in lymphocyte and granulocyte negative whole BM. Dusp1−/− mice have more abundant dOCP- and less dOCPlo at baseline. Plots and graphs are representative of at least n=4 biological replicates. Gated plots are based off of whole distribution of single stained populations and negative controls. (*P<0.05)
3.2 MKP-1 Regulates LPS-induced OC Formation and Fusion
Previous research has shown that Dusp-1 deficient mice present with an enhanced inflammatory phenotype and subsequent bone loss in response to LPS stimulation [14]. Moreover, these data suggest that the bone loss associated with LPS-stimulation was due to enhanced OC formation. To further address the role of MKP-1 in LPS-induced OC formation, we utilized a unique protocol for isolation of defined OC progenitors within the whole bone marrow macrophage population. In Figure 2A, our data shows that within the bone marrow population exists multiple sub-populations which respond differentially to pathogenic stimuli. As these data show, Dusp1−/− CD11bhi and dOCPlo formed very large multinucleated OC in response to 50ng/ml LPS compared to matched WT cells. Enumeration was conducted using calibrated evaluators blinded to the treatment conditions from each group. In Figure 2B pre-OC, defined as mononuclear trap positive cells were enumerated using an arbitrary trap positivity rating scale (0–4) based on percent of trap positive cells. As these results indicate, Dusp1 differentially regulates the ability of the CD11bhi population to respond to RANKL, yet no differences exist in the dOCPlo population between genotypes. Figures 2C and 2D show that in response to LPS, MKP-1 activity differentially regulates OC formation in primed and defined CD11bhi and dOCPlo populations. On both days 2 and 4, Dusp1−/− CD11bhi and dOCPlo had 4- and 3-fold more OC than matched WT cells (Day 2: P<0.05 and P<0.01, respectively, Day 4: P<0.05 each). To measure OC morphology at least n=4 samples/group were measured for size and multi-nucleation using Adobe Photoshop. Consistent with the results depicted in Figure 2A, CD11bhi and dOCPlo cells from Dusp1−/− mice were significantly larger (on orders of magnitude) than WT counterparts on both days 2(Figure 2E) and 4 (Figure 2F). As a whole, these results suggest that MKP-1 regulates OC formation and ability to fuse.
Figure 2. MKP-1 controls LPS-Induced OC formation in defined HSC populations.

(A) TRAP assay from Cd11b-sorted, RANKL-primed cells stimulated with 10ng/ml LPS for 48 and 96hr. (B) Pre-OC enumeration following RANKL priming. (C) OC enumeration 48h post-LPS stimulation. (D) OC enumeration 96h post-LPS stimulation. Values indicate that Dusp1−/− from CD11bhi and dOCPlo form significantly more OC than WT counterparts. (E) Day 2 OC area (F) Day 2 OC area. Area is presented as mean and range. Box indicates 25th to 75th percentile. Values indicate that Dusp1−/− sorted populations form larger OC than WT counterparts. Data (B–D) are plotted as mean ± SD of at least n=3 biological replicates. Photographs are presented at 10x magnification (*P<0.05, **P<0.01, ***P<0.001)
3.3 Dusp1 Regulates MAPK Phosphorylation in MCSF and RANKL-Primed dOCP stimulated with LPS
In order to mimic the bone microenvironment, cells were primed with MCSF and RANKL prior to stimulation with LPS. Since LPS is known to activate the MAPK pathways in naïve innate immune cells, we next aimed to see if our priming modulated the activity of the MAPK pathway in response to LPS. As shown in Figure 3, Dusp1 deficiency resulted in elevated and sustained induction of phosphorylated p38, pJNK and pERK relative to WT treated cells. Interestingly, MCSF alone and MCSF + RANKL stimulated phosphorylation of p38 relative to the other MAPKs, yet this was enhanced upon LPS stimulation.
Figure 3. MKP-1 Regulates MAPK Activation in MCSF and RANKL-Primed pre-OC Stimulated With LPS.

MAPK expression from cells primed with MCSF and RANKL for 48-hours and stimulated with A.a. LPS (50ng/ml) for up to 120 minutes. 20ug of total protein from each time point were separated by electrophoresis and probed with antibodies against phospho (p)-p38, p38, pJNK (p-p46/54), JNK (p46/54), pERK1, ERK1 and GAPDH (all 1:1000 in 5% BSA-TBST overnight). Blots were developed using anti-rabbit secondary (1:1000) for 1 hour followed by ECL for visualization. Dusp1 deficient dOCP had more and prolonged activation of pp38, pJNK and pERK relative to WT counterparts. Blot is a representative of n=3 biological replicates.
3.4 LPS-Treated Conditioned Media from Dusp1 Deficient OCP Drive Excessive OC formation in WT Cells
Since our investigation has shown excessive and larger OC formed in Dusp1−/− sorted CD11bhi and dOCPlo, we next aimed to look at the autocrine/paracrine activity of these chemokines using a conditioned media approach. Sorted WT cells were treated with conditioned media from WT and Dusp1−/− cells stimulated with LPS or blocking antibodies + LPS for 4 days. In a similar manner to cells stimulated directly with LPS, CM from Dusp1−/− CD11bhi and dOCPlo yield significantly more OC than matched, sorted WT counterparts (P <0.01 and P <0.0001, respectively) (Figure 4). While this assay did not generate robust OCs, the differential formation was observed across at least seven biological replicates. From these data, we conclude that MKP-1 regulates the expression and secretion of other factors that may influence OC fusion.
Figure 4. MKP-1 Regulates OC Formation in a Local Paracrine manner.

(A) TRAP assay from primed WT cells treated with WT or Dups1−/− conditioned media (CM). (B) enumeration of primed WT cells treated with WT or Dups1−/− conditioned media from LPS-stimulated cells. Data are plotted as mean ± SD (n=3, **P<0.01, ***P<0.001).
3.5 Dusp1 Deficiency Results in Excessive Cytokine and Chemokine Expression in Response to LPS
Given our current data that indicates that MKP-1 deficient dOCPs generate larger and more numerous OC in response to LPS, yet form independently of NFATc1, we next aimed to look at other factors involved in OC fusion. Recently, Ha et al showed that the chemokine CXCL2 could be directly involved in OC formation in response to LPS stimulation, yet the mechanism of chemokine induced OC formation and control has not been elucidated [19]. Using defined progenitors primed with RANKL and stimulated with LPS, we investigated the role of MKP-1 in chemokine-mediated OC formation in response to LPS. Using a pre-built mouse inflammatory cytokine/chemokine panel, code sets corresponding to over 179 genes were individually measured from each sample by NanoString technology to determine the actual number of mRNA transcripts present in each gene (without amplification). Data was then plotted against internal controls for genes showing the largest fold-change relative to WT control. Of interest was the chemokine portion of the panel, particularly Cxcl1 and Cxcl2 mRNA, which were up at day 4 by 15- and 20-fold, respectively (Table 1).
Table 1.
Chemokine Expression Using Nanostring Technology
| Female | Day 0 WT | Day 0 KO | Day 0 KO:WT | Day 2 WT | Day 2 KO | Day 2 KO:WT | Day 4 WT | Day 4 KO | Day 4 KO:WT |
|---|---|---|---|---|---|---|---|---|---|
| Cxcl1 | 18 | 9 | 0.48245614 | 42 | 135 | 3.229181665 | 69 | 1054 | 15.3712828 |
| Cxcl10 | 183 | 107 | 0.583755792 | 91 | 72 | 0.786341678 | 36 | 488 | 13.50886427 |
| Cxcl2 | 33 | 31 | 0.926337943 | 172 | 677 | 3.948107982 | 474 | 9683 | 20.42041884 |
| Cxcl3 | 23 | 7 | 0.292942743 | 211 | 1320 | 6.250437911 | 212 | 4115 | 19.42882772 |
| Cxcl5 | 12 | 10 | 0.808474576 | 49 | 41 | 0.81970862 | 114 | 154 | 1.346978046 |
| Cxcl9 | 24 | 7 | 0.311016949 | 11 | 14 | 1.289868668 | 7 | 21 | 2.962603878 |
| Cxcr4 | 5648 | 1518 | 0.268678675 | 1482 | 1483 | 1.001025959 | 2930 | 12318 | 4.203348246 |
Number of mRNA chemokine transcripts from female WT and Dusp1−/− cell lysates harvested at day 0 (primed with M-CSF and RANKL), Day 2 and Day 4 (primed and stimulated with 50ng/ml LPS). Bolded headings are ratio of KO (Dusp1−/−) transcripts to WT at each time point. Rows highlighted in red indicate chemokines which signal through the same receptor. Values are presented as absolute number of transcripts and ratio normalized to internal reference controls.
To validate the findings from our NanoString array as well as to determine the specificity of gene expression between additional populations which can form OC, we utilized qPCR from CD11bhi and dOCPlo populations primed with M-CSF and RANKL (Day 0), primed and treated with LPS for 48 hrs (Day 2) or 96 hrs (Day 4) shown in Figure 5. Cxcl1 mRNA expression in CD11bhi and dOCPlo (Figure 5A and 5B, respectively), we see that only dOCPlo was significantly up regulated in the Dusp1−/− population (Day 4). In Figure 5C and D however, each Dusp1−/− sorted populations showed robust and significant induction in both Cxcl1 and Cxcl2 mRNA expression on all days following LPS stimulation (P<0.05 and P<0.01). In fact, the Dusp1−/− CD11bhi population showed the largest disparity, being 3-fold (P<0.05) and then 2-fold higher (P<0.01) in expression at days 2 and 4, respectively. These data validate and corroborate NanoString data shown in Table 1. In line with this data, previous work from our lab have shown that protein expression of CXCL1 was up-regulated nearly 4-fold (P<0.001) in whole bone marrow macrophage populations stimulated with LPS for 24 hours (Supplemental Figure 1).
Figure 5. MKP-1 Regulates CXCL1 and CXCL2 mRNA Expression in response to LPS stimulation.

Gene expression from cells primed with M-CSF and RANKL (Day 0) or primed then stimulated with LPS for 48 (Day 2) or 96hrs (Day 4). CXCL1 expression from CD11bhi (A) and dOCPlo (B), respectively, as determined by qPCR. CXCL2 expression from CD11bhi (C) and dOCPlo (D) as determined by qPCR. Data are written or plotted as a representative of at least n=3 biological replicates. (*P<0.05, **P<0.01, ***P<0.001).
Previous research has shown that LPS can induce RANKL in osteoblast lineage [24]. Based on this phenomenon, we nexted aimed to investigate the effect of LPS-stimulation on expression of RANKL, its receptor (RANK) and the decoy ligand osteoprogenerin (OPG). Results depicted in Supplemental Figure 2 reveal that in response to MCSF, MCSF/RANKL and LPS stimulation, RANK and OPG expression were not significantly regulated by treatment or genotype. Surprisingly, RANKL mRNA expression was elevated following 4-day LPS-stimulation. However, when RANKL:OPG ratio was compared, the increase in RANKL mRNA was lost. Moreover, the data reveals that during mid-OC formation, the ratio of RANKL:OPG was significantly lower in Dusp1−/− dOCP.
3.6 MKP-1 Regulates CXCR2 Expression in Response to LPS stimulation
In order to properly adjust to increasing inflammatory signals such as cytokines or other ligands, cells typically up-regulate the cognate receptors to maximize the signaling capacity. Based on this phenomenon, we next measured the surface expression and kinetics of CXCR2, the receptor specific for CXCL1 and CXCL2 signaling. Figure 6A represents flow cytometry analysis of WT and Dusp1−/− cells primed with M-CSF and RANKL, then stimulated with LPS for up to 24 hours. As these data indicate, Dusp1−/− cells stimulated with LPS has a significant increase in expression at 24 hours compared to WT counterpart. Enumeration of this data (Figure 6B) reveals that CXCR2 surface staining was up nearly 2.5-fold in Dusp1 deficient cells compared to WT (P<0.001).
Figure 6. MKP-1 Regulates chemokine Receptor CXCR2 Expression in Response to LPS Stimulation.
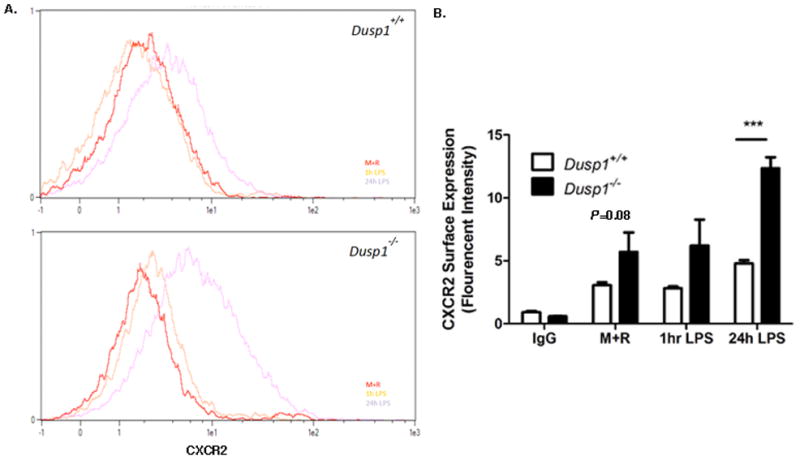
(A) Representative flow cytometry histograms of WT (top) and Dusp1−/− (bottom) cells treated with CXCR2 antibody (2ug/ml) after exposure to MCSF (not shown), M-CSF and RANKL (M+R) or M+R and LPS for 1hr or 24hr. (B) Enumeration of CXCR2 distribution in treated cells based on fluorescent intensity. Data is plotted as a representative of at least n=3 biological replicates. (***P<0.001).
3.7 Blocking Chemokines CXCL1 and CXCL2 Reduced OC Formation in Response to LPS Stimulation
Since our Nanostring data and RT-qPCR results indicated that chemokines Cxcl1 and Cxcl2 mRNA were significantly up-regulated in response to Dusp1 ablation, we next aimed to determine their role in LPS-induced OC formation using an antibody blocking strategy. As with our LPS-driven model, WT and Dusp1−/− cells were primed with M-CSF and RANKL then treated with CXCL1 or CXCL2 blocking antibodies prior to LPS stimulation. Blocking with both CXCL1 and CXCL2 ablated differences in OC formation between WT and Dusp1−/− CD11bhi and dOCPlo populations at day 4 (Fig. 7). In fact, αCXCL2 blockage resulted in significantly less OC from Dusp1−/− dOCP populations (P<0.001). Morphological analysis revealed that blocking these chemokines resulted in significantly smaller OC compared to those treated with LPS alone (Supplemental Figure 3). In fact, these OC were the size of negative (M-CSF) treated controls and there were no longer significant differences between genotypes of sorted populations. Additionally, using conditioned media, WT cells treated with media blocked with CXCL1 and CXCL2 blocking antibody again showed that these chemokines are driving the differential response between genotypes (Figure 7D). In both LPS-treated and conditioned media treated, neutralization of chemokines CXCL1 and CXCL2 abolished differences in OC formation between genotypes.
Figure 7. Blocking chemokine ligands abolished differences in LPS-Induced OC formation based on Dusp1 status.

(A) TRAP assay from dOCPlo, RANKL-primed cells pre-treaetd with CXCL1 and CXCL2 blocking antibodies (1ug/ml, respectively) or CXCR2 blocking antibody (30ug/ml), then stimulated with 10ng/ml LPS for 96 hours. (B) Enumeration of cells treated with IgG control or CXCL1 blocking antibody prior to stimulation with LPS. (C) Enumeration of cells treated with IgG control or CXCL2 blocking antibody prior to stimulation with LPS. (D) Enumeration of CM treated cells treated with CM alone or CM with CXCL1 and CXCL2 inhibitors. Photographs are presented at 10x magnification and are representative of n=3 biological replicates. (**P<0.01, ****P<0.0001).
3.8 MKP-1 Regulates OC Formation in Response to Direct Chemokine Stimulation
Finally, since we have shown that MKP-1 regulates LPS-induced OC formation through regulation of chemokines Cxcl1 and Cxcl2, we next aimed to bypass LPS-stimulation and observe OC formation in WT and Dusp1−/− cells stimulated with recombinant chemokines alone. Using RANKL-primed cells, our data presented in Fig. 8 shows that both CXCL1 and CXCL2 recombinant proteins (1μg/ml and 0.3μg/ml, respectively) induced OC independent of LPS. Moreover, these data reveal that Dusp1−/− formed more OC with CXCL1 (P<0.05) and CXCL2 (P<0.01) compared to WT controls. These observations are in line with data from Figure 6B which shows that in response to priming expression of CXCR2 was increased nearly 40% and was found to be just out of the range of detectable significant differences (P=0.08). Additionally, our data shows that the vehicle control stimulated with LPS generate more OC in the KO, which is consistent with previous findings. In all, these data reveal that in addition to regulating chemokine expression, MKP-1 regulates the response of dOCP to these chemokines, ultimately regulating the ability of the cell to form OC in response to inflammatory stimuli.
Figure 8. Treatment with recombinant CXCL1 and CXCL2 induces more OC in Dups1−/− dOCPs.

(A) TRAP assay from dOCPlo, RANKL-primed cells pre-treated with recombinant CXCL1 (1ug/ml) and CXCL2 (0.3ug/ml), or vehicle then stimulated with 10ng/ml LPS for 96 hours. (B) Enumeration of recombinant cytokine treatment. Photographs are presented at 10x magnification and are representative of n=3 biological replicates. (*P<0.05, **P<0.01).
4. Discussion
In the bone microenvironment cell fates are determined by integration of many signals to control resorption or deposition. Under inflammatory conditions, such as those seen in PD, it is the presence of virulent pathogens and the host response that determines the extent of pathological bone loss. Moreover, while periopathogens such as A. actinomycetemcomitans are found in diseased patients, they are also found in healthy patients. Therefore in understanding the role of signaling in progenitor cells that respond to these stimuli, we can address the hypothetical role of the host response by regulating aspects [1]. In the current study, we addressed the role of MKP-1 as a regulator of OC formation in response to periodontal pathogenic-derived LPS. Using a model system where defined OC progenitors were primed with RANKL prior to stimulation, we aimed to more accurately represent the microenvironment seen in periodontitis to better study pathogenic OC formation.
By far the most important finding uncovered from this study is that MKP-1 controls LPS-induced osteoclastogenesis through regulation of chemokines CXCL1 and CXCL2. It is well known that chemokines and their receptors are necessary for recruitment and adhesion of circulating leukocytes and pre-OC into the site of injury [18]. It is also known that some chemokines and their receptors may contribute to OC formation [17, 18, 20]. In the current study, we observed that MKP-1 regulates CXCL1, CXCL2 and CXCR2 in response to LPS. This finding is partially supported by previous work that showed that CXCL2 was significantly up-regulated in response to LPS (and RANKL) and blockade of CXCL2 or its receptor CXCR2 nearly ablated OC formation [19, 25]. Interestingly and in contrast to the previously reported studies [25], where RANKL was reported to induce CXCL2 expression, we observed that CXCL2 was only up-regulated in response to LPS and that this effect was amplified in the absence of MKP-1. Recent evidence has shown that second messenger activation of NFκB and AP-1 are capable of binding to promoter region on the Cxcl2 gene, thus regulating its expression [26]. While numerous studies have shown MAPK dysregulation in the absence of MKP-1, it was critical to determine what effect priming and subsequent LPS stimulation had on MAPK activation. Relative to MCSF-treated only and MCSF-RANKL-treated cells, LPS treated Dusp1−/− cells exhibited sustained phosphorylated levels of p38, JNK and surprisingly ERK relative to WT counterparts. These results indicate that stimulating the cells with MCSF and RANKL (priming) does not disrupt the known function of MKP-1 in response to pathogenic stimulation.
Given the observed signal dysregulation associated with the absence of MKP-1 in response to LPS as well as evidence that shows chemokine dysregulation, it was predictable that Cxcl2 mRNA would be elevated. Indeed, others have previously shown that CXCL2 was increased by MKP-1 null macrophages [27]. In addition to CXCL2 dysregulation, our study is the first to show that CXCL1 and the cognate receptor CXCR2 are significantly elevated in response to LPS and amplified in the absence of MKP-1. CXCL1, a heteroisomer of CXCL2 was also shown to be a by-product of endotoxin stimulation with similar function [28, 29]. Physiologically, both CXCL1 and -2 function as chemoattractants during inflammation and potentially as inducers of pre-OC fusion. Since mRNA levels of Cxcl1, Cxcl2 and Cxcr2 were all 9–11 fold elevated in Dusp1−/− preosteoclasts, it was necessary to determine the relative contribution of these chemokines towards osteoclastogenesis using CXCL1 and CXCL2 specific neutralizing antibodies. Results from this study are consistent with one study showing CXCL2 was necessary for OC formation[25]. Additionally, our results are the first reported which show that CXCL1 also contributes to OC formation in response to pathogenic stimuli. One explanation for this may be that both ligands signal through CXCR2 that is up regulated only in Dusp1−/− cells treated with LPS. In fact, LPS did not significantly increase Cxcr2 mRNA expression in WT cells. Not surprisingly, we also found that blocking CXCR2 reduced OC formation and size in Dusp-1 KO osteoclasts to WT levels (data not shown). Based on these results, it is conceivable that the observed increase in OC formed may be in the enhanced capability of Dusp1−/− cells is directly in respond to elevated levels of CXCL1 and CXCL2. Moreover, our data indicates that upon priming with M-CSF and RANKL, Dusp1−/− dOCP have enhanced receptor expression, indicating that these cells have the capacity to respond more robustly than WT counterparts to the elevated cognate chemokines. In a recent study, it was shown that CXCR2 resulted in a significant reduction in both osteoblast and osteoclast formation, resulting in reduced coupled bone turnover in response to wound healing [29].
Finally, to confirm the assertion that CXCL1 and CXCL2 are directly contributing to OC formation we treated primed WT and Dusp1−/− cells with equal amounts of recombinant chemokines. Interestingly, we found that the same concentration of chemokine elicited enhanced OC formation in Dusp1 deficient cells, albeit not to the extent as LPS-induced OC formation. Thus, independent of LPS, dOCP signaling in response to chemokines is further modified by the same chemokines already excessively produced in the absence of MKP-1. Indeed, it can be further speculated CXCR2 signaling is also in part regulated by MKP-1. Furthermore, given the excessive amount of chemokines produced in Dusp1−/− we can predict that proportional treatment with recombinant proteins may yield what we observe in LPS-stimulated cells alone, larger and more OC.
Previously work from our lab has shown that MKP-1 is necessary for proper OC formation in response to RANKL by regulating NFATc1 nuclear translocation and downstream OC gene expression [6]. In contrast, the current study found that NFATc1 gene expression was not dysregulated between genotypes following LPS stimulation (data not shown). While interesting, the lack of difference in known mediators of OC formation does not allow for an adequate explanation of the excessive and large OC observed in Dusp1 deficient dOCP in response to RANKL. In addition to up regulation of noted chemokines, an additional explanation for this may be due to the permissive nature of RANKL priming. Under non-primed conditions, we observed that LPS alone does not induce OC formation. However, pre-OC priming with RANKL allows for the machinery of OC formation (NFATc1 and DC-STAMP) to be expressed. Following priming, pathogenic insult from LPS likely commandeers the process of OC formation seen within the macrophage and monocyte/dOCP lineages lacking MKP-1 as a result of increased cytokines regulated by MKP-1. Interestingly, upon LPS stimulation, gene expression of NFATc1 and DCSTAMP were down regulated in both WT and Dusp1 deficient cells. Further supporting this explanation, data from Nanostring analysis confirmed previous observations in Dusp1 deficient cells, showing that cytokines including TNF and iNOS were significantly increased (data not shown). TNF is a direct product of LPS-engagement of TLR4 and has been shown to be involved in OC formation independent of RANKL [17, 30, 31]. In this mechanism, TNF (and LPS) specifically activate NF-κB and JNK-dependent c-Fos (a subunit of AP-1) contributing to OC formation [30]. Previously published work in our lab has shown that in response to LPS, MKP-1 deficient mouse tissues and in vitro cell cultures express significantly more TNF than WT counterparts [32]. Also of interest, iNOS (another direct product of LPS stimulation) was reported to aid in OC formation in response to TNF stimulation by up-regulating OC survival once formed. Moreover, that study showed that iNOS dependent nitric oxide (NO) production enhanced the size of the OC formed [33]. In fact, we have found that in response to LPS, both the iNOS gene and NO production were significantly upregulated in Dusp1−/− cells compared to WT controls (data not shown).
In conclusion, we show that CXCL1 and CXCL2 contribute to OC formation by a mechanism that is negatively regulated through MKP-1. Based on these findings, we hypothesize that chemokines including CXCL1 and CXCL2 contribute to OC formation in RANKL-primed cells by enhanced fusion in vitro. Under inflammatory conditions where p38 signaling is significantly elevated, including periodontal disease [16], it is conceivable that monocyte/dOCP progenitors have increased capacity to synthesize and respond to chemokines produced in the local environment and allow for pre-OC normally out of range to be in proximity to form OC. Based on our results, we may be able to develop targeted therapeutics strategies to attenuate chemokine driven OC-induced bone resorption.
Supplementary Material
Unsorted bone marrow macrophages were stimulated with LPS (100ng/ml) for 24hr. Cell supernants were measured for protein expression using a CXCL1 ELISA kit. Dups1−/− (KO) cells expressed nearly 3-fold more CXCL1 than matched WT cells. Data is plotted as a representative of at least n=3 biological replicates. (***P<0.001).
Gene expression from MCSF, MCSF + RANKL (M+R) and M+R primed cells stimulated with LPS (50ng/ml) for 48- and 96-hours. (A) RANK (Tnfrsf11a) (B) RANKL (Tnfsf11), (C) OPG (Tnfrsf11b). RANKL:OPG ration (D) based upon gene expression. All values were normalized to MCSF only treated controls. Data are written or plotted as a representative of at least n=3 biological replicates. (*P<0.05, ****P<0.001).
Sorted WT and Dusp1−/− cells were primed and blocked with CXCL1 or CXCL2 blocking antibodies prior to stimulation with LPS (as described previously). Cells were noticeably smaller following chemokine blocking. Measurements of OC area reveals that WT and Dusp1−/− cells treated with blocking media did not form OC larger than M-CSF only (Con) treated controls. Area is presented as mean and range. Box indicates 25th to 75th percentile. Data are plotted as a representation of at least n=3 biological replicates.
Highlights.
Dusp1−/− dOCPs are hyper-osteoclastogenic in response to LPS through a mechanism independent of NFATc1
CXCL1 and CXCL2 are necessary for LPS-induced OC formation
Cxcl1 and Cxcl2 expression is regulated by MKP-1
Dusp1−/− dOCPs are hyper-osteoclastogenic in response to equivalent amounts of recombinant chemokine
Acknowledgments
We would like to thank John Kurtz and for his assistance in data collection. This work was supported by grants from the NIH 5R01DE018290, 5R01DE021423, P30GM103331, R25DE022677, and T32DE017551-05.
Contributor Information
Michael S. Valerio, Email: valeriom@musc.edu.
Bethany A. Herbert, Email: herbertb@musc.edu.
Dimitrios S. Basilakos, Email: basilako@musc.edu.
Hong Yu, Email: yuho@musc.edu.
Keith L. Kirkwood, Email: klkirk@musc.edu.
References
- 1.Kajiya M, Giro G, Taubman MA, Han X, Mayer MP, Kawai T. Role of periodontal pathogenic bacteria in RANKL-mediated bone destruction in periodontal disease. J Oral Microbiol. 2010:2. doi: 10.3402/jom.v2i0.5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacquin C, Gran DE, Lee SK, Lorenzo JA, Aguila HL. Identification of multiple osteoclast precursor populations in murine bone marrow. J Bone Miner Res. 2006;21:67–77. doi: 10.1359/JBMR.051007. [DOI] [PubMed] [Google Scholar]
- 3.Jacome-Galarza CE, Lee SK, Lorenzo JA, Aguila HL. Identification, characterization, and isolation of a common progenitor for osteoclasts, macrophages, and dendritic cells from murine bone marrow and periphery. J Bone Miner Res. 2013;28:1203–13. doi: 10.1002/jbmr.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacquin C, Gran DE, Lee SK, Lorenzo JA, Aguila HL. Identification of multiple osteoclast precursor populations in murine bone marrow. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2006;21:67–77. doi: 10.1359/JBMR.051007. [DOI] [PubMed] [Google Scholar]
- 5.Hayashi H, Nakahama K, Sato T, Tuchiya T, Asakawa Y, Maemura T, et al. The role of Mac-1 (CD11b/CD18) in osteoclast differentiation induced by receptor activator of nuclear factor-kappaB ligand. FEBS letters. 2008;582:3243–8. doi: 10.1016/j.febslet.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 6.Valerio MS, Herbert BA, Griffin AC, 3rd, Wan Z, Hill EG, Kirkwood KL. MKP-1 signaling events are required for early osteoclastogenesis in lineage defined progenitor populations by disrupting RANKL-induced NFATc1 nuclear translocation. Bone. 2013 doi: 10.1016/j.bone.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xing L, Schwarz EM, Boyce BF. Osteoclast precursors, RANKL/RANK, and immunology. Immunol Rev. 2005;208:19–29. doi: 10.1111/j.0105-2896.2005.00336.x. [DOI] [PubMed] [Google Scholar]
- 8.Takayanagi H. The role of NFAT in osteoclast formation. Ann N Y Acad Sci. 2007;1116:227–37. doi: 10.1196/annals.1402.071. [DOI] [PubMed] [Google Scholar]
- 9.Yagi M, Miyamoto T, Sawatani Y, Iwamoto K, Hosogane N, Fujita N, et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202:345–51. doi: 10.1084/jem.20050645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Q, Valerio MS, Kirkwood KL. MAPK usage in periodontal disease progression. J Signal Transduct. 2012;2012:308943. doi: 10.1155/2012/308943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Q, Valerio MS, Kirkwood KL. MAPK usage in periodontal disease progression. Journal of signal transduction. 2012;2012:308943. doi: 10.1155/2012/308943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qi X, Pramanik R, Wang J, Schultz RM, Maitra RK, Han J, et al. The p38 and JNK pathways cooperate to trans-activate vitamin D receptor via c-Jun/AP-1 and sensitize human breast cancer cells to vitamin D(3)-induced growth inhibition. The Journal of biological chemistry. 2002;277:25884–92. doi: 10.1074/jbc.M203039200. [DOI] [PubMed] [Google Scholar]
- 13.Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol. 2006;176:1899–907. doi: 10.4049/jimmunol.176.3.1899. [DOI] [PubMed] [Google Scholar]
- 14.Sartori R, Li F, Kirkwood KL. MAP kinase phosphatase-1 protects against inflammatory bone loss. J Dent Res. 2009;88:1125–30. doi: 10.1177/0022034509349306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu H, Li Q, Herbert B, Zinna R, Martin K, Junior CR, et al. Anti-inflammatory effect of MAPK phosphatase-1 local gene transfer in inflammatory bone loss. Gene Ther. 2011;18:344–53. doi: 10.1038/gt.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Travan S, Li F, D’Silva NJ, Slate EH, Kirkwood KL. Differential expression of mitogen activating protein kinases in periodontitis. Journal of clinical periodontology. 2013;40:757–64. doi: 10.1111/jcpe.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Comerford I, McColl SR. Mini-review series: focus on chemokines. Immunol Cell Biol. 2011;89:183–4. doi: 10.1038/icb.2010.164. [DOI] [PubMed] [Google Scholar]
- 18.Miyamoto K, Ninomiya K, Sonoda KH, Miyauchi Y, Hoshi H, Iwasaki R, et al. MCP-1 expressed by osteoclasts stimulates osteoclastogenesis in an autocrine/paracrine manner. Biochemical and biophysical research communications. 2009;383:373–7. doi: 10.1016/j.bbrc.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 19.Ha J, Lee Y, Kim HH. CXCL2 mediates lipopolysaccharide-induced osteoclastogenesis in RANKL-primed precursors. Cytokine. 2011;55:48–55. doi: 10.1016/j.cyto.2011.03.026. [DOI] [PubMed] [Google Scholar]
- 20.Xing Q, de Vos P, Faas MM, Ye Q, Ren Y. LPS promotes pre-osteoclast activity by up-regulating CXCR4 via TLR-4. J Dent Res. 2011;90:157–62. doi: 10.1177/0022034510379019. [DOI] [PubMed] [Google Scholar]
- 21.Hoshino A, Ueha S, Hanada S, Imai T, Ito M, Yamamoto K, et al. Roles of chemokine receptor CX3CR1 in maintaining murine bone homeostasis through the regulation of both osteoblasts and osteoclasts. Journal of cell science. 2013;126:1032–45. doi: 10.1242/jcs.113910. [DOI] [PubMed] [Google Scholar]
- 22.Cicek M, Vrabel A, Sturchio C, Pederson L, Hawse JR, Subramaniam M, et al. TGF-beta inducible early gene 1 regulates osteoclast differentiation and survival by mediating the NFATc1, AKT, and MEK/ERK signaling pathways. PLoS One. 2011;6:e17522. doi: 10.1371/journal.pone.0017522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffin AC, 3rd, Kern MJ, Kirkwood KL. MKP-1 is essential for canonical vitamin D-induced signaling through nuclear import and regulates RANKL expression and function. Mol Endocrinol. 2012;26:1682–93. doi: 10.1210/me.2012-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossa C, Jr, Liu M, Kirkwood KL. A dominant function of p38 mitogen-activated protein kinase signaling in receptor activator of nuclear factor-kappaB ligand expression and osteoclastogenesis induction by Aggregatibacter actinomycetemcomitans and Escherichia coli lipopolysaccharide. Journal of periodontal research. 2008;43:201–11. doi: 10.1111/j.1600-0765.2007.01013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ha J, Choi HS, Lee Y, Kwon HJ, Song YW, Kim HH. CXC chemokine ligand 2 induced by receptor activator of NF-kappa B ligand enhances osteoclastogenesis. J Immunol. 2010;184:4717–24. doi: 10.4049/jimmunol.0902444. [DOI] [PubMed] [Google Scholar]
- 26.Kim CH, Kim JH, Lee J, Hsu CY, Ahn YS. Thiol antioxidant reversal of pyrrolidine dithiocarbamate-induced reciprocal regulation of AP-1 and NF-kappaB. Biological chemistry. 2003;384:143–50. doi: 10.1515/BC.2003.015. [DOI] [PubMed] [Google Scholar]
- 27.Hammer M, Mages J, Dietrich H, Servatius A, Howells N, Cato AC, et al. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J Exp Med. 2006;203:15–20. doi: 10.1084/jem.20051753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Locati M, Bonecchi R, Corsi MM. Chemokines and their receptors: roles in specific clinical conditions and measurement in the clinical laboratory. Am J Clin Pathol. 2005;123 (Suppl):S82–95. doi: 10.1309/M6U4B8L6TNAK4G9L. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bischoff DS, Sakamoto T, Ishida K, Makhijani NS, Gruber HE, Yamaguchi DT. CXC receptor knockout mice: characterization of skeletal features and membranous bone healing in the adult mouse. Bone. 2011;48:267–74. doi: 10.1016/j.bone.2010.09.026. [DOI] [PubMed] [Google Scholar]
- 30.Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106:1481–8. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou W, Bar-Shavit Z. Dual modulation of osteoclast differentiation by lipopolysaccharide. J Bone Miner Res. 2002;17:1211–8. doi: 10.1359/jbmr.2002.17.7.1211. [DOI] [PubMed] [Google Scholar]
- 32.Yu H, Sun Y, Haycraft C, Palanisamy V, Kirkwood KL. MKP-1 regulates cytokine mRNA stability through selectively modulation subcellular translocation of AUF1. Cytokine. 2011;56:245–55. doi: 10.1016/j.cyto.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee SK, Huang H, Lee SW, Kim KH, Kim KK, Kim HM, et al. Involvement of iNOS-dependent NO production in the stimulation of osteoclast survival by TNF-alpha. Exp Cell Res. 2004;298:359–68. doi: 10.1016/j.yexcr.2004.04.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Unsorted bone marrow macrophages were stimulated with LPS (100ng/ml) for 24hr. Cell supernants were measured for protein expression using a CXCL1 ELISA kit. Dups1−/− (KO) cells expressed nearly 3-fold more CXCL1 than matched WT cells. Data is plotted as a representative of at least n=3 biological replicates. (***P<0.001).
Gene expression from MCSF, MCSF + RANKL (M+R) and M+R primed cells stimulated with LPS (50ng/ml) for 48- and 96-hours. (A) RANK (Tnfrsf11a) (B) RANKL (Tnfsf11), (C) OPG (Tnfrsf11b). RANKL:OPG ration (D) based upon gene expression. All values were normalized to MCSF only treated controls. Data are written or plotted as a representative of at least n=3 biological replicates. (*P<0.05, ****P<0.001).
Sorted WT and Dusp1−/− cells were primed and blocked with CXCL1 or CXCL2 blocking antibodies prior to stimulation with LPS (as described previously). Cells were noticeably smaller following chemokine blocking. Measurements of OC area reveals that WT and Dusp1−/− cells treated with blocking media did not form OC larger than M-CSF only (Con) treated controls. Area is presented as mean and range. Box indicates 25th to 75th percentile. Data are plotted as a representation of at least n=3 biological replicates.