

Abstract
Macrophages are involved in a number of diseases, such as HIV infection/AIDS, tuberculosis, tumor development and atherosclerosis. Macrophages possess several cell surface receptors (e.g., the mannose receptor, MR) that may serve as drug delivery cellular portals for nanocarriers (NCs). In this study, the optimal structural configuration for cell uptake of mannosylated poly(ethylene glycol)-conjugate type NCs was determined. A series NCs was synthesized to systematically evaluate the effects of the number of mannose units (Man), the PEG carrier size and the mPEG spacer length between adjacent mannose units on NC uptake into MR-expressing J774.E murine macrophage-like cells. Among NCs with 0, 1, 2 or 4 units of mannose, the uptake of (Man)2-NC was the highest, suggesting a trade-off between avidity and NC-MR clustering on the cell surface that sterically hinders endocytosis. This optimal (Man)2-NC configuration was built into subsequent NCs to optimize the other two parameters, PEG carrier size and spacer length. NCs with 0, 5, 12, 20, 30 or 40 kDa linear PEG carriers showed an inverse relationship between PEG size and uptake. The 12 kDa PEG carrier was chosen for investigating the third parameter, the Man-Man distance, since it may represent the best trade off (i.e., tissue penetration vs. systemic clearance) for in vivo macrophage targeting. Three (Man)2-PEG12kDa NCs with different Man-Man distances (39, 56 or 89 Å) were synthesized. The uptake of the NC with the 56 Å distance between mannoses was four- and two-fold higher than NCs with 39 Å and 89 Å distances, respectively. Confocal microscopy confirmed that the optimized (Man)2-PEG12kDa NC with the 56 Å Man-Man distance was internalized via endocytosis consistent with temperature-dependent active uptake. In conclusion, the optimal NC structural parameters for targeting the MR on macrophage-like J774.E cells are (i) a small PEG polymer carrier, (ii) two mannose units per NC and (iii) a 56 Å distance between adjacent mannose units.
Keywords: Mannose receptor, nanocarrier, HIV-1, drug delivery, ligand design
Introduction
Macrophages are primarily involved with innate immune response and tissue homeostasis in mammals. Most tissue macrophages are in the resting state under normal conditions [1]. Upon insults initiated by invading microbes, tissue injuries or tissue stress, they can be activated into different functional phenotypes according to the tissue microenvironment [2-3]. Activated macrophages can be broadly classified into two main groups: classically activated macrophages (or M1) and alternatively activated macrophages (or M2) [4]. The M2 phenotype may be further divided into M2a, b, c subtypes [1]. M1, often regarded as pro-inflammatory, exhibit potent microbicidal properties and promote strong IL-12-mediated Th1 responses, while M2, often regarded as anti-inflammatory, support Th2-associated effector functions and resolution of inflammation and tissue injuries. Activated macrophages remain plastic and respond to the changing environment to modulate their activities [5]. The body response usually results in the clearance of the insult and the return of macrophages to their resting state. However, if these insults persist, pathological conditions can develop. For example, chronic M1 activity can cause collateral damage to normal tissues while chronic M2 activity can lead to tissue fibrosis. Thus, macrophages are involved in various pathological conditions, including obesity, tumor development, atherosclerosis, inflammation and infection with Mycobacterium tuberculosis, Leishmania protozoa and HIV [1, 6-10]. The idea of targeting activated macrophages to convert them to a normal phenotype has been proposed, however in many cases the cell surface biomarkers for abnormal activity and the regulatory effector molecules to revert activated macrophages are yet to be identified [9, 11-13]. By contrast, delivery of drugs to infected macrophages using nanocarriers (NCs) or cell carriers has been extensively explored [14-19].
Liposomes and their variants have been extensively used as drug NCs [20-23]. Other commonly used nanoconstructs include micelles, dendrimers, nanospheres, solid lipid particles and polymeric particles. All of these forms of NCs suffer some shortcomings [24]. Conjugation with polyethylene glycol (PEG), or PEGylation, has been used to overcome some of the shortcomings of non-PEG-based NCs, such as low solubility, low stability, immunogenicity and recognition by the phagocytes of the reticuloendothelial system (RES) [25-27]. However, in the case of drug delivery to macrophages, PEGylation can reduce the delivery efficiency due to reduced recognition by macrophages. Therefore, it is desirable to endow PEG NCs with a macrophage-targeting moiety to compensate for the reduced recognition. The combined feature of PEGylation and targeting in a NC can offer prolonged circulation times, cell type-specific delivery and reduction in dosage &/or dose frequency.
There are a number of receptors that can serve as potential cell surface targets on macrophages including scavenger receptors, formyl peptide receptors, integrins, mannose receptors, galactose receptors and Fc-receptors [28]. Among them, the mannose receptor (MR) (also known as MMR, CD206, or MRC1) has attracted the most interest [29-30]. MR is expressed on most tissue macrophages, dendritic cells, hepatic endothelial cells, as well as on selected lymphatic endothelial cells, kidney mesangial cells, tracheal smooth muscle cells and retinal pigment epithelium [29, 31]. However, it is not significantly expressed on monocytes, the precursor cells to macrophages. MR mainly recognizes terminal mannose, fucose and N-acetylglucosamine sugars of many glycoproteins on various bacteria and viruses, including that on gp120 of HIV-1. MR also recognizes sugar and non-sugar parts of endogenous ligands, including sulfated glycoprotein hormones, collagen, gelatin, lysosomal hydrolases, tissue plasminogen activator and neutrophil myeloperoxidase. There are other mannose-recognizing receptors, including Endo180, DC-SIGN, L-SIGN, and SIGNR [29], whose cell type expression patterns are similar to that of MR.
Previously, we reported nanoparticles with surface-displayed mannose were taken up into MR-expressing J774.E cells consistent with active receptor-mediated uptake that was specific and inhibitable [32]. Maximum NC association was attained with 9% mannoside-terminated PEG chains, increasing uptake more than 3-fold compared to non-targeted NCs. However, in view of the lack of information regarding the optimal ligand display pattern properties (i.e, configuration) of a mannosylated PEG NC for targeting the macrophage MR, the present study was undertaken to systematically investigate the effect of the number of mannose units per NC, the distance between the mannose units and the PEG carrier size on NC uptake. A novel NC design was employed to precisely control the geometry of the mannose-targeting moiety on NC. It is found that the optimal NC uptake occurred for a NC displaying two mannose units that are spaced 56 Å apart on a small sized PEG carrier. The polymeric NCs described in this report can either be used as stand alone NCs carrying one or more copies of drug/cargo or they can be used as optimized targeting ligands on other carriers such as nanoparticles.
Materials and Methods
Materials
NovaSyn Sieber resin was purchased from Millipore (Bellerica, MA), Fmoc-γ-Abu-OH, Fmoc-Ser-OH, Fmoc-Ser(OtBu)-OH, Fmoc-Cys(Trt)-OH from Chem-impex (Wood Dale, IL), mPEGx-maleimide (x=5kDa, 12kDa, 20kDa, 30kDa, 40kDa) from NOF (Tokyo, Japan), α-D-Mannose pentaacetate, boron trifluoride diethyl etherate, N,N-Diisopropylethylamine (DIPEA), NO production kit and NBT reduction kit from Sigma Aldrich (Saint Louis, MO), Trifluoroacetic acid (TFA) from Fisher Chemical (Fair Lawn, NJ). Fmoc-amino-dPEG6-acid, Fmoc-amino-dPEG12-acid and Fmoc-amino-dPEG20-acid were purchased from Quanta Biodesign (Powell, OH). Tetramethylrhodamine B dextran (10,000 M.W.) and 4’,6-diamidino-2-phenylindole, dihydrochloride (DAPI) were purchased from Life Technologies (Grand Island, NY).
Synthesis and Characterization of Nanocarriers
FITC-Gaba-PEG12-Ser(Man)-Gaba-Gaba-Cys-amide, FITC-Gaba-Ser(Man)-PEG12-Ser(Man)-Gaba-Gaba-Cys-amide, FITC-GABA-Ser(Man)-PEG12-Ser(Man)-PEG12-Ser(Man)-Gaba-Gaba-Cys-amide, and FITC-Gaba-Ser(Man)-PEG12-Ser(Man)-PEG12-Ser(Man)-PEG12-Ser(Man)-Gaba-Gaba-Cys-amide (Fig. 1) were synthesized following standard solid-supported peptide synthetic protocols using a Nautilus 2400 Automated Synthesizer. Fmoc-L-Ser-α-D-[Man(OAc)4]-OH was synthesized as previously described [33] NovaSynTG Sieber resin was used in the amino acid coupling with molar ratio 1:4:4:8 (resin: amino acid: coupling reagent (HATU, HOAt): base (DIPEA)) for four hrs. Fmoc-L-Ser-α-D-[Man(OAc)4]-OH and Fmoc-amino-PEG12 acid were conjugated to the peptidic core in the same way as the regular amino acid coupling in molar ratio of 1:2:2:4 (resin : amino acid : coupling reagents (HATU, HOAt): base (DIPEA)) for 12 hrs. Fmoc group was removed by 20% piperidine in NMP. MALDI TOF/MS and analytical reverse phase high-performance liquid chromatography (RP-HPLC) were performed after each amino acid coupling to confirm the molecular weight and purity. Five equivalents of fluorescein isothiocyanate (FITC) was dissolved in NMP and reacted to the N terminal of the peptide on solid support in the presence of 10 equivalents of DIPEA for 12 hours. The acetyl protecting groups of the mannose moieties were removed by sodium methoxide in anhydrous methanol on the solid support. Peptides were cleaved from the resin by TFA containing scavangers reagents and diluted with DCM (85% DCM, 2% DODT, 1% TIS, 12% TFA) for 1.5 hrs. The cleaved peptides were then precipitated with cold ether-hexane mixture (50%-50% v/v). Semi-preparative RP-HPLC was used to purify the NCs. The fractions were analyzed (HPLC), pooled and lyophilized providing high purity samples (purity >90%). PEGylation was performed in the liquid phase by reacting 5 equivalent mPEGx-maleimide (x= 5, 12, 20, 30, or 40 kDa) to the cysteine moiety of the NCs in 1 mL of phosphate-buffered saline (PBS, pH 7.4). The reaction was stirred overnight at room temperature. Sephadex G-50 or Sephadex G-15 column was used to purify the PEG NCs. Aliquots of the purified PEG NCs were collected and characterized using MALDI-TOF/MS. The collected PEG NCs were then lyophilized. NCs possessing three different Man-Man distances (39, 56 and 89 Å, cf. Fig. 1) were synthesized. The corresponding PEG amino acids were Fmoc-PEG6-OH, Fmoc-PEG12-OH and Fmoc-PEG20-OH respectively. The mannosylated and labeled building blocks were fully characterized and the PEG carriers were obtained from a commercial source. The molecular weights of the PEG carriers were confirmed by MALDI-TOF mass spectrometry in our laboratory. After conjugation, the products were purified and structural integrity was confirmed by MALDI-TOF MS. Monitoring the differences of the centroid of the m/z signals (non-conjugated vs. conjugated) is an accepted method in bioconjugate chemistry for following conjugation reactions of polydisperse precursors. Since the conjugation steps have always been carried out using a 1:1 stoichiometry, the loading efficiency is determined by the chemical reaction, thiol-maleimide coupling. The MALDI-TOF MS spectra provided further verification for the conjugate structures.
Figure 1.

General structure of mannosylated PEG nanocarriers (NCs). The bracketed section depicts the Man-Man spacer distance, which consists of a short linear monodisperse PEG chain (mPEG) of 6, 12 and 20 PEG units corresponding to 25, 46, or 75 Å (extended lengths) and a mannose attached to Ser resulting in Man-Man distances of 39, 56 or 89 Å. The PEG carrier in the NC is a much larger polydisperse linear PEG (5, 12, 20 or 30 kDa).
Cell Uptake of NC
J774.E cells were a gift from Dr. Philip D. Stahl, Washington University (St. Louis, WA). The cells were cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS), 1 X penicillin/streptomycin at 37°C in 5% CO2/95% air atmosphere. In uptake experiments, J774.E cells were plated at 2.5 × 104 cells/well to poly-D-Lysine-coated 24-well plate to achieve better macrophage differentiation. The cells were incubated with test and control NCs at specified concentrations in culture condition for 1 hr at either 37 or 4 °C. The attached cells were then washed with phosphate buffered saline (PBS) once, exposed to acid wash solution (0.5 M sodium chloride and 0.2 M acetic acid, pH 2.5) at room temperature to remove cell surface associated and non-internalized NCs. The cells were washed three times with PBS followed by fluorescence microscopy to observe cellular uptake. After microscopy, cells were lysed with 1 N NaOH overnight and neutralized with 1 N HCl the next day. The fluorescence in the neutralized cell lysate was quantified on a fluorescence microplate reader at Ex485/Em517 nm. The fluorescence intensity in arbitrary units was normalized against the cell lysate protein amount in each well as determined by the Bradford protein assay (using the reagent from Pierce). Normalization minimizes well-to-well variation in uptake caused by the differences in cell mass and allows for the statistical pooling of different experiments with slightly different plating densities. Free FITC was tested as a control of NC uptake and showed no detectable uptake due to its negative charge (not shown).
In the uptake inhibition experiments, the cells were pre-incubated, or not pre-incubated, for 40 minutes with mannan, a yeast-derived natural MR ligand that is a mannose polymer in structure, followed by incubation for 1 hr with NCs in the presence or absence of mannan, respectively. Mannan was used at a concentration of 5 mg/ml, which was determined by the MTT cytotoxicity assay to be non-toxic (data not shown).
Fluorescence Microscopy and Confocal Microscopy
For fluorescent microscopy, J774.E cells were seeded in 24-well plates while for confocal microscopy the cells were seeded in Laboratory-Tek II 4-chamber cover glasses slides. When the cells reached ~ 80% confluence they were incubated with 80 nM NCs in 20 mM HEPES-buffered Hanks’ balanced saline solution (HBSS) for 1 hour at 37°C. The incubation media included a general fluid endocytosis marker tetramethylrhodamine B dextran (10,000 MW) at 50 μM and the nuclear dye DAPI at 5 μg/ml. The cells were then washed with PBS five times. NC uptake into live J774.E cells was examined under an inverted Olympus fluorescence microscope (model IX71S1-F3) at the magnification of 40x objective. Confocal microscopy was performed on a Leica TCS SP2 Spectral confocal microscope using the XYZ mode. A sequential image acquisition mode was used to avoid possible partial light spillover from one color to another. The middle cell sections of selected Z-stacks are shown in Fig. 8. Quantification of green fluorescence within individual cells and in a middle section of an acquired Z-stack was performed using Leica’s software, LAS AF, version 2.4.1.
Figure 8.
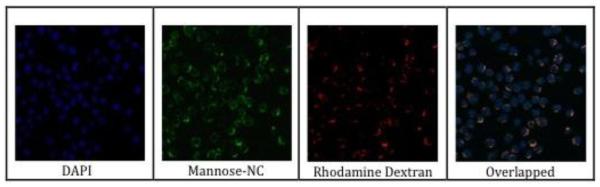
Colocalization of FITC-labeled, optimized NC with the fluid phase endocytosis marker, tetramethylrhodamine B dextran, in J774.E cells. The images of middle sections of cells were acquired using the same procedure as used in the confocal microscopy-based uptake experiments (Fig. 5 – 7), except that a nuclear dye and a fluid phase endocytosis marker were also used. DAPI: image aquired in the blue channel. Mannose-NC: image acquired in the green channel. Rhodamine Dextran: image acquired in the red channel. Overlapped: merged image of the blue, green and red images.
Isolation of rat peritoneal resting macrophages and their use in NO production and NBT reduction assays
Rats were allowed to acclimate in an animal facility at Rutgers, the State University of NJ for a minimum of 5 days prior to use. The animals were euthanized using CO2 asphyxiation. Ice-cold HBSS perfusion medium (0.5 mM EGTA added, pH 7.2, Ca2+ Mg2+ free) was injected into the peritoneal cavity. The peritoneum of the rat was massaged for 30 seconds and the solution inside the abdominal cavity containing peritoneal macrophages was withdrawn. Injection, massage and withdrawl were repeated twice and the collected macrophages were pooled, washed three times with HBSS using centrifugation at 300 × g/4 °C for 8 minutes. The settled macrophages were suspended and seeded in 96-well plates at 1 × 105 cells/well in RPMI medium containing 10% FBS. After 4 hrs of culture, cells were used for NO production and NBT reduction assays using the respective kits from Sigma Aldrich.
Statistics analysis
One-way ANOVA followed by Dunnett’s multiple comparison test was used for multiple group comparison. In the case of two-group comparisons (Fig. 7), the method was equivalent to t-test and the t-test was used. Statistical significance is indicated as P < 0.05.
Figure 7.
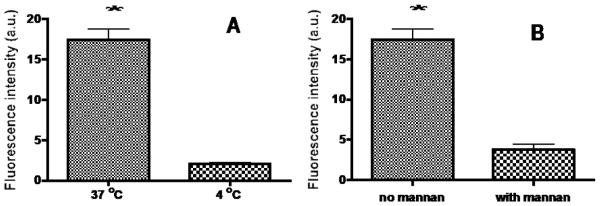
(A) Temperature-dependent uptake into J774.E cells. The cells were incubated with 2 mannose units/12 kDa PEG-NC for 1 hr at 37 and 4 °C, respectively, followed by PBS wash. The cells were immediated examined by confocal microscopy. Fluorescence intensity of middle sections of randomly selected cells was quantified using the confocal microscope’s software. (B) MR-mediated uptake into J774.E cells in the presence of 5 mg/ml of the inhibitor mannan. Values are reported as mean +/− s.d. of three independent experiments. * p < 0.001.
Results
Nanocarrier (NC) synthesis and characterization
The general structure of the mannosylated PEG NC depicted in Fig.1 was designed to optimize the NC for targeting macrophages through the MR. All individual NCs as well as their precursor intermediates were successfully synthesized and characterized. As representative examples, Fig. 2 shows the structure and MALDI-TOF/MS spectrum of the NC with 4 mannose units but without a PEG carrier moiety while Fig. 3 shows the polydispersity patterns of five NCs with 2 mannose units and different sizes of PEG carriers. Comparison of Figures 2 and 3 reveals that the polydispersity of PEG NCs is due to the intrinsic polydispersity of their PEG carrier not the mPEG spacer.
Figure 2.
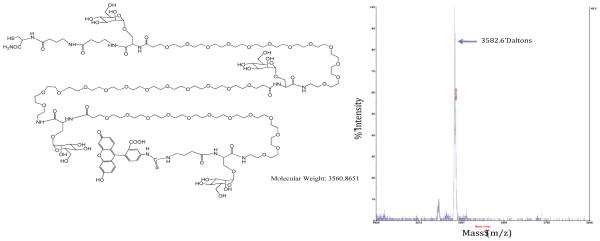
The structure and MALDI-TOF/MS spectrum of a NC displaying 4 mannose units (n = 3 and Man-Man distance of 56 Å) showing the correct molecular ion as [M+Na].
Figure 3.
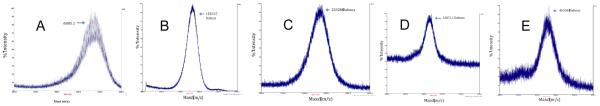
MALDI-TOF MS Spectra of PEG5kDa-NC (A), PEG12kDa-NC (B), PEG20kDa-NC (C) and PEG30kDa-NC (D) and PEG40kDa-NC (E), respectively. All NCs display 2 mannose units and with a Man-Man distance of 56 Å.
Determination of Optimal Mannose Copy Number
The systematic NC optimization followed a sequential testing strategy with the mannose copy number per NC the first parameter examined. NCs with different mannose unit numbers, a fixed Man-Man distance (cf, Fig. 1) of 56 Å and without a PEG carrier were evaluated. The fluorescent FITC moiety served as a label for quantification of NC cellular uptake. NCs contained 0 (control), 1, 2, 3, or 4-units of mannose (cf. Fig. 1 for the general structure and Fig. 2 for the structure of a 4-unit mannose NC).
NC cellular uptake was investigated using murine MR-expressing J774.E cells since human macrophage-like cell lines do not express MR [34]. Although human monocyte-derived macrophages express MR, they were not used because they are intrinsically variable often leading to conflicting results [35]. Preparations of monocyte-derived macrophages can vary in many biological aspects, such as the level of T cell contamination, macrophage phenotye, the ability to support HIV replication and HIV co-receptor expression [35]. The high variability was also confirmed in the current study (not shown). Pilot experiments determined that a NC concentration of 80 nM resulted in measurable intracellular fluorescence. The cells in triplicate wells were incubated with 80 nM of each of the NCs for 1 hr, stripped of cell surface-bound NC with an acid buffer wash and washed with PBS. The FITC-fluorescence intensity of the cell lysate was read and normalized based on protein amount in the lysate to minimize well-to-well variation in cell mass. Fig. 4 shows the combined results of three independent experiments. Uptake of all mannosylated NCs was higher than that of non-mannosylated control NCs, suggesting MR-dependent uptake. As the mannose unit number increased from 1 to 2, the uptake increased. However, as the mannose unit number increased further from 2 to 3 and from 3 to 4, uptake reduced. Uptake of 2-unit mannose NC showed the highest uptake (taken as 100%), followed by the 1-unit (50%), 3-unit (40.6%) and 4-unit (23.4%) NCs (Fig. 4). Fluorescence and confocal microscopy were carried out. The fluorescence intensity of cells incubated with different mannose unit numbers of the testing NCs followed the same pattern as the uptake result (microscopic images not shown). Therefore, it was determined that the optimal mannose unit number for MR targeting was two.
Figure 4.
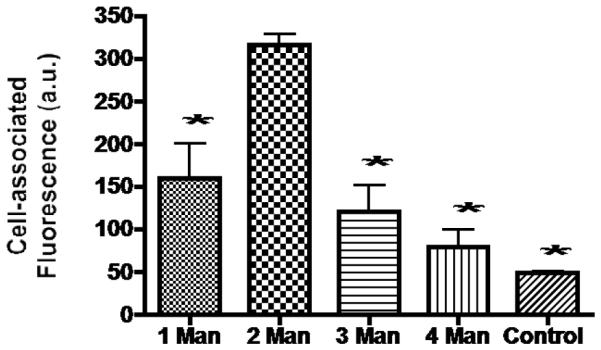
Effect of the number of mannose units per NC on NC uptake into J774.E cells. Cells were incubated with each of the five FITC-labeled NCs for 1 hr then sequentially washed. The cells were lysed with 1 N NaOH and neutralized with 1 N HCl. The fluorescence was measured and the readings were normalized by the amount of protein lysates. Data are reported as the mean +/− s.d. of three independent experiments. *: p < 0.05 = difference between the uptake level of the 2 unit mannose NC and that of a NC with a different mannose unit number. Control: non-mannosylated NC (n = 0).
Determination of Optimal Size of the PEG Carrier
Having established that the optimal mannose unit number per NC was two, the second parameter, the PEG carrier size was evaluated. The cellular uptake of NCs with two units of mannose but different PEG carrier sizes (5, 12, 20, 30, and 40 kDa, respectively) (cf. Fig.1 for NC general structure) was investigated. Quantification of cellular fluorescence was determined by confocal microscopy. This method of quantification represents internalized NCs almost exclusively since there was little cell surface fluorescence. Fluorescence in the middle sections in XYZ mode of randomly picked up cells was quantitatively determined using the software built into the confocal microscope. Fig. 5 shows that there is an inverse relationship between the size of the PEG carrier and the level of NC internalization. The highest was the NC with the smallest 5 kDa PEG moiety (100%), followed by NCs with 12 kDa PEG (41.7%), 20 kDa (27.7%), 30 kDa (26.4%) and 40 kDa (19.7%) PEG moiety, respectively.
Figure 5.
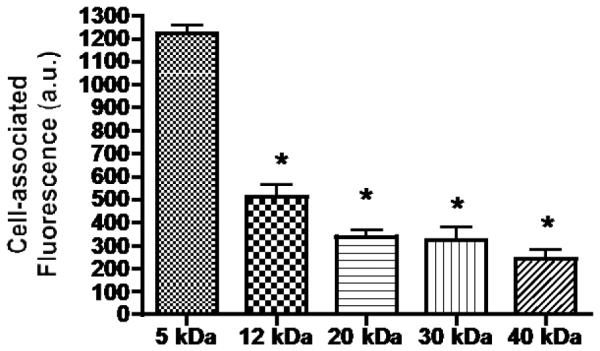
Effect of PEG carrier size on NC uptake into J774.E cells. Cells were incubated with each of the five FITC-labeled, two copy mannose NCs with difference sized PEG carriers for 1 hr and PBS-washed. The cells were examined under a confocal microscope. Intracellular fluorescence in middle sections of randomly picked up cells was quantified using confocal microscopy. *: p < 0.05 is for the difference between the uptake of the 5 kDa PEG NC and the other NCs shown.
Determination of optimal length between two adjacent mannose units (Man-Man distance)
The third parameter, the Man-Man spacer distance, was evaluated using NCs with two mannose units and a 12 kDa PEG carrier but a variable Man-Man distance of 39, 56, or 89 Å. Based on a structure model of the MR [36] and the 3D structure of the mannose-binding protein A whose corresponding structure resembles the dimeric CRD4 domain of MR [37], molecular docking was used to estimate the optimal Man-Man distance (~56 Å) (data not shown; Discovery Studio 3.1 (Accelrys Inc., San Diego, CA). The 12 kDa PEG size was chosen considering the trade-off between ability to penetrate tissues and reach macrophages (the smaller the better) and the half-life in the circulation (the higher the better), as described in detail in the Discussion. Uptake of the three NCs into J774.E cells with different Man-Man distances was evaluated in the same way as previously described. Fig. 6 shows that NC internalization with the Man-Man distance of 56 Å (taken as 100%) is higher than that of the NC with a shorter (39 Å, at 21.3%) or longer spacer distance (89 Å, at 58.6%).
Figure 6.
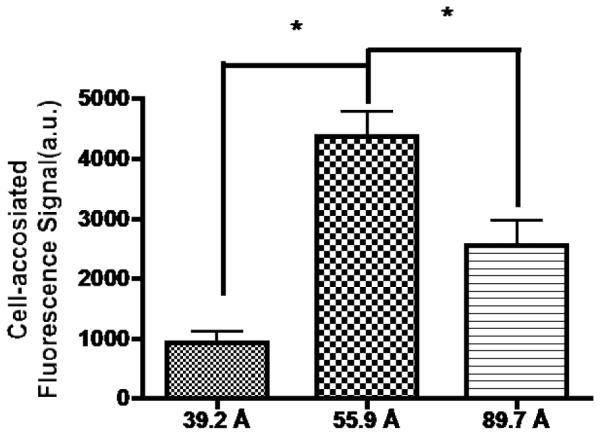
Effect of Man-Man distance on NC uptake into J774.E cells. Cells were incubated with 80 nM of each of the three FITC-labeled NCs with different Man-Man distances for 1 hr and PBS-washed. The cells were examined under a confocal microscope. Intracellular fluorescence in middle sections of randomly selected cells was quantified using the software accompanying the confocal microscope. *: p < 0.05 is for the difference between the uptake level of the NC with a 56 Å Man-Man distance and that of a NC with a different Man-Man distance.
Active and MR-mediated uptake of the optimized mannosylated and PEG NC
The optimized NC with two mannose units spaced 56 Å apart and a 12 kDa PEG carrier was further studied for MR-targeting. Internalization in J774.E cells of the optimized NC was in a temperature- and MR-dependent manner as shown by quantification of intracellular fluorescence of confocal microscopic images (Fig. 7A & B; images not shown). The uptake at 37 °C was 8-fold higher than the uptake at 4 °C while it was reduced 5-fold in the presence of the mannan ligand that competed with the NC for MR. The results suggest that the internalization of the mannosylated and PEG NC is active consistent with a receptor- and MR-mediated mechanism. A similar pattern of uptake was seen in separate uptake experiments, in which the uptake was quantified by intracellular fluorescence in cell lysates (not shown). It is known that macrophages internalize MR-bound, non-opsinized ligand by endocytosis. To confirm that the optimized NC enters cells by endocytosis, a confocal study using fluid phase endocytosis marker (Tetramethylrhodamine B dextran) was carried out (Fig. 8). The result showed that FITC-labeled, optimized NC was colocalized with the marker, confirming that the NC was taken in by endocytosis.
Optimized NCs do not activate rat peritoneal resting macrophages in vitro
The optimized mannosylated PEG NC, under uptake conditions, was not cytotoxic to J774.E cells based on the MTT cell viability assay [38] (data not shown). To ascertain if the engagement of MR by NC would activate macrophages, rat peritoneal resting macrophages were collected and tested for nitric oxide production (NO, a signaling molecule in macrophage activation) and nitro blue tetrazolium (NBT) reduction (measuring reactive oxygen intermediates (ROI), the phenotype of the classically activated macrophages). Fig. 9 shows that, under the uptake conditions utilized in this study, the optimized NC is negative for both activities suggesting that the NC is unlikely to activate macrophages in vivo.
Figure 9.
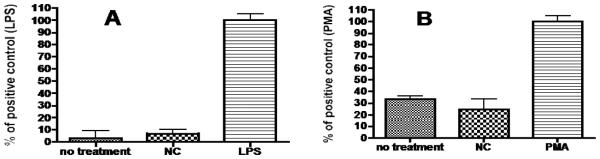
Optimized NC does not trigger (A) NO production or (B) reactive oxygen intermediate (ROI) production. Triplicates of rat peritoneal resting macrophages were plated at 105/well in 96-well plates and the cells were left in culture for 4 hrs for attachment to take place. The cells were then treated with the optimized 2-unit mannose/12 kDa PEG-NC (NC), NO positive control lipopolysaccharides (LPS) at 500 ng/ml, or ROI positive control PMA at 500 ng/ml. The group of cells with no treatment was used as the negative control. The cells in wells were then assayed for NO production using the Sigma NO kit or for ROI production using Sigma NBT kit. Data is expressed as % of positive control. Each graph represents the mean +/− s.d. of three independent experiments. The difference between positive control and NC is statistically significant (p < 0.001) for both assays.
Discussion
In this study, the architectural features of targeting ligand display were optimized for polymeric conjugate-type NCs. The ligand used in this investigation was mannose and three parameters were serially optimized including mannose ligand copy number per NC (five levels), polymeric carrier (six levels) and the space between two adjacent mannose ligands (three levels). The blinded, grid-wise multi-parameter statistical design (e.g., usually three level Box-Benhken) commonly used for drug and chemical formulation [39-40] could not be used because of the time and cost of synthesizing the vast number of required NCs (a total of 90 excluding the controls). However, the discrete nature of the structural descriptors (i.e., Man unit number, the length of the mPEG spacer) and the commercial availability of the building blocks (mPEG, polydisperse PEG) alternatively led a serial sequential design strategy. In this strategy, one parameter is tested at a time. In the testing of the first parameter, the other untested parameters are either absent or at an optimal level by estimation that must be confirmed later. The first parameter investigated was the mannose ligand unit number per NC, the PEG carrier was absent and the optimal Man-Man distance was estimated as described before. The calculated Man-Man distance was confirmed upon evaluation in macrophage-like cells (cf. Fig. 6). It was observed that the optimal configuration of mannosylated PEG NCs for actively targeting macrophages through the MR possesses a small PEG carrier (e.g., 5 kDa) displaying 2-units of mannose spaced 56 Å apart. Overall, fourteen NCs were synthesized and tested.
The optimal mannose copy number is governed by the interaction between the mannose ligand and the MR. Compared to protein-protein interactions, the carbohydrate-protein interaction is weak with the monosaccharide-protein binding Kd in the mM to μM range [41] with low specificity [42]. A survey of the known structures of carbohydrate-lectin complexes suggests that the underlying mechanism is a shallow binding pocket on lectin proteins and a limited variety of chemical groups on the sugars [43]. To achieve high affinity binding to carbohydrates, nature has evolved a strategy of multivalency binding (avidity) for lectins, enzymes, receptors and other carbohydrate-binding proteins [44-45]. The mammalian MR family of receptors has four members: MR, Endo180, DEC-205 and phospholipase A2 receptor. Although each member of the family has 8 or 10 C-type lectin-like domains (CTLDs, previously known as carbohydrate recognition domains (CRDs)), only one domain exhibits measurable binding affinity for mannose [36]. In the MR this domain is CTLD4. By contrast, each of the carbohydrate-binding domains of lectins binds to a cognate monocarbohydrate with measurable binding affinity [46]. In addition, in contrast to oligomeric nature of lectins in the plasma membrane [46-47], the MR exists mostly as a monomer [47-48]. This suggests that the 2 unit NC can form a 2-dimensional lattice with multiple MR molecules on the cell surface (clustering). The two opposing effects of NC internalization may explain why two mannose units are required for optimal receptor interactions. As the unit number increases from 0 to 1 and from 1 to 2, the avidity increases resulting in increased binding of the NC to the MR resulting in internalization while the clustering is negligible. However, when there are more than 2-copies of mannose on a single NC it clusters two or more MR on the cell surface reducing endocytosis of the NC [49].
The microscopic images (Fig. 8) as well as the active uptake of the optimized NC suggest that the mechanism of internalization is endocytosis. This is consistent with the fact that many natural ligands for MR are internalized by macrophages using endocytosis [29, 50]. Macrophages also avidly phagocytose opsinized particles (pathogens and NCs) through FcRs and CRs receptors [51]. Although the MR can mediate phagocytosis in an opsin-independent manner [31, 51], the phagocytosis capability is likely to be very low [52]. This suggests that in vivo the optimized NC will likely enter macrophages by endocytosis, not by phagocytosis. If the pharmacological target is in the vesicular compartment (endosomes and lysosomes) pathogens that cause tuberculosis or leishmaniasis can be targeted and the optimized NC is a suitable delivery vehicle. However, if the pharmacological target is in the nucleus or cytosol of the cell, as are most of the HIV drug targets, the optimized NC may need to incorporate an endosomal escape mechanism [53].
This study found an inverse relationship between the PEG carrier size in the NC and NC uptake in vitro, which is not surprising. However, to make a decision on PEG carrier size for in vivo delivery, other aspects of drug delivery must also be considered. They include non-brain endothelial permeability, half-life in the circulation, tissue interstices penetration and biosafety of the PEG NC. By definition, macrophages reside in tissue interstices. An IV-administered NC must pass endothelium to reach this space. Studies in rabbits [54] and rats [55] have showed that the interstitial concentrations of the two most abundant plasma proteins (albumin, m.w. 67 kDa, and transferrin, m.w. 80 kDa) are about 40% of that in plasma. In humans, transcapillary albumin escape rate (TERalb) is a practical measure of colloid protein and particle permeability through systemic capillary endothelium [56]. Therefore, albumin can be used as a proxy for the size and magnitude of a circulating PEG that can leak into the tissue interstices; that is, the apparent PEG carrier size should be no larger than 67 kDa for leakage into tissue interstices. Since the apparent size of linear PEG is 3 to 10 times its molecular weight (it behaves 3 to 10 times bigger in size than a globular protein with a comparable molecular weight [57]), the size cut-off for linear PEG to enter tissue interstices is estimated to be about 12 kDa. At this size, renal clearance is significant and half-life is not very long [57]. From a biosafety perspective, the PEG size should be above the ~ 0.4 kDa threshold for acute PEG toxicity due to the catabolic products from oxidative degradation in the body [58], but not much higher than 30 kDa. Above 30 kDa, PEG conjugates are neither significantly degraded inside cells, nor exocytosed out of the cells and end up accumulating in lysosomes [59-60]. This is a risk similar to a “lysosomal storage disease” [61], which can be prevented by using degradable PEG carriers to facilitate elimination [62]. In this study, the optimized NC does not activate rat peritoneal resting macrophages (Fig. 9). This check is necessary because the MR can initiate signaling and participate in macrophage activation [63-64]. Taken together, the PEG size in an IV-administered, macrophage-targeted NC should be close to 12 kDa and not much larger. Alternatively, a macrophage-targeted and PEG NC may be administered by other routes and the PEG carrier size in the NC can be smaller or larger than 12 kDa, depending on the route. The alternative routes include local injection to target the regional lymph nodes, vaginal and rectal topical application to target the mucosa and cell-based delivery [65] to target systemic organs/tissues or lymph nodes. In the latter strategy, NCs can be packed in ghost red blood cells or phagocytosed by monocytes in vivo or ex vivo.
The current study found that the optimal space between two neighboring mannose units in NC is 56 Å. This length is an approximation based both on molecular modeling of the related Endo180 X-ray structure and the availability of mPEG building blocks to synthesize NC with three different testing Man-Man distances (i.e., short PEG chains, 25, 46, or 75 Å in length). At present, a high-resolution structure of the entire MR molecule (or the extracellular portion of MR) has not been reported. However, a 2.3 Å-resolution structure of a dimeric CTLD4 of MR has been reported [37] that resembles the corresponding domain in the high-resolution structure of rat mannose-binding protein A. Therefore, this structure was used in molecular modeling. In the docking study, two CTLD4 domains consist of two mannose binding sites (data not shown). Compared to a shorter spacer of 39 Å and a longer spacer of 89 Å, the 56 Å distance proved to be optimal (Fig. 6). It should be emphasized that the spacer lengths are hypothetical values since all of the building blocks constituting the ligand (monodisperse PEG spacer, Ser-Man units) are highly flexible. Figure 10 shows two distinct conformations, a fully extended and a folded shape of the ligand. Man-Man distances were measured as interatomic distances between the oxygen atoms of the (axial) 2-OH groups of the mannose units, since the chirality of C-2 atom differentiates mannose from the rest of the ubiquitous monosacharides (e.g. glucose, galactose).
Figure 10.
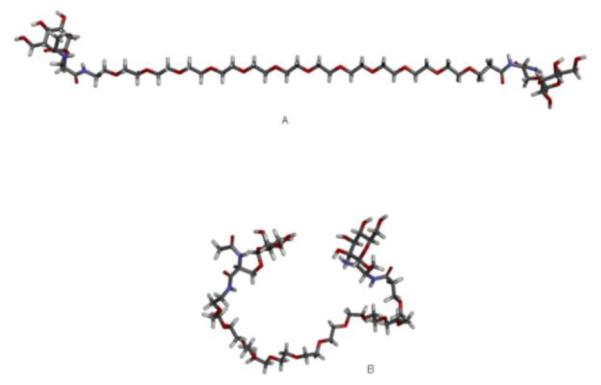
Extended and folded conformations of the Ser(Man)-mPEG12-Ser(Man) ligand on the optimized NC. The Man-Man distances can vary between 56 Å (Fig. 10A) in a fully extended conformation and 8 Å (Fig. 10B) in a folded conformation.
The optimum spacing of 56 Å may also be explained in terms of thermodynamics. Avidity is the result of a greater binding free energy (ΔG) than the sum of individual monovalent binding free energy. The length of a flexible spacer usually plays a pivotal role in determining avidity, as it exerts opposite effects on the two components of the ΔG, namely the change in enthalpy and the change in entropy [45]. The flexibility of a spacer relieves the strains of slight mismatches, thus minimizing a potential penalty on enthalpy change. However, at the same time a flexible spacer can cost a lot in entropy. For example, upon the binding of a bivalent NP with two targeting ligands separated by a flexible spacer made of three mPEG units, the conformational entropy cost of the linker can be as high as 10 kcal/mol [45]. This entropy cost is underappreciated. One can find examples of nanoparticles in the literature that designed flexible spacers that are too long, which are almost guaranteed to fail for entropic reasons [45]. All three of spacers reported in the current studies are flexible, made of mPEG6, mPEG12, mPEG20 with 39, 56 and 89 Å in length, respectively (Fig. 1 and 10). The respective conformational entropy costs for the three spacers would be roughly 10, 20 and 30 kcal/mol. Since the conformational entropy cost of the 39 Å spacer is lower than that of the 56 Å spacer but the uptake level is only 21.3% of that of the later (Fig. 6 and 7), the conformational entropy cost is not the reason for the reduced uptake of the 39 Å spacer NC. This suggests that the 39 Å spacer is too short for a two-unit mannose NC to engage two CTLD4 domains extended from two closely aligned MR molecules on a J774.E cell as the docking exercise demonstrates. On the other hand, the uptake of the 89 Å spacer NC is at 58.6% of that of the 56 Å spacer NC, suggesting that the extra conformational entropy cost of about 10 kcal/mole was significant but not overly damaging to the total binding energy.
Conclusions
To our knowledge, the precise configuration of a mannosylated PEG NC for targeting macrophages via the mannose receptor has not been determined. In the current study, systematic optimization was carried out; in which a series of NCs with a variety of PEG carrier sizes, different numbers of mannose ligands separated by three Man-Man distances were synthesized and tested. It was found that the optimized NC possesses a small PEG carrier and two mannose units that are 56 Å apart in order to penetrate cells most efficiently. The in vitro optimized parameters thus provide guidelines for designing mannosylated PEG NCs for in vivo drug delivery to macrophages.
Acknowledgement
This research is supported by the the National Institute of Allergy and Infectious Diseases (Award AI051214) and the National Institutes of Health CounterACT Program through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (Award U54AR055073).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cassol E, Cassetta L, Alfano M, Poli G. Macrophage polarization and HIV-1 infection. J. Leukoc. Biol. 2010;87:599–608. doi: 10.1189/jlb.1009673. [DOI] [PubMed] [Google Scholar]
- 2.Gordon S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 4.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front. Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 5.Laskin DL, Vasanthi RS, Carol RG, Laskin JD. Macrophage and tissue injury: agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 2011;51:267–288. doi: 10.1146/annurev.pharmtox.010909.105812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benoit M, Dennues B, Mege JL. Macrophage polarization in bacterial infections. J. Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 7.Shimada K. Immune system and antherosclerotic disease – heterogeneity of leukocyte subsets participating in the pathogenesis of antherosclerosis. Circ. J. 2009;73:994–1001. doi: 10.1253/circj.cj-09-0277. [DOI] [PubMed] [Google Scholar]
- 8.Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistence in human obesity. Curr. Pharm. Des. 2008;14:1225–1230. doi: 10.2174/138161208784246153. [DOI] [PubMed] [Google Scholar]
- 9.Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. Macrophage polarization in tumour progression. Semin. Cancer Biol. 2008;18:349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Herbein G, Varin A. The macrophage in HIV-1 infection: from activation to deactivation? Retrovirology. 2010;7:33. doi: 10.1186/1742-4690-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson HM, Barker RN, Erwig LP. Macrophages: promising targets for the treatment of atherosclerosis. Curr. Vasc. Pharmacol. 2009;7:234–243. doi: 10.2174/157016109787455635. [DOI] [PubMed] [Google Scholar]
- 12.Thepen T, Huhn M, Melmer G, Tur MK, Barth S. Fcgamma receptor 1 (CD64), a target beyond cancer. Curr. Pharm. Des. 2009;15:2712–2718. doi: 10.2174/138161209788923967. [DOI] [PubMed] [Google Scholar]
- 13.Vielhauer V, Kulkarni O, Reichel CA, Anders HJ. Targeting the recruitment of monocytes and macrophages in renal disease. Semin. Nephrol. 2010;30:318–333. doi: 10.1016/j.semnephrol.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Brynskikh AM, Zhao Y, Mosley RL, Li S, Boska MD, Klyachko NL, Kabanov AV, Gendelman HE, Batrakova EV. Macrophage delivery of therapeutic nanozymes in a murine model of Parkinson’s disease. Nanomedicine (Lond.) 2010;5:379–396. doi: 10.2217/nnm.10.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dou H, Grotepas CB, McMillan JM, Destache CJ, Chaubal M, Werling J, Kipp J, Rabinow B, Gendelman HE. Macrophage delivery of nanoformulated antiretroviral drugs to the brain in a murine model of neutoAIDS. J. Immunol. 2009;183:661–669. doi: 10.4049/jimmunol.0900274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magnani M, Rossi L, Fraternale A, Silvotti L, Quintavalla F, Piedimonte G, Matteucci D, Baldinotti F, BEndinelli M. Feline immunodeficiency virus infection of macrophages: in vitro and in vivo inhibition by dideoxycytidine-5’-triphosphate-loaded erythrocytes. AIDS Res. Hum. Retroviruses. 1994;10:1179–1186. doi: 10.1089/aid.1994.10.1179. [DOI] [PubMed] [Google Scholar]
- 17.Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E, Ostroff GR, Czech MP. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009:1180–1184. doi: 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rossi L, Serafini S, Pierige F, Antonelli A, Cerasi A, Fraternale A, Chiarantini L, Magnani M. Erythrocyte-based drug delivery. Expert Opin. Drug Deliv. 2005:311–322. doi: 10.1517/17425247.2.2.311. [DOI] [PubMed] [Google Scholar]
- 19.Pierige F, Serafini S, Rossi L, Magnani M. Cell-based drug delivery. Adv. Drug Deliv. Rev. 2008;60:286–295. doi: 10.1016/j.addr.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 20.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Descov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 21.Pasquetto MV, Vecchia L, Covini D, Digilio R, Scotti C. Targeted drug delivery using immunoconjugates: principles and applications. J. Immunother. 2011;34:611–628. doi: 10.1097/CJI.0b013e318234ecf5. [DOI] [PubMed] [Google Scholar]
- 22.Mufamadi MS, Pillay V, Choonara YE, Du Toit LC, Modi G, Naidoo D, Ndesendo VM. A review on composite liposomal technologies for specialized drug delivery. J. Drug Deliv. 2011 doi: 10.1155/2011/939851. 2011. article ID: 939851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huynh NT, Passirani C, Saulnier P, Benoit JP. Lipid nanocapsules: a new platform for nanomedicine. Int. Pharm. 2009;379:201–209. doi: 10.1016/j.ijpharm.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 24.Rawat M, Singn D, Saraf S, Saraf S. Nanocarriers: promising vehicle for bioactive drugs. Biol. Pharm. Bull. 2006;29:1790–1798. doi: 10.1248/bpb.29.1790. [DOI] [PubMed] [Google Scholar]
- 25.Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs. 2008;22:315–329. doi: 10.2165/00063030-200822050-00004. [DOI] [PubMed] [Google Scholar]
- 26.Hamidi M, Azadi A, Rafiei P. Pharmacokinetic consequences of pegylation. Drug Deliv. 2006;13:399–409. doi: 10.1080/10717540600814402. [DOI] [PubMed] [Google Scholar]
- 27.monghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. 2001;53:283–318. [PubMed] [Google Scholar]
- 28.Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu. Rev. Immunol. 2005;23:901–944. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- 29.Irache JM, Salman HH, Gamazo C, Espuelas S. Mannose-targeted systems for the delivery of therapeutics. Expert Opin. Drug Deliv. 2008;8:703–724. doi: 10.1517/17425247.5.6.703. [DOI] [PubMed] [Google Scholar]
- 30.Shukla RK, Tiwari A. Carbohydrate molecules: an expanding horizon in drug delivery and biomedicine. Crit. Rev. Ther. Drug Carrier Syst. 2011;28:255–292. doi: 10.1615/critrevtherdrugcarriersyst.v28.i3.20. [DOI] [PubMed] [Google Scholar]
- 31.Kerrigan AM, Brown GD. C-type lectins and phagocytosis. Immunobiology. 2009;214:562–575. doi: 10.1016/j.imbio.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D’Addio SM, Baldassano S, Shi L, Cheung L, Adamson DH, Bruzek M, Anthony JE, Laskin DL, Sinko PJ, Prud’homme RK. Optimization of cell receptor-specific targeting through muntivalent surface decoration of polymeric nanocarriers. J. Controlled Release. 2013;168:41–49. doi: 10.1016/j.jconrel.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kragol G, Otvos L. Orthogonal solid-phase synthesis of tetramannosylated peptide constructs carrying three independent branched epitopes. Tetrahedron. 2001;57:957–966. [Google Scholar]
- 34.J. Vigerust D, Vick S, L. Shepherd V. Characterization of functional mannose receptor in a continuous hybridoma cell line. BMC Immunol. 2012;13:51. doi: 10.1186/1471-2172-13-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cassol E, Alfano M, Biswas P, Poli G. Monocyte-derived macrophages and myeloid cell lines as targets of HIV-1 replication and persistence. J Leukoc Biol. 2006;80:1018–1030. doi: 10.1189/jlb.0306150. [DOI] [PubMed] [Google Scholar]
- 36.Llorca O. Extended and bent conformations of the mannose receptor family. Cell Mol Life Sci. 2008;65:1302–1310. doi: 10.1007/s00018-007-7497-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feinberg H, Park-Snyder S, R. Kolatkar A, T. Heise C, E. Taylor M, I. Weis W. Structure of a C-type carbohydrate recognition domain from the macrophage mannose receptor. J Biol Chem. 2000;275:21539–21548. doi: 10.1074/jbc.M002366200. [DOI] [PubMed] [Google Scholar]
- 38.T. Vistica D, Skehan P, Scudiero D, Monks A, Pittman A, Boyd MR. Tetrazolium-based assays for cellular viability: a critical examination of selected parameters affecting formazan production. Cancer Res. 1991;51:2515–2520. [PubMed] [Google Scholar]
- 39.Box GEP, Behnken DW. Some New Three Level Designs for the Study of Quantitative Variables. Technometrics. 1960;2:455–475. [Google Scholar]
- 40.Gannu R, Palem CR, Yamsani SK, Yamsani VV, Yamsani MR. Enhanced bioavailability of buspirone from reservoir-based transdermal therapeutic system, optimization of formulation employing Box-Behnken statistical design. AAPS PharmSciTech. 2010;11:976–985. doi: 10.1208/s12249-010-9451-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeng X, Andrade CA, Oliveira MD, Sun XL. Carbohydrate-protein interactions and their biosensing applications. Anal Bioanal Chem. 2012;402:3161–3176. doi: 10.1007/s00216-011-5594-y. [DOI] [PubMed] [Google Scholar]
- 42.Lee RT, Hsu TL, Huang SK, Hsieh SL, Wong CH, Lee YC. Survey of immune-related, mannose/fucose-binding C-type lectin receptors reveals widely divergent sugar-binding specificities. Glycobiology. 2011;21:512–520. doi: 10.1093/glycob/cwq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weis WI, Drickamer K. Structural basis of lectin-carbohydrate recognition. Annu Rev Biochem. 1996;65:441–473. doi: 10.1146/annurev.bi.65.070196.002301. [DOI] [PubMed] [Google Scholar]
- 44.Sansone F, Casnati A. Multivalent glycocalixarenes for recognition of biological macromolecules: glycocalyx mimics capable of multitasking. Chem Soc Rev. 2013;42:4623–4639. doi: 10.1039/c2cs35437c. [DOI] [PubMed] [Google Scholar]
- 45.Mammen M, Choi S, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 46.Fred Brewer C. Binding and cross-linking properties of galectins. Biochim Biophys Acta. 2002;1572:255–262. doi: 10.1016/s0304-4165(02)00312-4. [DOI] [PubMed] [Google Scholar]
- 47.Itano MS, Steinhauer C, Schmied JJ, Forthmann C, Liu P, Neumann AK, Thompson NL, Tinnefeld P, Jacobson K. Super-resolution imaging of C-type lectin and influenza hemagglutinin nanodomains on plasma membranes using blink microscopy. Biophys J. 2012;102:1534–1542. doi: 10.1016/j.bpj.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su Y, Bakker T, Harris J, Tsang C, Brown GD, Wormald MR, Gordon S, Dwek RA, Rudd PM, Martinex-Pomares L. Glycosylation influences the lectin activities of the macrophage mannose receptor. J Biol Chem. 2005;280:32811–32820. doi: 10.1074/jbc.M503457200. [DOI] [PubMed] [Google Scholar]
- 49.Bourguignon LY, Singer SJ. Transmembrane interactions and the mechanism of capping of surface receptors by their specific ligands. Proc Natl Acad Sci U S A. 1977;74:5031–5035. doi: 10.1073/pnas.74.11.5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 51.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 52.Le Cadec V, Emorine LJ, Toesca I, Cougoule C, Maridonneau-Parini I. The human macrophage mannose receptor is not a professional phagocytic receptor. J Leukoc Biol. 2005;77:934–943. doi: 10.1189/jlb.1204705. [DOI] [PubMed] [Google Scholar]
- 53.El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11:13–22. doi: 10.1208/s12248-008-9071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rutili G, Arfors KE. Protein concentration in interstitial and lymphatic fluids from the subcutaneous tissue. Acta Physiol Scand. 1977;99:1–8. doi: 10.1111/j.1748-1716.1977.tb10345.x. [DOI] [PubMed] [Google Scholar]
- 55.Barber BJ, Schultz TJ, Randlett DL. Comparative analysis of protein content in rat mesenteric tissue, peritoneal fluid, and plasma. Am J Physiol. 1990;258:714–718. doi: 10.1152/ajpgi.1990.258.5.G714. [DOI] [PubMed] [Google Scholar]
- 56.Pedrinelli R, Dell'Omo G, Bandnelli S, Penno G, Mariani M. Transvascular albumin leakage and forearm vasodilatation to acetylcholine in essential hypertension. Am J Hypertens. 2000;13:256–261. doi: 10.1016/s0895-7061(00)00250-8. [DOI] [PubMed] [Google Scholar]
- 57.Pasut G, Veronese FM. State of the art in PEGylation: the great versatility achieved after forty years of research. J Control Release. 2012;161:461–472. doi: 10.1016/j.jconrel.2011.10.037. [DOI] [PubMed] [Google Scholar]
- 58.Herold DA, Keil K, Bruns DE. Oxidation of polyethylene glycols by alcohol dehydrogenase. Biochem Pharmacol. 1989;38:73–76. doi: 10.1016/0006-2952(89)90151-2. [DOI] [PubMed] [Google Scholar]
- 59.Bendele A, Seely J, Richey C, Sennello G, Shopp G. Short communication: renal tubular vacuolation in animals treated with polyethylene-glycol-conjugated proteins. Toxicol Sci. 1998;42:152–157. doi: 10.1006/toxs.1997.2396. [DOI] [PubMed] [Google Scholar]
- 60.Cho WS, Cho M, Jeong J, Choi M, Cho HY, Han BS, Kim SH, Kim HO, Lim YT, Chung BH. Acute toxicity and pharmacokinetics of 13 nm-sized PEG-coated gold nanoparticles. Toxicol Appl Pharmacol. 2009;236:16–24. doi: 10.1016/j.taap.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 61.Duncan R, Richardson SC. Endocytosis and intracellular trafficking as gateways for nanomedicine delivery: opportunities and challenges. Mol Pharm. 2012;9:2380–2402. doi: 10.1021/mp300293n. [DOI] [PubMed] [Google Scholar]
- 62.Gunaseelan S, Pooyan S, Chen P, Samizadeh M, Palombo MS, Stein S, Zhang X, Sinko PJ. Multimeric peptide-based PEG nanocarriers with programmable elimination properties. Biomaterials. 2009;30:5649–5659. doi: 10.1016/j.biomaterials.2009.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cambi A, Netea MG, Mora-Montes HM, Gow NA, Hato SV, Lowman DW, Kullberg BJ, Torensma R, Williams DL, Figdor CG. Dendritic cell interaction with Candida albicans critically depends on N-linked mannan. J Biol Chem. 2008;283:20590–20599. doi: 10.1074/jbc.M709334200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feriotti C, Flavio VL, de Araujo EF, da Costa TA, Calich VL. Mannosyl-Recognizing Receptors Induce an M1-Like Phenotype in Macrophages of Susceptible Mice but an M2-Like Phenotype in Mice Resistant to a Fungal Infection. PLoS One. 2013;8:e54845. doi: 10.1371/journal.pone.0054845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gunaseelan S, Gunaseelan K, Deshmukh M, Zhang X, Sinko PJ. Surface modifications of nanocarriers for effective intracellular delivery of anti-HIV drugs. Adv Drug Deliv Rev. 2010;62:518–531. doi: 10.1016/j.addr.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]