

Abstract
Angiotensin-(1-7) (Ang-(1-7)/AT7-Mas receptor axis is an alternative pathway within the renin angiotensin system (RAS) that generally opposes the actions of Ang II/AT1 receptor pathway. Advanced glycated end product (AGEs) including glucose- and methylglyoxal-modified albumin (MGA) may contribute to the development and progression of diabetic nephropathy in part through activation of the Ang II/AT1 receptor system; however, the influence of AGE on the Ang-(1-7) arm of the RAS within the kidney is unclear. The present study assessed the impact of AGE on the Ang-(1-7) axis in NRK-52E renal epithelial cells. MGA exposure for 48 hours significantly reduced the intracellular levels of Ang-(1-7) approximately 50%; however, Ang I or Ang II expression was not altered. The reduced cellular content of Ang-(1-7) was associated with increased metabolism of the peptide to the inactive metabolite Ang-(1-4) [MGA: 175 ± 9 vs. Control: 115 ± 11 fmol/min/mg protein, p<0.05, n=3] but no change in the processing of Ang I to Ang-(1-7). Treatment with Ang-(1-7) reversed MGA-induced cellular hypertrophy and myofibroblast transition evidenced by reduced immunostaining and protein expression of α-smooth muscle actin (α-SMA) [0.4±0.1 vs. 1.0±0.1, respectively, n=3, p<0.05]. Ang-(1-7) abolished AGE-induced activation of the MAP kinase ERK1/2 to a similar extent as the TGF-β receptor kinase inhibitor SB58059; however, Ang-(1-7) did not attenuate the MGA-stimulated release of TGF-β. The AT7-Mas receptor antagonist D-Ala7-Ang-(1-7) abolished the inhibitory actions of Ang-(1-7). In contrast, AT1 receptor antagonist losartan did not attenuate the MGA-induced effects. We conclude that Ang-(1-7) may provide an additional therapeutic approach to the conventional RAS blockade regimen to attenuate AGE-dependent renal injury.
1. Introduction
Angiotensin-(1-7) is an alternative product of the renin-angiotensin system (RAS) expressed in the circulation, brain and various peripheral tissues including the heart, vasculature and kidney (10; 42). The peptide is derived either from Ang I by direct processing of endopeptidases including neprilysin and thimet oligopeptidase or from Ang II by the monocarboxypeptidases such as angiotensin converting enzyme 2 (ACE2) (9; 44). Conversely, ACE which processes Ang I to Ang II is the predominant metabolizing enzyme for Ang-(1-7) in the circulation (9; 44). Ang-(1-7) primarily recognizes the Mas receptor, a protein distinct from either the AT1 or AT2 receptors that bind and transduce the actions of Ang II (42). Indeed, accumulating evidence over the past 25 years now functionally partitions the RAS into two divergent pathways that comprise the ACE-Ang II-AT1 receptor and ACE2/NEP-Ang-(1-7)-Mas receptor (9;10). In general, the Ang-(1-7) system exhibits biological effects that are opposite from those of the Ang II-AT1 receptor axis; chronic administration of Ang-(1-7) is associated with a reduction in blood pressure, natriuresis and diuresis, anti-proliferative and anti-fibrotic effects, as well as reduced inflammation (10; 42; 54). Moreover, pharmacological approaches that block the RAS including ACE inhibitors or AT1 receptor antagonists increase circulating levels of Ang-(1-7); thus, circulating levels of the peptide may contribute to the beneficial actions of RAS blockade (9).
Although most experimental studies demonstrate beneficial actions of Ang-(1-7) treatment, Ang-(1-7) may induce deleterious effects similar to Ang II that are particularly evident in the kidney (54). Walther and colleagues reported that Mas receptor null mice exhibit a lower degree of renal injury than wildtype littermates when exposed to a high salt diet (21) Moreover, chronic administration of Ang-(1-7) into wildtype mice was associated with an increased incidence of injury, including a pronounced extent of inflammatory markers (21). In this regard, Burns et al. reported that Ang-(1-7) induced epithelial to mesenchymal or myofibroblast transition (MT) in the normal rat kidney (NRK-52E) proximal tubule cell line (5). In these studies, Ang II-stimulated MT was abolished by the Ang-(1-7) antagonist D-Ala7-Ang-(1-7) (DAL or A779) and the ACE2 inhibitor MLN4760 (5). The results of the Burns study implies that either Ang II was directly converted to Ang-(1-7) by ACE2 or that Ang II stimulated Ang-(1-7) synthesis to induce the myofibroblast phenotype via the Mas receptor (5). Additionally, Liu et al. find that Ang-(1-7) stimulated proliferation of human mesangial cells that was associated with an activation of the mitogen activated kinase (MAPK) ERK (31). Diabetic nephropathy (DN) in particular, has been linked to activation of intrarenal RAS where suppression of this hormonal system by AT1 blockers (ARBs) or ACE inhibitors attenuates proteinuria and declining renal function (4; 20). A growing body of evidence indicates that advanced glycated end products (AGEs) are increased in diabetes and are associated with diabetic nephropathy (15; 32). AGEs induce MT and consequently tubulointerstitial fibrosis through activation of specific receptors for AGEs (RAGE) (46; 47). However, the role of Ang-(1-7) in AGE-mediated effects within the tubular epithelium has not been established. The current study tested the hypothesis that the beneficial or deleterious actions of Ang-(1-7) may reflect the underlying pathological conditions. We assessed the influence of Ang-(1-7) on MT and associated signaling pathways induced by the AGE product methylglyoxal albumin (MGA) in the rat kidney NRK-52E cells, a well-characterized cell model that expresses an intracellular RAS (1;7). Additionally, we determined the extent that chronic MGA exposure influences the Ang-(1-7) processing pathways in this epithelial cell line. We report that MGA significantly reduced the cellular expression of Ang-(1-7) which was associated with the increased metabolism of the peptide directly to Ang-(1-4), but not the conversion of Ang I to Ang-(1-7). Exogenous Ang-(1-7) treatment abrogated the MGA-induced MT which was reversed by the Mas receptor antagonist DAL. Moreover, Ang-(1-7) attenuated both the MGA- and TGF-β-dependent stimulation of the extracellular regulated kinase 1/2 (ERK1/2) pathway, but did not reduce the MGA-induced release of TGF-β. The present studies suggest that Ang-(1-7) abrogates the MGA-stimulated myofibroblast phenotype by inhibiting the chronic stimulation of the TGF-β-ERK pathway in NRK-52E cells.
2. Methods
2.1. Cell Culture
Normal kidney proximal epithelial cells (NRK-52E) cells were obtained from American Tissue Type Culture (Arlington VA; passage 8) and maintained at 37°C in plastic 75 cm2 flasks in Dulbecco's modified Eagle's medium (DMEM/F12, Invitrogen) containing 5% fetal bovine serum (FBS), L-Glutamine and 15 mM HEPES buffer (1). The culture flasks were kept in a 95% air and 5% CO2 humidified environment at 37°C. At confluence, the cells were washed and maintained in serum free DMEM/F12 without supplements for 24 hours prior to the MGA exposure and subsequent biochemical studies.
2.2. Cell Treatments
Glycated albumin (MGA) was prepared as described (50). Briefly, 500 μM methylglyoxal (Sigma, St. Louis, MO, US) was incubated with 100 uM BSA (Sigma) dissolved in phosphate buffered saline (PBS) for 24 hours, then washed on 10 kDa filters (Macrosep® Advance Device, Pall Life Sciences, MI, USA) to remove excess methyl glyoxal, reconstituted with DMEM/F12 serum free media and passed through a 0.2 μmicron filter. TGF-β (5 nanograms (ng)/ml, Peprotech, NJ, USA) was prepared according to manufacturer to treat cells in a subset of experiments. Cells were co-treated with one or combinations of the following: Ang-(1-7) (100 nM, Bachem, CA, USA), D-Ala7-Ang-(1-7) (10 μM, DAL, Bachem), ERK1/2 kinase inhibitor, PD 98059 (1 μM, PD, Sigma), TGF- β receptor kinase inhibitor; SB525334 (1 μM, SB, Selleckchem, TX, USA), the AT1 receptor antagonist losartan (1 μM, Merck Pharmaceuticals, NJ, USA), the renin inhibitor aliskerin (1 μM, Tocris) and the ACE inhibitor lisinopril (1 μM, Sigma).
2.3. 3H-Leucine incorporation
Cellular hypertrophy was determined by 3H-leucine incorporation (18). Cells were incubated for 48 hours in serum-free media with or without 100 μM MGA in 24 well plates. The MGA cells were treated with 100 nM Ang-(1-7), 10 μM DAL, 1 μM PD98059 and 1 μM Losartan. The cells were pulsed with 0.5 μCi of 3H-Leucine (L-[4, 5-3H (N)], Perkin Elmer, Boston, MA, USA) for the last 24 hours at 37°C. The cells were washed twice with PBS, fixed in ice cold 10% trichloroacetic acid (TCA) and kept on ice for 15 minutes, and then washed twice in 5% TCA. The acid insoluble proteins were dissolved in 0.05 N NaOH and 0.1% sodium dodecyl sulfate (SDS) at 37°C and incorporation was determined by liquid scintillation counting. All experiments were performed in triplicate. Values were expressed as the percentage (%) of control per well for each experiment.
2.4. Western blotting
After 24 hours starvation of the cells in serum free medium, cells were incubated for 48 hours with MGA (100 uM) or 15 min with TGF-β (5 ng/ml), followed by immunoblot assays for phosphorylation or protein expression. The phosphorylation of the ERK1/2 was measured by western blotting as described (17). Briefly, cells were lysed in a Triton-lysis buffer consisting of 100 mM NaCl, 50 mm NaF, 5 mM EDTA, 1 % tritonX-100, 50 mM Tris-HCl, pH 7.4, with 0.01 mM NaVO4, 0.1mM phenylmethylsulfonylfluoride (PMSF), and 0.6 μM leupeptin. The lysates were then sonicated for 5 seconds, and centrifuged at 10,000 g for 5 min to remove insoluble debris. Supernatants (10-50μg) were diluted in Laemmli buffer with β-mercaptoethanol and boiled for 5 min, separated on 10% SDS polyacrylamide gels for 1 h at 120V in Tris-glycine SDS and transferred to a polyvinylidene difluoride membrane (PVDF). Blots were blocked with 5% Bio-Rad Dry Milk and TBS with Tween and probed overnight at 4°C with primary antibodies for phospho-p44/42 MAPK (ERK1/2) (1:2000; rabbit polyclonal, Cell Signaling, Cambridge, MA), 44/42 MAPK (ERK1/2) (1:3000, rabbit poly clonal, Cell Signaling), α-SMA (1:5000, mouse monoclonal, Sigma),
Membranes were treated with HRP-labeled polyclonal anti rabbit secondary antibodies (1:5000) or anti-mouse secondary antibodies (1:3000) for 1 hour and detected with chemiluminescent substrates (Pierce Biotechnology, Rockford, IL). For ERK phosphorylation assays, membranes probed with P-ERK1/2 were stripped and re-probed for total ERK1/2. Membranes for α-SMA were stripped and re-probed with rabbit polyclonal anti-EFα1 (1:3000) antibody as a loading control and bands were quantified using MCID densitometry software (InterFocus Imaging, Linton, England).
2.5. Immunofluorescent microscopy
NRK-52E cells were grown in 8 chamber slides for two days in DMEM/F12 containing 5% FBS which was replaced with serum free media for 24 hr (1). Cells were treated with either serum free media as control conditions or 100 μM MGA with or without Ang-(1-7), DAL, PD, SB, LOS, TGF-β (5 ng) or their combination for 72 hours. Cells were washed with PBS and fixed with 2% paraformaldehyde for 15 minutes. Following a PBS rinse, cells were permeabilized with 0.2% Triton and then blocked with 3% BSA (Sigma #A-8022). The fixed cells were probed with a primary antibody for α-SMA (1:200, mouse monoclonal, Sigma). Antibodies were diluted in 3% normal donkey serum. After overnight incubation with the primary antibody at 4°C, cells were rinsed with PBS twice, incubated with fluorescent anti-mouse Alexa Fluor 568 secondary antibody (1:400; Invitrogen, Carlsbad, CA), and the slides were mounted with Molecular Probes ProLong mounting media with DAPI (Invitrogen) to stain the nuclei.
2.6. Cell Area
Cells area was assessed utilizing ImageJ 1.4 software (http://rsb.info.nih.gov/ij/) in a subset of immunofluorescent images (section 2.5) to demonstrate the influence of MGA on cellular hypertrophy in the absence or presence of Ang-(1-7) or the ERK inhibitor PD98059. Fields were chosen at random and all cells were measured in each field. The data was expressed as a percentage of the control cell area.
2.7. Peptide Assays
Cells in serum free medium for 24 hours were treated with 100 uM MGA for an additional 48 hours. Cells were washed twice in ice-cold PBS, harvested, and the cells pellets snap-frozen and stored at −80°C (1). The cell pellet was reconstituted in MilliQ water on ice and immediately placed in a boiling water bath for 15 minutes. The homogenate was then sonicated and acidified with trifluoroacetic acid (TFA) to a final concentration of 0.2%, and centrifuged at 20,000g for 20 min at 4°C. The resultant supernatant was applied to an activated Sep-Pak C18 extraction column, washed with 0.2% TFA, and the peptide fraction eluted with 3 ml 80% methanol/0.2% TFA. Blank solutions contained only the MillQ water and TFA. Their extracted values were subtracted from those determined for the cells. Measurement of immunoreactive Ang I, Ang II and Ang-(1-7) in the extracted cells was assessed by three distinct RIAs (1). The Ang-(1-7) RIA fully recognizes Ang-(1-7) and Ang-(2-7), but cross-reacts less than 0.01% with Ang-(3–7), Ang II, Ang I, and their fragments. The Ang II RIA equally recognizes Ang III, Ang-(3-8), and Ang-(4-8), but cross-reacts less than 0.01% with Ang I and Ang-(1-7). The Ang I RIA fully recognizes Ang-(2–10) and Ang-(3–10), but cross-reacts with Ang II and Ang -(1–7) less that 0.01%. The limits of detection for each RIA were as follows: Ang-(1–7), 4 femtomoles (fmol)/tube; Ang II, 0.5 fmol/tube; and Ang I, 5 fmol/tube.
For TGF-β quantification in the media, cells were seeded in 12-well plates. The cells were placed in serum free for 24 hours before being treated with MGA and the following: Ang-(1-7), DAL, PD, or LOS. Cells maintained in the serum free media served as the controls. The cell media was collected on ice and the TGF-β content was quantified by Quantikine® ELIZA (R&D Systems, MN, US) immunoassay according to manufacturer instructions. The sensitivity of the assay was 15 pg/ml and the release data expressed as ng/ml.
2.8. Angiotensin metabolism
To characterize the processing of the peptides in vitro, metabolism assays were conducted on cells homogenates as described previously (1; 34). Confluent cells were starved for 24 hrs, and treated with either serum free or MGA containing media (100 uM) for 48 hrs. Cells then were washed with cold PBS, harvested and immediately frozen at -80°C. The cell pellets were homogenized in metabolism buffer (10 mM HEPES, 125 mM NaCl, 10 μM ZnCl2, pH 7.4), then centrifuged at 100,000g for 10 min. Either 125I-Ang-I or Ang-(1-7) (0.5 nM) was incubated with 2 μg protein of the cell supernatant in a final assay volume of 0.5 ml at 37C°. The reaction was stopped by addition of ice-cold 1.0% phosphoric acid, centrifuged at 16,000 g, and the supernatants stored at −20°C. Samples were separated by reverse-phase high-performance liquid chromatography (HPLC) and the 125I-products were detected by a Bioscan flow-through γ detector (44). Products were identified by comparison of their retention times to 125I-angiotensn standards. We employed gradient elution for Ang I metabolism (1), and isocratic elution for Ang-(1-7) metabolism (34). Peptides were iodinated by the chloramine T method and purified by HPLC (specific activity > 2000 Ci/mmol). Enzyme activities were expressed as fmol product of Ang-(1-4) or Ang-(1-7) per mg protein per minute (fmol/mg/min). Total protein content was determined in the cell supernatant by Bradford protein assay with a standard of BSA.
2.9. Statistical analysis
The data were expressed as mean ± standard error (SEM). Differences between the groups were analyzed by one-way ANOVA and Newman-Keuls multiple comparison analysis. Differences between control and MGA treated cells for peptides content and enzyme activities were analyzed by paired Student's t test. A probability value of P<0.05 was required for statistical significance. All figures were constructed with GraphPad Prism V plotting and statistical software.
3. Results
3.1. Angiotensin expression and metabolism
As previous studies indicated the NRK-52E cells express a complete RAS, we determined the influence of chronic MGA exposure on the intracellular expression of angiotensin peptides (1; 7; 53). As shown in Figure 1, Ang-(1-7) peptide content was significantly decreased in the MGA-exposed cells at 48 hours as compared to the control cells; 396 ± 59 vs. 219 ± 54 fmol/mg protein, respectively (P<0.05; n=3). In contrast, the peptide levels of Ang I and Ang II were not different between the control and MGA treatment (Figure 1).
Figure 1. MGA exposure reduces intracellular levels of Ang-(1-7) in the in the NRK-52E cells.
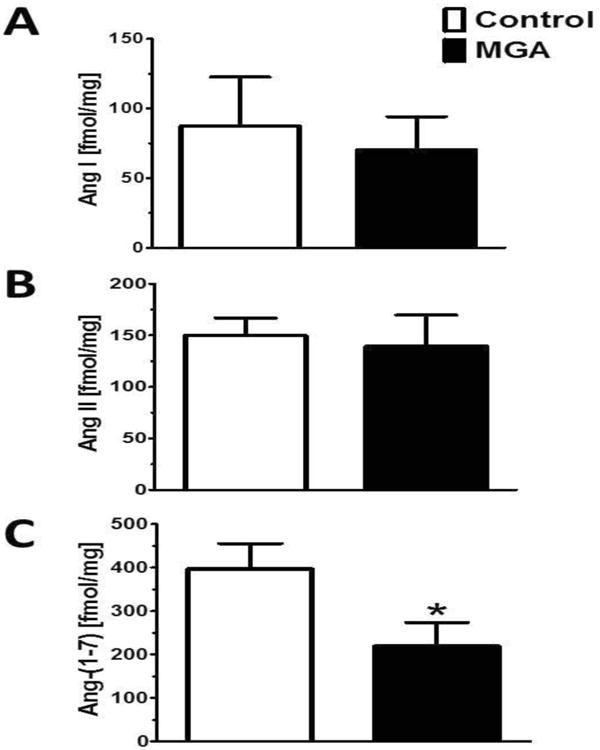
Panel A: Ang I peptide levels are not different between control and MGA treated cells. Panel B: There are no difference in Ang II peptide levels between MGA treated cells and control cells. Panel C: Ang-(1-7) peptide levels were significantly lower in the MGA treated cells than control cells. Intracellular peptide content was quantified by separate RIAs to Ang I, Ang II and Ang-(1-7). Data are the mean ± SEM from 3 different cell passages. *P< 0.05.
We then assessed whether the reduced cellular content of Ang-(1-7) with MGA reflects alterations in the metabolism or synthesis of the peptide. Using a 100,000 × g supernatant fraction, we determined both the rate of Ang-(1-7) metabolism and the conversion of Ang I to Ang-(1-7) (Figures 2 and 3, respectively). As shown in the chromatographs from control and MGA cells, Ang-(1-7) was exclusively hydrolyzed to a single peak corresponding to Ang-(1-4) as assessed under isocratic elution conditions (34). However, Ang-(1-7) was metabolized at a greater rate in the MGA-treated cells as compared to the control cells [175 ± 9 vs. 115 ± 11 fmol//mg/min, P<0.05, n=3] (Figure 2). As shown in Figure 3, Ang I was processed to Ang-(1-7) in the cell supernatant as assessed under gradient elution conditions (1). Although the generation of Ang-(1-7) from Ang I tended to decline in the MGA-treated cells [56 ± 7 vs. 66 ± 8 fmol/mg/min, n=3], this did not reach statistical significance (Figure 3). Note that the different peak shapes for Ang-(1-7) in Figures 2 and 3 reflect the two separation methods utilized for the Ang-(1-7) and Ang I metabolism studies.
Figure 2. MGA exposure increases Ang-(1-7) metabolism in the NRK-52E cells.
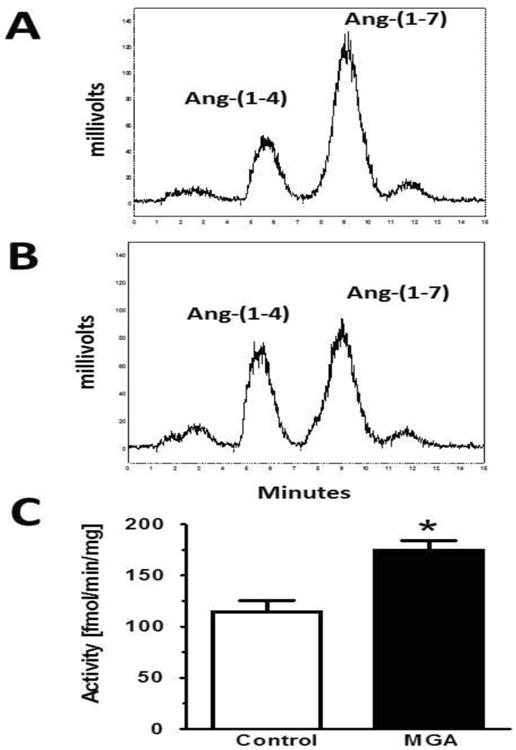
Peptide metabolism was assessed in the 100,000 × g supernatant fraction from control and MGA-treated (48 hours) NRK-52E cells for 30 minutes at 37°C. Peptide products were determined by HPLC separation with isocratic elution conditions (38). Panel A: Ang-(1-7) conversion to Ang-(1-4) in control cells. Panel B: Ang-(1-7) conversion to Ang-(1-4) in MGA-treated cells. Panel C: Quantification of Ang-(1-4) generation from Ang-(1-7) revealed a higher rate of Ang-(1-7) conversion with MGA exposure. Data are the mean ± SEM of 3 different cell passages. *P< 0.05 vs. control.
Figure 3. MGA exposure does not influence Ang I to Ang-(1-7) formation in the NRK-52E cells.
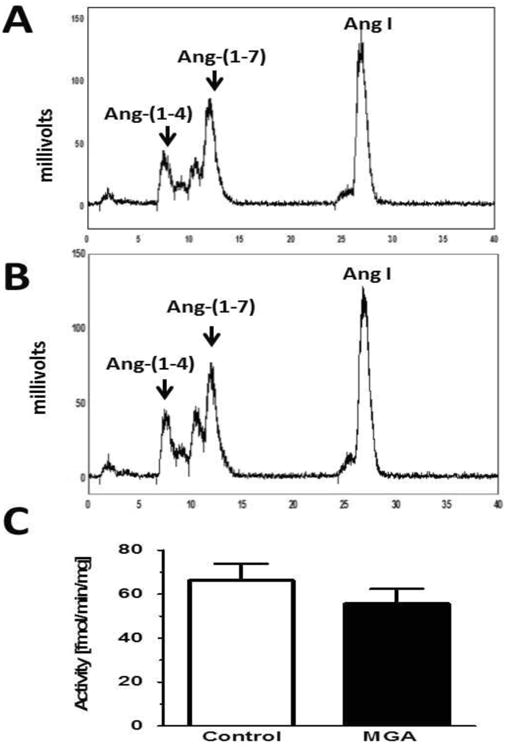
Peptide metabolism was assessed in the 100,000 × g supernatant fraction from control and MGA-treated (48 hours) NRK-52E cells for 30 minutes at 37°C. Peptide products were determined by HPLC separation under gradient conditions (1). Panel A: Ang I conversion to Ang-(1-7) in control cells. Panel B: Ang I conversion to Ang-(1-7) in MGA treated cells. Panel C: Quantification of Ang-(1-7) generation from Ang I revealed no differences between control and MGA treated cells. Data are the mean ± SEM of 3 different cell passages.
3.2. Cellular hypertrophy
Tubular hypertrophy is a common and early response in the diabetic kidney (39; 40). Since AGEs directly induce cellular hypertrophy, we assessed the influence of MGA on hypertrophy in confluent monolayers of NRK-52E cells by determining the cellular incorporation of 3H-leucine (17; 28). As shown in Figure 4, MGA treatment for 48 hours significantly increased 3H-leucine incorporation approximately 2-fold [176 ± 10% of control; P< 0.05, n=5]. Co-treatment with 100 nM Ang-(1-7) abolished the extent of hypertrophy induced by MGA [87± 7% of control, n=5]. Co-treatment with the specific antagonist to the Mas receptor DAL (10 μM) reversed the inhibitory effects of Ang-(1-7) on MGA-induced hypertrophy [162 ± 18% vs. 87± 7%]. Treatment with the antagonist DAL alone did not influence the extent of MGA-dependent hypertrophy (162 ± 18%, Figure 4)
Figure 4. Ang-(1-7) attenuates MGA-induced cell hypertrophy in the NRK-52E cells.

Left: MGA treatment for 48 hours significantly increased 3H-leucine incorporation approximately 180% of control. Ang-(1-7) (A7, 100 nM) significantly reduced the extent of hypertrophy induced by MGA. The Ang-(1-7) receptor antagonist D-Ala7-Ang-(1-7) (DAL, 10 μM), reversed the inhibitory effects of Ang-(1-7) on MGA-induced hypertrophy. The ERK 1/2 inhibitor PD98059 (PD, 1 μM), abolished the cellular hypertrophy induced by MGA. Ang-(1-7) and PD98059 exhibit no additive effects on hypertrophy. The DAL antagonist alone or the AT1 antagonist losartan (LOS) alone did not influence the MGA-dependent hypertrophy. 3H-leucine incorporation is expressed as percentage (%) from control cells for each experiment. Data are the mean ± SEM from 5 different cell passages. Inset: MGA treatment for 48 hours increased cell area as measured in immunofluorescent images. Ang-(1-7) (A7, 100 nM) and PD98059 (PD, 1 μM), reversed the increase in cell area induced by MGA.*P < 0.05 vs. control, #P < 0.05 vs. MGA-treated cells.
The MAP kinase ERK1/2 constitutes one of the signaling pathways activated by AGEs involved in the hypertrophic response (20; 36). As shown in Figure 4, co-treatment with the selective ERK1/2 inhibitor PD98059 (PD, 1μM) also abolished MGA-induced hypertrophy [67 ± 11% vs. 176 ± 10%, p < 0.05]. Co-treatment of both Ang-(1-7) and PD did not reveal additive effects [69 ± 10%] to PD or Ang-(1-7) treatment alone. Finally, treatment with different RAS blockers including the AT1 receptor antagonist Losartan (LOS, 1 μM), the renin inhibitor Aliskiren (1 μM), and the ACE inhibitor lisinopril (1 μM) did not significantly reduce cellular hypertrophy induced by MGA [159 ± 22%; 143 ± 32%, 182 ± 26%, respectively]. In addition, MGA treatment for 48 hours increased cell area approximately 4-fold [467 ± 70% of control, p < 0.05, n=3] (Figure 3 inset). Ang-(1-7) significantly decreased the MGA-induced cell area [204 ± 15% vs. 467 ± 70%; respectively, p<0.05, n=3]. Treatment with PD98059 (PD, 1μM) also significantly reduced the effect of MGA on cell area [149 ± 12% vs. 467 ± 70%, p < 0.05, n=3].
3.3. Myofibroblast transition
The NRK-52E cell line is a well-characterized model of epithelial to mesenchymal or myofibroblast transition (MT) (6; 30). AGEs induce a myofibroblast phenotype in these cells that is dependent on TGF-β expression (29). We examined the effect of Ang-(1-7) on MGA-induced MT by immunofluorescent staining and protein expression of α–SMA (Figures 5 and 6). MGA induced an apparent increase in α-SMA staining compared to the control cells that was accompanied by an altered cellular morphology characterized by extensive actin filament crosslinking (Figures 5A and 5B- high magnification; 5C and 5D- low magnification). Ang-(1-7) (100 nM) treatment appeared to attenuate the MGA-induced phenotype (Figure 5E). Moreover, the inhibitory effects of Ang-(1-7) on MT appeared to be reversed by the Ang-(1-7) receptor antagonist DAL (10 μM) (Figure 5F). Furthermore, both the ERK inhibitor PD98059 (PD, 1 μM) and the TGF-β receptor kinase inhibitor SB525334 (SB, 1 μM) appeared to prevent this transition (Figure 5G and 5H). Finally, the addition of the AT1 receptor antagonist losartan did not appear to attenuate α-SMA in comparison to treatment with Ang-(1-7), PD or SB (Figure 5, I). As a positive control, cells treated with TGF- β (5 ng/ml) demonstrate extensive α-SMA staining similar to that with MGA (Figure 5J).
Figure 5. Ang-(1-7) appears to inhibit MGA-induced fluorescent staining for de novo expression of α-SMA in NRK 52-E cells.
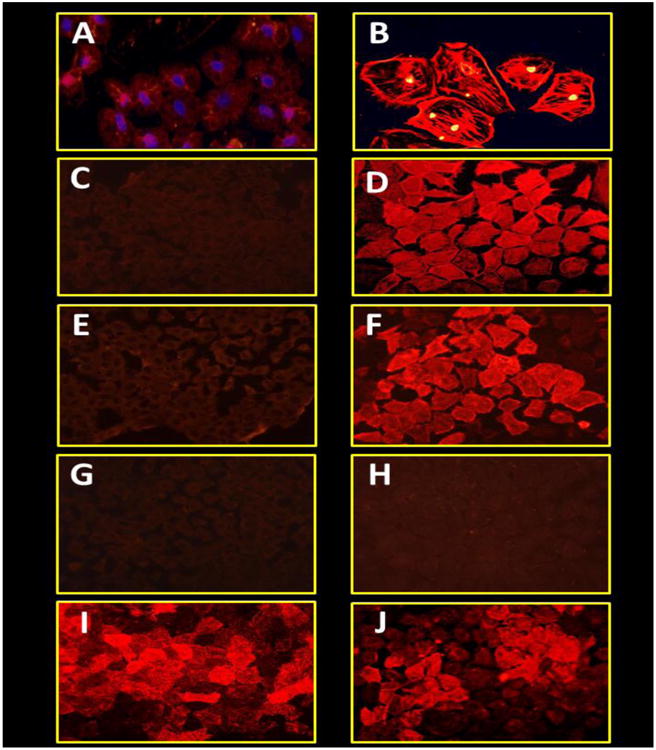
Cells were incubated with serum free media (A and C) or MGA (100 μM, B, D-I) or TGF-β (5 ng, J) for 72 hrs. MGA-treated cells were treated with either Ang-(1-7) (100 nM, E), Ang-(1-7) and DAL (10 μM, F), ERK1/2 inhibitor PD98059 (1 μM, G), TGF-β receptor kinase inhibitor SB525334 (1 μM, H) or AT1 receptor antagonist losartan (1 μM, I). The immunofluorescent images are representative of 3 different cell passages.
Figure 6. Ang-(1-7) inhibits MGA-induced protein expression of expression of α-SMA in NRK 52-E cells.
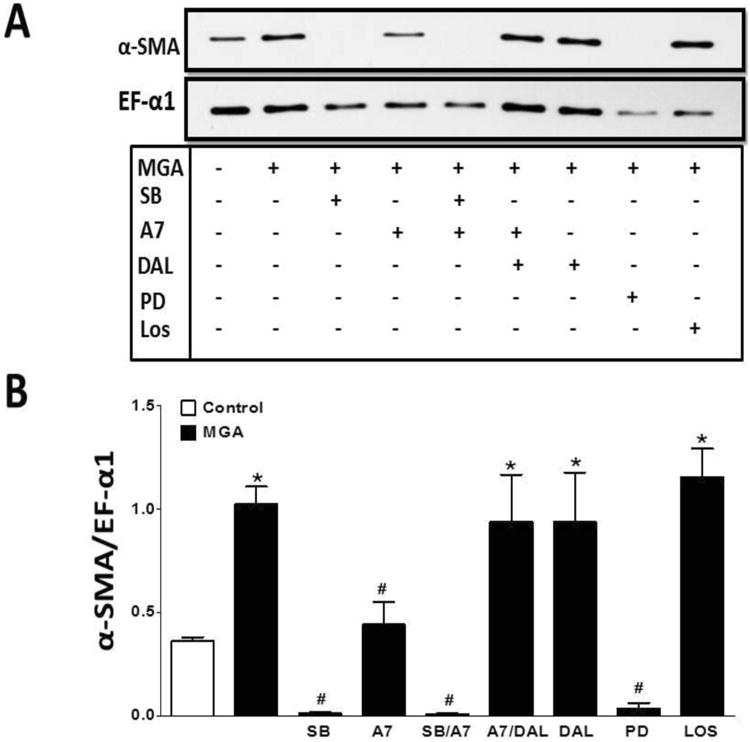
Western blot analysis of MGA (100 μM) induced α-SMA expression after 48 hours. Ang-(1-7) (A7, 100 nM) attenuated the increase in α-SMA, which was blocked by the Mas receptor antagonist D-Ala7-Ang-(1-7) (DAL, 10 μM). Both the ERK1/2 inhibitor PD98059 (PD, 1 μM) and TGF-β receptor kinase inhibitor SB525334 (SB, 1 μM) abolished the increase in α-SMA expression. The AT1 antagonist losartan (LOS, 1 μM) did not influence the α-SMA expression. Data are the mean ± SEM from 3-4 different cell passages. *P < 0.05 vs. control, #P < 0.05 vs. MGA.
As shown in Figure 6, MGA significantly induced α-SMA protein expression 3-fold as compared to the control cells [1.0 ± 0.1 vs. 0.3 ± 0.1, respectively, n=3, P<0.05]. Consistent with the immunofluorescent staining, Ang-(1-7) significantly reduced the MGA-induced expression of α-SMA (Figure 6) [0.4 ± 0.1 vs. 1.0 ± 0.1, respectively, n=3, P<0.05]. The inhibitory effects of Ang-(1-7) were blocked by DAL (Figure 6). We further show that the MGA-dependent stimulation of α-SMA expression was abolished by both the ERK1/2 inhibitor PD98059 (PD) and the TGF-β receptor kinase inhibitor SB525334 (SB) [0.04 ± 0.02 and 0.01 ± 0.01, respectively, n=3, P<0.05 versus MGA]. In agreement with the immunofluorescent studies, losartan (LOS) treatment did not significantly reduce α-SMA expression (Figure 6).
3.4. TGF-β release
Since TGF-β may be a key mediator for MT in the NRK-52E cells we determined whether Ang-(1-7) reduces TGF-β release. As shown in Figure 7, MGA significantly increased TGF-β release approximately 3-fold compared to control [1.16 ± 0.1 vs. 0.4 ± 0.1 ng/ml, respectively; P<0.05, n=6], and consistent with previous studies on AGE-induced stimulation of TGF-β (36; 44). However, co-treatment with Ang-(1-7) did not influence the release of TGF- β. We noted a trend for reduction in TGF-β release with PD or the combination of PD and Ang-(1-7), and these values were not significantly different than control. Treatment with the AT1 receptor antagonist losartan (LOS) did not influence the MGA-dependent release of TGF-β.
Figure 7. Ang-(1-7) does not influence MGA-induced release of TGF-β in NRK-52E cells.

Cells were exposed to MGA for 48 hours and TGF-β in the cell media determined by ELISA. MGA exposure increased TGF-β release approximately 3-fold. There were no significant effects on MGA-induced TGF-β release by Ang-(1-7) (A7, 100 nM), D-Ala7-Ang-(1-7) (DAL, 10 μM), or the AT1 receptor antagonist losartan (LOS, 1 μM). TGF-β release was not significantly different between the ERK1/2 inhibitor PD98059 (PD, 1 μM) or combined Ang-(1-7) and PD (A7/PD) treatment to control. Data are the mean ± SEM from 4-5 different cell passages. *P < 0.05 vs. control.
3.5. ERK activation
Previous studies suggest that AGEs release TGF-β to activate ERK1/2 signaling and stimulate MT (30). Therefore, we examined whether Ang-(1-7) targets activation of the ERK1/2 pathway following MGA or TGF-β treatment (Figures 8 and 9, respectively). As shown in Figure 8, treatment of the NRK-52E cells with MGA for 48 hours resulted in a sustained activation of ERK1/2. Quantitation of the immunoblot data revealed a 2.5- and 4-fold increase in the density of phosphorylated ERK 1 and 2, respectively (Figure 8). Ang-(1-7) abolished the MGA-induced phosphorylation of both ERK isoforms. The inhibitory effects of Ang-(1-7) were likely mediated by the Mas receptor as the DAL antagonist completely blocked the Ang-(1-7) effect. Additionally, both the ERK1/2 inhibitor PD98059 (PD) and TGF-β receptor kinase inhibitor SB525334 (SB) abolished the MGA-induced stimulation of ERK1/2 phosphorylation (Figure 8). In contrast, the AT1 receptor antagonist losartan (LOS) did not attenuate AGE mediated ERK1/2 phosphorylation. Finally, we show that Ang-(1-7) reduced the TGF-β-dependent phosphorylation of ERK1/2 in the MGA-exposed NRK-52E cell (Figure 9). The inhibitory effect of Ang-(1-7) was reversed by Mas-receptor antagonist DAL (Figure 9).
Figure 8. Ang-(1-7) inhibits MGA-induced phosphorylation of ERK 1/2 in NRK 52-E cells.
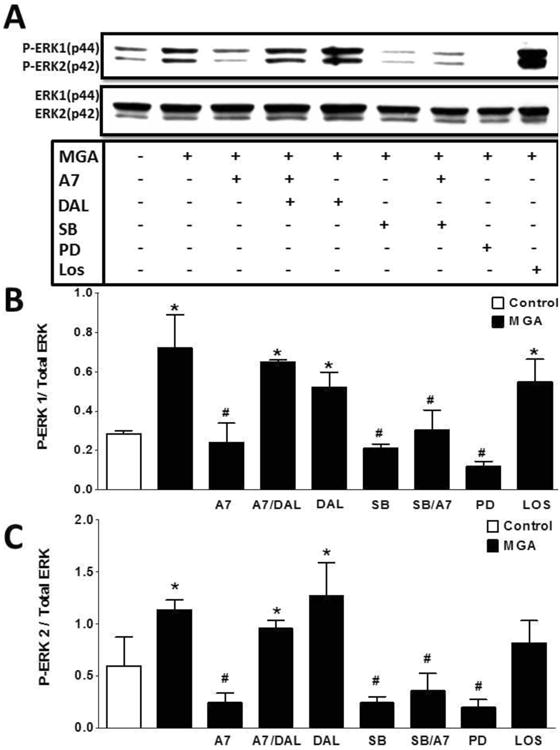
Western blot analysis of ERK1/2 with MGA treatment for 48 hours. MGA increased the phosphorylation of ERK1/2. Ang-(1-7) attenuated that MGA-induced phosphorylation which was blocked by D-Ala7-Ang-(1-7) (DAL, 10 μM). Inhibitors to TGF-β1 receptor kinase (SB, 1 μM) and ERK1/2 (PD, 1 μM) abolished ERK1/2 phosphorylation. The AT1 receptor antagonist losartan (LOS, 1 μM) did not inhibit the MGA-induced ERK1/2 activation. Data are the mean ± SEM from 3 different cell passages. *P < 0.05 vs. control, #P < 0.05 vs. MGA.
Figure 9. Ang-(1-7) inhibits TGF-β induced phosphorylation of ERK 1/2 in NRK-52E cells.
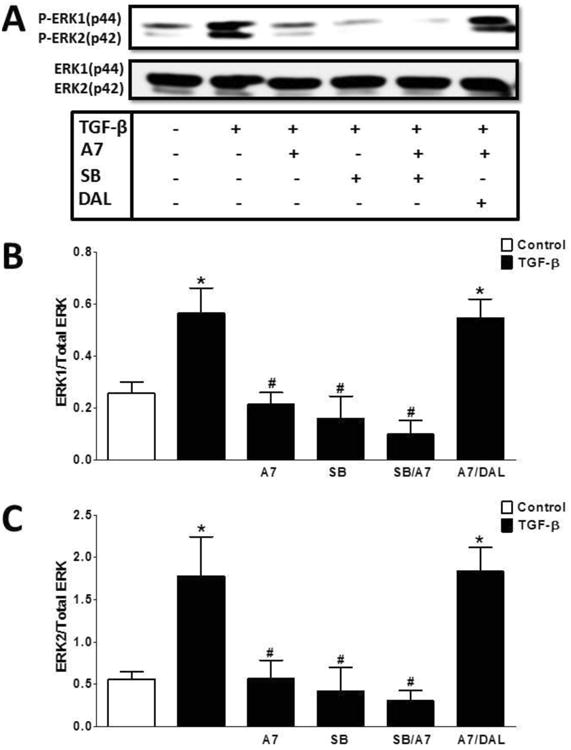
Western blot analysis of TGF-β-induced ERK1/2 phosphorylation. TGF-β (5 ng/ml) stimulated ERK1/2 phosphorylation after a 15 min incubation. Ang-(1-7) (A7, 100 nM) pretreatment inhibited TGF-β induced ERK1/2 phosphorylation. The inhibitory effect of Ang-(1-7) was reversed by the antagonist D-Ala7-Ang-(1-7) (DAL, 10 μM). Pretreatment with TGF-β receptor kinase inhibitor SB525334 (SB, 1 μM) abolished ERK1/2 phosphorylation and there was no additive effect of Ang-(1-7) and SB. Data are the mean ± SEM from 3 different cell passages. *P < 0.05 vs. control, #P < 0.05 vs. MGA treated cells.
4. Discussion
The present study demonstrates that the angiotensin heptapeptide Ang-(1-7) attenuates AGE-induced cellular hypertrophy and the myofibroblast phenotype likely through activation of the Mas receptor in NRK-52E epithelial cell line. Furthermore, we report that Ang-(1-7) abolished the chronic activation of the ERK1/2 pathway following the sustained exposure to the advanced glycated albumin product MGA. Although Ang-(1-7) did not attenuate the MGA-induced release of the cytokine TGF-β, the peptide abolished the direct stimulation of ERK by TGF-β. These data are consistent with the outcome of previous studies that TGF-β is a key component of AGE-induced cellular signaling (14; 28; 51). The current results revealed an apparent regulatory pathway between MGA and the intracellular expression of Ang-(1-7). Chronic MGA exposure significantly reduced the intracellular levels of Ang-(1-7) but did not influence the expression of either Ang II or Ang I. The selective effects of MGA on the Ang-(1-7) axis of the RAS appear to reflect an increase in the metabolism of the peptide rather than alterations in the formation of Ang-(1-7) from Ang I. Finally, we could not demonstrate a protective influence of RAS blockade with the AT1 receptor antagonist losartan suggesting that the Ang-(1-7)-Mas receptor axis of the RAS may constitute a more relevant therapeutic target for the AGE-mediated effects than the Ang II-AT1 receptor pathway in the NRK-52E cells.
As recently reviewed, the beneficial influence of the Ang-(1-7)-Mas receptor pathway of the RAS within the kidney is somewhat controversial (54). Although studies by Benter's group as well as others demonstrate renoprotective effects of Ang-(1-7) (2; 3; 42), there is additional evidence that the Ang-(1-7) axis may exacerbate renal damage (54). Utilizing the Mas knockout mouse, Walther and colleagues demonstrated this transgenic model exhibited reduced renal injury following a high salt diet (21). Moreover, administration of Ang-(1-7) to wildtype mice on the high salt diet exhibited enhanced injury that was absent in the Mas null mice (21). Shao et al reported that exogenous Ang-(1-7) exacerbated diabetic injury in streptozotocin-treated rats (45). In this regard, Ang-(1-7) stimulated cellular proliferation in human primary mesangial cells through both ERK and p38 kinase pathways that were sensitive to the Ang-(1-7) antagonist DAL (54). Scholey and colleagues further show that Ang-(1-7) stimulated the ERK1/2 pathway via cAMP/protein kinase A in human mesangial cells (31). Moreover, Burns et al reported that the Ang II-induced MT in the NRK-52E cell line was abolished by both an ACE2 inhibitor and the DAL antagonist, but not the AT1 receptor antagonist irbesartan (5). The Burns study further showed that Ang-(1-7) directly induced MT in the NRK-52E cells, as well as enhanced α-SMA expression in rat kidney cortex following a 10 day infusion of the peptide (5). In contrast to the latter study, we treated the NRK-52E cells with Ang-(1-7) in the presence of MGA to provide a pathological stimulus. It is possible that the higher dose of Ang-(1-7) may explain the marked cellular differences apparent in the two studies. Indeed, the latter study reported that 10 μM Ang-(1-7) increased the release and mRNA levels of TGF-β while we found no effect of 100 nM Ang-(1-7) on cytokine release in MGA-exposed cells (5). It is worth noting that both studies demonstrate that the Ang-(1-7)/Mas antagonist DAL blocked the cellular actions of Ang-(1-7) suggesting the Mas receptor may transduce dose-dependent effects of the peptide. In this regard, de Mello-Aires and colleagues reported dose-dependent biphasic effects of Ang-(1-7) on the proximal tubule NHE3 exchanger that were completely blocked by DAL (8). Ang II also exhibits biphasic effects on sodium reabsorption that are mediated through the AT1 receptor and multiple signaling pathways, albeit the tubular actions of Ang II are opposite to those of Ang-(1-7) (22). The cellular effects of Ang-(1-7) following MGA exposure are consistent with a recent report that the peptide attenuated MT induced by high glucose conditions in the NRK52 cells (52). In this regard, Zhou et al demonstrated that Ang-(1-7) blocked the acute stimulation of both ERK1/2 and p38 kinase, as well as reduced TGF-β release in response to the hyperglycemic conditions (52). Gava et al also find that Ang-(1-7) attenuated the release of TGF-β and p38 phosphorylation, as well as reduced 3H-leucine incorporation in the porcine epithelial LLPK1 cell line following high glucose exposure (18). In the present study, we failed to demonstrate a significant effect of Ang-(1-7) to attenuate the MGA-induced release of TGF-β. Since the cellular actions of MGA are closely linked to TGF-β, the current results suggest that Ang-(1-7) may influence the TGF-β dependent phosphorylation of ERK1/2 rather than the induced release of the cytokine. Although activation of SMAD proteins is considered the canonical TGF-β signaling pathway, TGF-β stimulates ERK1/2 through phosphorylation of the adaptor protein SRC homology 2 domain-containing-transforming A (SHCA) and subsequent formation of a SHCA/GRB2/SOS complex (28). Since Ang-(1-7) blocked both MGA and TGF-β stimulation of ERK1/2, it is unclear whether the peptide directly influences the TGF-β receptor or the downstream target ERK. The Gava study reported that Ang-(1-7) increased the activity of the cellular phosphatase SHP-1 and that the tyrosine phosphatase inhibitor phenylarsine oxide blocked the inhibition of p38 phosphorylation by Ang-(1-7) (18). Tallant and colleagues demonstrate that Ang-(1-7) activates the dual specificity phosphatases (DUSPs) that targets phosphorylated forms of both tyrosine and serine/threonine residues, and inhibits MAP kinase pathways including ERK1/2 (35). Additional studies are necessary to identify whether cellular phosphatases are responsible for the inhibitory effects of Ang-(1-7) on both the acute and chronic ERK activation in the NRK-52E cells.
A growing body of experimental evidence suggests that AGEs contributes to the development of diabetic injury and other renal and non-renal pathologies (14; 15; 37; 38; 46-48). Myofibroblast transdifferentiation may contribute to tubular injury that accompanies diabetic nephropathy, although the extent that MT contributes to fibrosis is controversial (25; 27). Both AGEs and TGF-β are reported to stimulate MT and fibrosis (14; 29; 30; 37). AGEs reportedly induce MT by interacting with their protein receptors RAGE through both TGF-β dependent and independent pathways (29; 48). Treatment with TGF-β1 receptor kinase inhibitor SB525334 abolished the MGA-induced MT and the chronic activation of ERK1/2 implying the predominance of the TGF-β pathway in the NRK-52E cells. Functional links between AGEs, TGF-β and the differential activation of the renal RAS are becoming increasingly evident as well (48). Cao and colleagues find that AGEs (hydoxychloride-treated rat albumin) increased the expression of the precursor protein angiotensinogen, ACE and AT1 receptors in the NRK-52E cells (8). Chou et al also reported that chronic treatment of NRK-52E cells with high glucose or TGF-β reduced the mRNA and protein expression of ACE2 and the Ang-(1-7)/Mas receptor (12). In human mesangial cells, AGEs (glutaraldehyde-treated BSA) reduced the mRNA expression of ACE2 and the secretion of Ang-(1-7) in the media of the cells (24). We did not assess the synthetic components of the RAS; however, the intracellular levels of Ang I and Ang II were not changed following MGA exposure. In contrast, exposure to MGA was associated with reduced cellular content of Ang-(1-7), and this may reflect the enhanced metabolism of the peptide to Ang-(1-4) as opposed to alterations in the synthesis of Ang-(1-7) from Ang I. The identity of this metabolizing peptidase induced by MGA in the NRK-52E cells is currently unknown. We recently characterized an Ang-(1-7) metalloendopeptidase in sheep cerebrospinal fluid (CSF) and brain medulla that metabolized Ang-(1-7) to Ang-(1-4) (34). The peptidase activity was significantly higher in glucocorticoid-exposed animals and inversely correlated to the CSF levels of Ang-(1-7) consistent with a reduced “Ang-(1-7) tone” in this model of fetal programming (34). Studies are in progress to identify the Ang-(1-7) endopeptidase in the NRK-52 cells, as well as the mechanism for the increased expression of activity following MGA exposure.
The present studies in the NRK-52E cells support the renoprotective effects of Ang-(1-7); however, we could not demonstrate the benefit of blockade of the ACE-Ang II-AT1 receptor axis in the NRK-52E cells. Indeed, the three treatment regiments that included the ACE inhibitor lisinopril, the renin inhibitor aliskiren and the AT1 receptor antagonist losartan failed to attenuate the cellular hypertrophy induced by MGA. We also found no effect of AT1 receptor blockade on α-SMA, TGF-β release or chronic ERK1/2 phosphorylation by MGA. Although we did not perform an extensive characterization of the RAS components in these cells; the NRK-52E cells express a complete RAS capable of expressing the active peptides Ang II and Ang-(1-7), as well as their respective receptors and associated signaling pathways (1; 8; 52; 53). It is important to emphasize that the current findings do not imply that RAS inhibition is not beneficial in the treatment of diabetic renal disease, although the current therapeutic regimen of RAS blockade does not abolish or reverse diabetic nephropathy (41). Moreover, recent clinical trials with dual or combined RAS blockade may not provide optimal renal protection and may increase deleterious outcomes (33; 44). In this regard, these therapies may actually reduce the renal expression of Ang-(1-7), and RAS blockade may not provide the potential benefit to the kidney due to the loss of local Ang-(1-7) expression (39; 48). Clearly, our studies utilizing an in vitro cell approach in the NRK-52E cells require further validation in a diabetic animal model or following chronic in vivo AGE treatment.
In conclusion, we demonstrate that the alternative peptide product of the RAS Ang-(1-7) attenuates AGE-induced hypertrophy, chronic ERK activation, TGF-β-induced ERK, and MT of the proximal tubule NRK-52E cell line. The reduced expression of intracellular levels of Ang-(1-7) by AGE may contribute to the cellular responses associated with AGE exposure in the NRK-52E. Hence, supplementation of an orally active form of Ang-(1-7) with either an AT1 receptor antagonist or ACE inhibitor may potentially provide a more effective therapeutic approach to attenuate renal injury associated with increased expression of advanced glycated products.
Highlights.
AGE product methylglyoxal albumin (MGA) induces myofibroblast transition (MT)
MGA reduced intracellular Angiotensin-(1-7) [Ang 7] but increased TGF-β release.
Ang 7 treatment abolished TGF-β activation of ERK1/2 but not TGF-β release.
Ang 7 treatment also abolished MGA-induced MT and ERK1/2 stimulation.
Ang 7 limits MT by inhibiting ERK1/2 in MGA-exposed renal epithelial cells.
Acknowledgments
This present studies represent partial fulfillment of the requirements for the degree of Doctorate of Philosophy in the Department of Physiology and Pharmacology Wake Forest University School of Medicine for Ebaa M. Alzayadneh. The authors acknowledge Nancy T. Pirro for her technical contribution to these studies.
Funding: These studies were supported by the National Institutes of Health (HL-51952, HL-56973) and the Wake Forest Venture Fund.
Footnotes
Conflict of Interests: None declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Alzayadneh EM, Chappell MC. Nuclear expression of renin-angiotensin system components in NRK-52E renal epithelial cells. J Renin Angiotensin Aldosterone Syst. 2014 doi: 10.1177/1470320313515039. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benter IF, Yousif MH, Dhaunsi GS, Kaur J, Chappell MC, Diz DI. Angiotensin-(1-7) prevents activation of NADPH oxidase and renal vascular dysfunction in diabetic hypertensive rats. Am J Nephrol. 2008;28:25–33. doi: 10.1159/000108758. [DOI] [PubMed] [Google Scholar]
- 3.Benter IF, Yousif MHM, Anim JT, Cojocel C, Diz DI. Angiotensin-(1-7) prevents development of severe hypertension and end-organ damage in spontaneously hypertensive rats treated with L-NAME. Am J Physiol Heart Circ Physiol. 2006;290:H684–H691. doi: 10.1152/ajpheart.00632.2005. [DOI] [PubMed] [Google Scholar]
- 4.Brenner BM, Cooper ME, de ZD, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 5.Burns WC, Velkoska E, Dean R, Burrell LM, Thomas MC. Angiotensin II mediates epithelial-to-mesenchymal transformation in tubular cells by ANG 1-7/MAS-1-dependent pathways. Am J Physiol Renal Physiol. 2010;299:F585–F593. doi: 10.1152/ajprenal.00538.2009. [DOI] [PubMed] [Google Scholar]
- 6.Burns WC, Twigg SM, Forbes JM, Pete J, Tikellis C, Thallas-Bonke V, Thomas MC, Cooper ME, Kantharidis P. Connective Tissue Growth Factor plays an important role in advanced glycation end product - induced tubular epithelial-to-mesenchymal transition: Implications for diabetic renal disease. J Am Soc Nephrol. 2006;17:2484–2494. doi: 10.1681/ASN.2006050525. [DOI] [PubMed] [Google Scholar]
- 7.Cao W, Xu J, Zhou ZM, Wang GB, Hou FF, Nie J. Advanced oxidation protein products activate intrarenal renin-angiotensin system via a CD36-mediated, redox-dependent pathway. Antioxid Redox Signal. 2013;18:19–35. doi: 10.1089/ars.2012.4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castelo-Branco RC, Leite-Delova DC, de Mello-Aires M. Dose-dependent effects of angiotensin-(1-7) on the NHE3 exchanger and [Ca(2+)](i) in in vivo proximal tubules. Am J Physiol Renal Physiol. 2013;304:F1258–F1265. doi: 10.1152/ajprenal.00401.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chappell MC. Emerging evidence for a functional angiotesin-converting enzyme 2-angiotensin-(1-7)-Mas receptor axis; more than regulation of blood pressure? Hypertension. 2007;50:596–599. doi: 10.1161/HYPERTENSIONAHA.106.076216. [DOI] [PubMed] [Google Scholar]
- 10.Chappell MC, Marshall AC, Alzayadneh EM, Shaltout HA, Diz DI. Update on the Angiotensin Converting Enzyme 2-Angiotensin (1-7)-Mas Receptor Axis: Fetal Programing, Sex Differences, and Intracellular Pathways. Front Endocrinol (Lausanne) 2014;4:201. doi: 10.3389/fendo.2013.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Chen JK, Neilson EG, Harris RC. Role of EGF receptor activation in angiotensin II-induced renal epithelial cell hypertrophy. J Am Soc Nephrol. 2006;17:1615–1623. doi: 10.1681/ASN.2005111163. [DOI] [PubMed] [Google Scholar]
- 12.Chou CH, Chuang LY, Lu CY, Guh JY. Interaction between TGF-beta and ACE2-Ang-(1-7)-Mas pathway in high glucose-cultured NRK-52E cells. Mol Cell Endocrinol. 2013;366:21–30. doi: 10.1016/j.mce.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Christiane R, Tzvetanka B, Sybille F, Martin F, Gunter W. Advanced glycation end-products induce cell cycle arrest and hypertrophy in podocytes. Nephrology Dialysis Transplantation. 2008;23:2179. doi: 10.1093/ndt/gfn085. [DOI] [PubMed] [Google Scholar]
- 14.Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY. Transforming growth factor-β regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999;56:1455–1467. doi: 10.1046/j.1523-1755.1999.00656.x. [DOI] [PubMed] [Google Scholar]
- 15.Forbes JM, Cooper ME, Oldfield MD, Thomas MC. Role of advanced glycation end products in diabetic nephropathy. J Am Soc Nephrol. 2003;14:S254–S258. doi: 10.1097/01.asn.0000077413.41276.17. [DOI] [PubMed] [Google Scholar]
- 16.Fujita H, Omori S, Ishikura K, Hida M, Awazu M. ERK and p38 mediate high-glucose-induced hypertrophy and TGF-β expression in renal tubular cells. American Journal of Physiology - Renal Physiology. 2003;286:F120–F126. doi: 10.1152/ajprenal.00351.2002. [DOI] [PubMed] [Google Scholar]
- 17.Gallagher PE, Ferrario CM, Tallant EA. MAP kinase/phosphatase pathway mediates the regulation of ACE2 by angiotensin peptides. Am J Physiol Cell Physiol. 2008;295:C1169–C1174. doi: 10.1152/ajpcell.00145.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gava E, Samad-Zadeh A, Zimpelmann J, Bahramifarid N, Kitten GT, Santos RA, Touyz RM, Burns KD. Angiotensin-(1-7) activates a tyrosine phosphatase and inhibits glucose-induced signalling in proximal tubular cells. Nephrol Dial Transplant. 2009;24:1766–1773. doi: 10.1093/ndt/gfn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giani JF, Munoz MC, Pons RA, Cao G, Toblli JE, Turyn D, Dominici FP. Angiotensin-(1-7) reduces proteinuria and diminishes structural damage in renal tissue of stroke-prone spontaneously hypertensive rats. Am J Physiol Renal Physiol. 2011;300:F272–F282. doi: 10.1152/ajprenal.00278.2010. [DOI] [PubMed] [Google Scholar]
- 20.Gross ML, El-Shakmak A, Szabo A, Koch A, Kuhlmann A, Munter K, Ritz E, Amann K. ACE-inhibitors but not endothelin receptor blockers prevent podocyte loss in early diabetic nephropathy. Diabetologia. 2003;46:856–868. doi: 10.1007/s00125-003-1106-8. [DOI] [PubMed] [Google Scholar]
- 21.Heringer-Walther S, Gembardt F, Perschel FH, Katz N, Schultheiss HP, Walther T. The genetic deletion of Mas abolishes salt induced hypertension in mice. Eur J Pharmacol. 2012;689:147–153. doi: 10.1016/j.ejphar.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 22.Houillier P, Chambrey R, Achard JM, Froissart M, Poggioli J, Paillard M. Signaling pathways in the biphasic effect of angiotensin II on apical Na/H antiport activity in proximal tubule. Kidney Int. 1996;50:1496–1505. doi: 10.1038/ki.1996.464. [DOI] [PubMed] [Google Scholar]
- 23.Huang JS, Chuang LY, Guh JY, Yang YL, Hsu MS. Effect of taurine on advanced glycation end products-induced hypertrophy in renal tubular epithelial cells. Toxicol Appl Pharmacol. 2008;233:220–226. doi: 10.1016/j.taap.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Ishibashi Y, Matsui T, Yamagishi S. Olmesartan Blocks Advanced Glycation End Products-induced VCAM-1 Gene Expression in Mesangial Cells by Restoring Angiotensin-converting Enzyme 2 Level. Horm Metab Res. 2013 doi: 10.1055/s-0033-1361114. [DOI] [PubMed] [Google Scholar]
- 25.Jinde K, Nikolic-Paterson DJ, Huang XR, Sakai H, Kurokawa K, Atkins RC, Lan HY. Tubular phenotypic change in progressive tubulointerstitial fibrosis in human glomerulonephritis. Am J Kidney Dis. 2001;38:761–769. doi: 10.1053/ajkd.2001.27693. [DOI] [PubMed] [Google Scholar]
- 26.Kriz W, Kaissling B, Le HM. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest. 2011;121:468–474. doi: 10.1172/JCI44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM, Derynck R. TGF-βactivates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007;26:3957–3967. doi: 10.1038/sj.emboj.7601818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li JH, Huang XR, Zhu HJ, Oldfield M, Cooper M, Truong LD, Johnson RJ, Lan HY. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: implications for diabetic renal and vascular disease. FASEB J. 2004;18:176–178. doi: 10.1096/fj.02-1117fje. [DOI] [PubMed] [Google Scholar]
- 29.Li JH, Wang W, Huang XR, Oldfield M, Schmidt AM, Cooper ME, Lan HY. Advanced glycation end products induce tubular epithelial-myofibroblast transition through the RAGE-ERK1/2 MAP kinase signaling pathway. Am J Pathol. 2004;164:1389–1397. doi: 10.1016/S0002-9440(10)63225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu GC, Oudit GY, Fang F, Zhou J, Scholey JW. Angiotensin-(1-7)-induced activation of ERK1/2 is cAMP/protein kinase A-dependent in glomerular mesangial cells. Am J Physiol Renal Physiol. 2012;302:F784–F790. doi: 10.1152/ajprenal.00455.2011. [DOI] [PubMed] [Google Scholar]
- 31.Makita Z, Radoff S, Rayfield EJ, Yang Z, Skolnik E, Delaney V, Friedman EA, Cerami A, Vlassara H. Advanced glycosylation end products in patients with diabetic nephropathy. N Engl J Med. 1991;325:2836–842. doi: 10.1056/NEJM199109193251202. [DOI] [PubMed] [Google Scholar]
- 32.Mann JF, Schmieder RE, McQueen M, Dyal L, Schumacher H, Pogue J, Wang X, Maggioni A, Budaj A, Chaithiraphan S, Dickstein K, Keltai M, Metsarinne K, Oto A, Parkhomenko A, Piegas LS, Svendsen TL, Teo KK, Yusuf S. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008;372:547–553. doi: 10.1016/S0140-6736(08)61236-2. [DOI] [PubMed] [Google Scholar]
- 33.Marshall AC, Pirro NT, Rose JC, Diz DI, Chappell MC. Evidence for an Angiotensin-(1-7) Neuropeptidase Expressed in the Brain Medulla and CSF of Sheep. J Neurochem. 2014 doi: 10.1111/jnc.12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCollum LT, Gallagher PE, Ann Tallant E. Angiotensin-(1-7) attenuates angiotensin II-induced cardiac remodeling associated with upregulation of dual-specificity phosphatase 1. American Journal of Physiology - Heart and Circulatory Physiology. 2012;302:H801–H810. doi: 10.1152/ajpheart.00908.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moon JY, Tanimoto M, Gohda T, Hagiwara S, Yamazaki T, Ohara I, Murakoshi M, Aoki T, Ishikawa Y, Lee SH, Jeong KH, Lee TW, Ihm CG, Lim SJ, Tomino Y. Attenuating effect of angiotensin-(1-7) on angiotensin II-mediated NAD(P)H oxidase activation in type 2 diabetic nephropathy of KK-A(y)/Ta mice. Am J Physiol Renal Physiol. 2011;300:F1271–F1282. doi: 10.1152/ajprenal.00065.2010. [DOI] [PubMed] [Google Scholar]
- 36.Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, Atkins RC, Osicka T, Jerums G, Cooper ME. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE) J Clin Invest. 2001;108:1853–1863. doi: 10.1172/JCI11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, Atkins RC, Osicka T, Jerums G, Cooper ME. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE) J Clin Invest. 2001;108:1853–1863. doi: 10.1172/JCI11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parving HH, Brenner BM, McMurray JJ, de Zeeuw D, Haffner SM, Solomon SD, Chaturvedi N, Ghadanfar M, Weissbach N, Xiang Z, Armbrecht J, Pfeffer MA. Aliskiren Trial in Type 2 Diabetes Using Cardio-Renal Endpoints (ALTITUDE): rationale and study design. Nephrol Dial Transplant. 2009;24:1663–1671. doi: 10.1093/ndt/gfn721. [DOI] [PubMed] [Google Scholar]
- 39.Rasch R, Norgaard JO. Renal enlargement: comparative autoradiographic studies of 3H-thymidine uptake in diabetic and uninephrectomized rats. Diabetologia. 1983;25:280–287. doi: 10.1007/BF00279944. [DOI] [PubMed] [Google Scholar]
- 40.Ruggenenti P, Cravedi P, Remuzzi G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nature Reviews Nephrology. 2010;6:319–330. doi: 10.1038/nrneph.2010.58. [DOI] [PubMed] [Google Scholar]
- 41.Santos RA. Angiotensin-(1-7) Hypertension. 2014;63:1138–1147. doi: 10.1161/HYPERTENSIONAHA.113.01274. [DOI] [PubMed] [Google Scholar]
- 42.Seyer-Hansen K, Hansen J, Gundersen HJ. Renal hypertrophy in experimental diabetes. A morphometric study. Diabetologia. 1980;18:501–505. doi: 10.1007/BF00261707. [DOI] [PubMed] [Google Scholar]
- 43.Shaltout HA, Westwood B, Averill DB, Ferrario CM, Figueroa J, Diz DI, Rose JC, Chappell MC. Angiotensin metabolism in renal proximal tubules, urine and serum of sheep: Evidence for ACE2-dependent processing of Angiotensin II. Am J Physiol Renal Physiol. 2006;292:F82–F91. doi: 10.1152/ajprenal.00139.2006. [DOI] [PubMed] [Google Scholar]
- 44.Shao Y, He M, Zhou L, Yao T, Huang Y, Lu LM. Chronic angiotensin (1-7) injection accelerates STZ-induced diabetic renal injury. Acta Pharmacol Sin. 2008;29:829–837. doi: 10.1111/j.1745-7254.2008.00812.x. [DOI] [PubMed] [Google Scholar]
- 45.Soulis-Liparota T, Cooper ME, Dunlop M, Jerums G. The relative roles of advanced glycation, oxidation and aldose reductase inhibition in the development of experimental diabetic nephropathy in the Sprague-Dawley rat. Diabetologia. 1995;38:387–394. doi: 10.1007/BF00410275. [DOI] [PubMed] [Google Scholar]
- 46.Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A, Pischetsrieder M, Stern D, Schmidt AM, D'Agati VD. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol. 2000;11:1656–1666. doi: 10.1681/ASN.V1191656. [DOI] [PubMed] [Google Scholar]
- 47.Thomas MC, Tikellis C, Burns WM, Bialkowski K, Cao Z, Coughlan MT, Jandeleit-Dahm K, Cooper ME, Forbes JM. Interactions between renin angiotensin system and advanced glycation in the kidney. J Am Soc Nephrol. 2005;16:2976–2984. doi: 10.1681/ASN.2005010013. [DOI] [PubMed] [Google Scholar]
- 48.Tikellis C, Bialkowski K, Pete J, Sheehy K, Su Q, Johnston C, Cooper M, Thomas M. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57:1018–1025. doi: 10.2337/db07-1212. [DOI] [PubMed] [Google Scholar]
- 49.Westwood ME, Argirov OK, Abordo EA, Thornalley PJ. Methylglyoxal-modified arginine residues--a signal for receptor-mediated endocytosis and degradation of proteins by monocytic THP-1 cells. Biochim Biophys Acta. 1997;1356:84–94. doi: 10.1016/s0167-4889(96)00154-1. [DOI] [PubMed] [Google Scholar]
- 50.Yang J, Liu Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol. 2001;159:1465–1475. doi: 10.1016/S0002-9440(10)62533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou L, Xue H, Wang Z, Ni J, Yao T, Huang Y, Yu C, Lu L. Angiotensin-(1-7) attenuates high glucose-induced proximal tubular epithelial-to-mesenchymal transition via inhibiting ERK1/2 and p38 phosphorylation. Life Sci. 2012;90:454–462. doi: 10.1016/j.lfs.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 52.Zhou L, Xue H, Yuan P, Ni J, Yu C, Huang Y, Lu LM. Angiotensin AT1 receptor activation mediates high glucose-induced epithelial-mesenchymal transition in renal proximal tubular cells. Clin Exp Pharmacol Physiol. 2010;37:e152–e157. doi: 10.1111/j.1440-1681.2010.05421.x. [DOI] [PubMed] [Google Scholar]
- 53.Zimmerman D, Burns KD. Angiotensin-(1-7) in kidney disease: a review of the controversies. Clin Sci (Lond) 2012;123:333–346. doi: 10.1042/CS20120111. [DOI] [PubMed] [Google Scholar]