

Abstract
Mitochondria critically regulate cytoplasmic Ca2+ concentration ([Ca2+]c), but the effects of sensory neuron injury have not been examined. Using FCCP (1μM) to eliminate mitochondrial Ca2+ uptake combined with oligomycin (10μM) to prevent ATP depletion, we first identified features of depolarization-induced neuronal [Ca2+]c transients that are sensitive to blockade of mitochondrial Ca2+ buffering in order to assess mitochondrial contributions to [Ca2+]c regulation. This established the loss of a shoulder during the recovery of the depolarization (K+)-induced transient, increased transient peak and area, and elevated shoulder level as evidence of diminished mitochondrial Ca2+ buffering. We then examined transients in Control neurons and neurons from the 4th lumbar (L4) and L5 dorsal root ganglia after L5 spinal nerve ligation (SNL). The SNL L4 neurons showed decreased transient peak and area compared to control neurons, while the SNL L5 neurons showed increased shoulder level. Additionally, SNL L4 neurons developed shoulders following transients with lower peaks than Control neurons. Application of FCCP plus oligomycin elevated resting [Ca2+]c in SNL L4 neurons more than in Control neurons. Whereas application of FCCP plus oligomycin 2s after neuronal depolarization initiated mitochondrial Ca2+ release in most Control and SNL L4 neurons, this usually failed to release mitochondrial Ca2+ from SNL L5 neurons. For comparable cytoplasmic Ca2+ loads, the releasable mitochondrial Ca2+ in SNL L5 neurons was less than Control while it was increased in SNL L4 neurons. These findings show diminished mitochondrial Ca2+ buffering in axotomized SNL L5 neurons but enhanced Ca2+ buffering by neurons in adjacent SNL L4 neurons.
Keywords: Neuropathic pain, mitochondrial Ca2+, spinal nerve ligation, sensory neuron function
1. Introduction
Pain following peripheral nerve injury is in part the result of elevated sensory neuron excitability due to disordered Ca2+ signaling. Specifically, axonal trauma is followed by diminished Ca2+ influx through voltage-gated Ca2+ channels (Hogan et al., 2000; McCallum et al., 2006), in part attributable to reduced activation of Ca2+-calmodulin-dependent protein kinase II (Kawano et al., 2009; Kojundzic et al., 2010; Tang et al., 2012). The cytoplasmic Ca2+ signal initiated by Ca2+ influx is further shaped in sensory neurons by the simultaneous processes of Ca2+ extrusion, sequestration, and release from stores. We have identified dysfunction of these processes in axotomized neurons following painful nerve injury, including elevated function of the plasma membrane Ca2+-ATPase (PMCA) (Gemes et al., 2012b), accompanied by decreased resting cytoplasmic Ca2+ ([Ca2+]c) (Fuchs et al., 2005), reduced function of the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) (Duncan et al., 2013) that reduces ER Ca2+ stores (Gemes et al., 2009; Rigaud et al., 2009), with resulting elevation of store-operated Ca2+ entry (SOCE) (Gemes et al., 2011) and diminished release of Ca2+ from stores upon neuronal activity through the process of Ca2+-induced Ca2+ release. Together, these disturbances of Ca2+ signaling contribute to elevated generation and transmission of high-frequency trains of action potentials in the injured sensory neurons (Gemes et al., 2009; Gemes et al., 2012a; Hogan et al., 2008; Lirk et al., 2008; Sapunar et al., 2005; Tang et al., 2012).
Mitochondria are also recognized as a critical element in intracellular Ca2+ management (Nicholls, 2005). In sensory neurons, mitochondria serve as a nonsaturable buffer with a clearance rate in neurons that exceeds the PMCA and Na+/Ca2+ exchanger (Herrington et al., 1996). Mitochondrial sequestering of Ca2+ during periods of high [Ca2+]c after neuronal activation buffers peak [Ca2+]c, while the subsequent slow release of Ca2+ as the [Ca2+]c falls prolongs [Ca2+]c recovery and produces a characteristic shoulder in the Ca2+ transient (Werth and Thayer, 1994). Thus, mitochondrial Ca2+ buffering modulates the duration and peak levels of activity-induced Ca2+ transients, and protects the neuron from excessive elevations of [Ca2+]c. More recent findings indicate constitutive operation of mitochondrial Ca2+ cycling even at low levels of [Ca2+]c in resting sensory neurons (Colegrove et al., 2000; Kang et al., 2008). The ability of mitochondria to sequester Ca2+ is tied to their energy state, since Ca2+ influx through the mitochondrial Ca2+ uniporter is driven by the potential difference across the inner mitochondrial membrane (ΔΨm). In a reciprocal fashion, mitochondrial energy production in sensory neurons is upregulated by Ca2+ loading (Duchen, 1999), which provides a link between cell activity and energy production through elevations of [Ca2+]c.
There has been minimal exploration of mitochondrial function in sensory neurons subjected to models of painful traumatic neuropathy. We have previously noted a decrease in the incidence of a shoulder (also called a plateau) in the depolarization-induced transient trace of axotomized fifth lumbar (L5) neurons in the spinal nerve ligation (SNL) model of peripheral nerve injury (Fuchs et al., 2005), which suggests a deficit in mitochondrial function after injury. Furthermore, there is a growing recognition of a contributing role of mitochondrial dysfunction in painful neuropathy (Chu et al., 2011; Flatters and Bennett, 2006; Joseph and Levine, 2006). Accordingly, we hypothesized that nerve injury disrupts Ca2+ buffering by mitochondria in axotomized sensory neurons. Ideally, mitochondrial buffering of activity-induced Ca2+ influx would be gauged by isolating this process through blocking SERCA and PMCA, but this leads to inability of the neuron to maintain stable [Ca2+]c under resting conditions (Duncan et al., 2013). We therefore adopted the strategy of first identifying measurable features of mitochondrial buffering revealed by their sensitivity to ΔΨm elimination with FCCP. These features were then compared in injured and control neurons, including the L4 population of neurons that are intact after SNL but are exposed to inflammation that accompanies Wallerian degeneration of the degenerating L5 axonal fragments. Overall, our findings confirm a fundamental dependence of sensory neuron Ca2+ homeostasis on mitochondrial activity. Furthermore, we have identified extensive disruption of this role in divergent patterns for both axotomized (L5) and intact (L4) neuronal populations after SNL, such that the axotomized L5 neurons exhibit features suggesting reduced mitochondrial Ca2+ buffering while the L4 population shows amplified mitochondrial buffering activity.
2. Results
2.1 FCCP plus oligomycin for blockade of mitochondrial Ca2+ buffering
As an overall strategy for evaluating injury effects on mitochondrial Ca2+ buffering, we first sought to characterize which quantifiable features of the depolarization-induced Ca2+ transient (Fig. 1A and B) reliably reflect mitochondrial Ca2+ uptake and release. This requires selective blockade of mitochondrial Ca2+ buffering, for which we chose the protonophore FCCP (1μM) since this prevents Ca2+ accumulation by elimination of ΔΨm. However, protonophores also deplete ATP through reversal of ATP synthase function (Budd and Nicholls, 1996), which may disrupt ATP-dependent SERCA and PMCA pathways, resulting in nonspecific inhibition of Ca2+ sequestration and extrusion. We therefore co-administered the ATP synthase blocker oligomycin (10μM), which has been shown to prevent ATP depletion in dissociated neurons (Budd and Nicholls, 1996). To further confirm preservation of ATP levels in our dissociated sensory neurons, we assayed the level of ATP available for driving physiological functions by measuring the performance of the ATP-driven Ca2+ pump PMCA as an indictor of ATP availability (Schatzmann and Vincenzi, 1969), quantified as the time constant of recovery for transients during SERCA blockade (thapsigargin 1μM, Fig. 1C) (Gemes et al., 2012b). This was unaffected by this combination of FCCP and oligomycin (FCCP/Oligo, Fig. 1D), which confirms prior findings that FCCP/Oligo does not deplete ATP during the timeframe of our experiments (Budd and Nicholls, 1996). Using TMRM as an indicator of ΔΨm, we additionally confirmed the efficacy of FCCP/Oligo in collapsing ΔΨm (Fig. 1E), thereby validating FCCP/Oligo application as a means of blocking mitochondrial Ca2+ buffering.
Figure 1.

Measures and protocol validation. (A) Measured parameters for K+-induced Ca2+ transients are shown for resting level, amplitude, peak, shoulder level, and time for 80% recovery to resting level. (B) To determine shoulder level, [Ca2+]c traces (top panel) were differentiated (bottom panel, with the first positive and first negative maxima clipped), and the presence of two negative maxima (indicated by *) was considered as evidence of a shoulder. The level of the shoulder was measured at the inflection point (dashed arrows), identified as time during the recovery phase at which the slope of the [Ca2+]c vs. time trace transient shifted from becoming less negative to becoming more negative. (C) To test adequacy of ATP levels, plasma membrane Ca2+ ATPase (PMCA) activity is measured as the time constant (τ) for the exponential recovery of [Ca2+]c from small Ca2+ loads. The top panel is an example of repeated depolarization during application of Tyrode’s solution (Tyr) and after vehicle application in Tyrode’s. The bottom panel shows response to co-administration of FCCP (1μM) and oligomycin (10μM). (D) Average data show PMCA activity is unchanged. (E) Application of FCCP successfully reduces mitochondrial ΔΨm recorded by TMRM fluorescence, normalized as percent of baseline.
2.2 Effect of mitochondrial blockade on Ca2+ transients in Control and SNL neurons
To identify measurable traits of mitochondrial buffering, we compared Ca2+ transients in Control neurons before and after elimination of mitochondrial Ca2+ uptake through application of FCCP/Oligo (Fig. 2). Two durations of K+ depolarization were used, 0.5s and 1.3s, in order to encompass a range of Ca2+ loads, and findings were compared to Control neurons that received the same depolarization protocol but with application of only vehicle solution during the second pair of depolarizations. A shoulder during the transient recovery was evident in 20 of 41 (49%) baseline transients that followed depolarization by 0.5s K+ (hereafter referred to as “0.5s transients”), which decreased to 7 of 41 (17%, P < 0.01) after FCCP/Oligo. At baseline, 1.3s transients showed a shoulder phase in 39 of 41 neurons (95%), which decreased to 5 of 41 (12%, P < 0.001) after FCCP/Oligo. In contrast, vehicle controls showed that all 0.5s transients with shoulders at baseline (n=3) retained shoulders on repeat 0.5s transients, as did all but one of 1.3s transients with shoulders (n=41). These observations confirm the findings of others (Svichar et al., 1997; Thayer and Miller, 1990; Werth and Thayer, 1994) that blocking mitochondrial Ca2+ accumulation with FCCP interferes with the formation of a shoulder, from which we can infer that the shoulder is an indicator of prior neuronal mitochondrial Ca2+ buffering.
Figure 2.

Demonstration traces of the response of neuronal [Ca2+]c to depolarizations induced by K+ application of 0.5s and 1.3s duration in Tyrode’s solution (Tyr) and 1.5 minutes after switching to a bath solution containing either vehicle (A) or FCCP/Oligo (B). Response to FCCP/Oligo application is shown for neurons from Control dorsal root ganglion and from the fourth lumbar (L4) and L5 ganglia after L5 spinal nerve ligation (SNL). The second depolarization was initiated only after recovery from the preceding depolarization. During FCCP/Oligo application, the amplitude of the 1.3s transient increases by 2.10-fold for Control, 2.25-fold for SNL L4, and 1.53-fold for SNL L5, compared to baseline (Tyr).
Mitochondrial regulation of cytoplasmic Ca2+ accumulation during activation of Control neurons was tested by determining the fold change of the depolarization-induced peak [Ca2+]c caused by FCCP/Oligo application, normalized against the baseline peak (Table 1). Whereas the peak of 0.5s transients was not predictably affected by FCCP/Oligo (i.e. the one-sample Wilcoxon test showed that the fold change was not significantly different than 1), transient peaks during 1.3s transients were more than doubled by FCCP/Oligo, which indicates that mitochondrial buffering regulates the extent of [Ca2+]c rise, confirming prior findings in sensory neurons (Gover et al., 2007; Lu et al., 2006; Svichar et al., 1997; Thayer and Miller, 1990). A lack of a similar change in vehicle controls (Table 1) shows that the increased transient peak is not attributable to repetitive stimulation. To identify if injury affects mitochondrial regulation of the 1.3s transient peak, we compared the responses to FCCP/Oligo application of SNL L4 and L5 neurons that were studied concurrently with the Control neurons. This showed a significant main effect of Group, with a pattern of a greater increase for SNL L4 neurons and decrease for SNL L5 neurons, (Table 1), although paired comparisons were not significant.
Table 1.
Fold change in Peak and Area induced by FCCP/Oligo
| Peak | Area | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Depolarization | 0.5s | 1.3s | 0.5s | 1.3s | ||||||||
| Agent | Vehicle | FCCP/ Oligo | Vehicle | FCCP/Oligo | Vehicle | FCCP/ Oligo | Vehicle | FCCP/Oligo | ||||
| Group | C | C | C | C | L4 | L5 | C | C | C | C | L4 | L5 |
| n | 66 | 41 | 70 | 41 | 33 | 26 | 61 | 44 | 66 | 44 | 38 | 28 |
| Median | 1.25 | 1.10 | 1.05 | 2.04 | 2.47 | 1.76 | 1.84 | 2.38 | 1.30 | 2.92 | 4.30 | 2.71 |
| IQR | 1.08–1.52 | 0.59–1.61 | 0.93–1.14 | 1.56–2.84 | 1.69–4.20 | 1.24–3.10 | 1.18–2.72 | 1.07–3.98 | 0.99–1.60 | 1.86–5.56 | 2.55–6.26 | 1.70–5.70 |
| 1-sample Wilcoxon | <0.001 | 0.692 | 0.07 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | ||||
| Vehicle vs. FCCP/Oligo | <0.001 | 0.202 | <0.001 | |||||||||
| Group Main Effect | 0.046 | 0.273 | ||||||||||
| Paired Comparisons | ||||||||||||
Measured parameters include the peak and area of the transients produced by depolarization with K+. Depolarizations were of either 0.5s or 1.3s duration. Agents included either vehicle or FCCP 1μM combined with oligomycin 10μM (FCCP/Oligo). Groups included Control (C), and neurons from the 4th lumbar (L4) and L5 dorsal root ganglia after spinal nerve ligation. n is the number of neurons. The median and interquartile range (IQR) are shown for the fold change of the measured parameter produced by FCCP/Oligo application, calculated as the ratio of the second depolarization (following FCCP/Oligo) divided by the first depolarization (baseline) for each neuron. 1-sample Wilcoxon is the P of an effect of vehicle or FCCP/Oligo in that group using a one-sample Wilcoxon test (compared to a value of 1). Vehicle vs. FCCP/Oligo is the P for the comparison of vehicle vs. FCCP/Oligo in control neurons using Mann-Whitney test. Group Main Effect P is the probability of a main effect comparing the three groups (C, L4, L5) during FCCP/Oligo application, using Kruskal-Wallis. Paired Comparisons gives the P for significant paired comparisons between these groups (blank when none) using Dunn’s test.
P < 0.05.
P < 0.05,
P < 0.01.
Transient area (Table 1) was increased upon FCCP/Oligo application at both depolarization durations, but statistical comparison (Mann-Whitney) to the changes induced by vehicle application showed that only 1.3s transients were more affected by FCCP/Oligo than by vehicle alone. The selective effects of FCCP/Oligo further highlight the preferential influence of mitochondrial buffering on large Ca2+ loads. Comparing the effect of FCCP/Oligo on 1.3s transient area in SNL L4 and L5 neurons to Control showed that there was no effect of group on the regulation of this parameter by mitochondrial buffering.
Duration of the transient, measured as the time to achieve 80% recovery back to resting level (T80, Table 2), was prolonged in the second depolarizations during FCCP/Oligo for both 0.5s and 1.3s transients, but these prolongations were not different from the effects of vehicle alone. This indicates that T80 does not reflect mitochondrial function, and it was not examined further.
Table 2.
Fold change in T80 and τ induced by FCCP/Oligo
| T80 | Shoulder Level | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Depolarization | 0.5s | 1.3s | 0.5s | 1.3s | Combined | |||||||
| Agent | Vehicle | FCCP/ Oligo | Vehicle | FCCP/ Oligo | Vehicle | FCCP/ Oligo | Vehicle | FCCP/ Oligo | Vehicle | FCCP/Oligo | ||
| Group | C | C | C | C | C | C | C | C | C | C | L4 | L5 |
| n | 61 | 44 | 66 | 44 | 3 | 7 | 19 | 5 | 22 | 9 | 9 | 4 |
| Median | 1.39 | 1.32 | 1.33 | 1.78 | 1.73 | 1.26 | 1.06 | 2.27 | 1.06 | 1.51 | 2.79 | 0.77 |
| IQR | 1.01–1.98 | 0.88–2.46 | 1.07–1.91 | 1.00–8.09 | 0.05–2.69 | 0.96–2.33 | 0.86–1.33 | 1.00–7.24 | 0.85–1.36 | 0.91–2.54 | 2.32–9.55 | 0.55–1.46 |
| 1-sample Wilcoxon | <0.001 | 0.002 | <0.001 | <0.001 | 0.75 | 0.109 | 0.251 | 0.125 | 0.206 | 0.027 | ||
| Vehicle vs. FCCP/Oligo | 0.647 | 0.105 | 0.500 | 0.044 | 0.042 | |||||||
| Group Main Effect | 0.009 | |||||||||||
| Paired Comparisons | L4 vs. L5* | |||||||||||
Measured parameters include the transient duration measured as the time for 80% recovery back to baseline from the peak, and the [Ca2+]c level of the shoulder during the recovery phase of transients produced by depolarization with K+ of either 0.5s or 1.3s duration. Agents included either vehicle or FCCP 1μM combined with oligomycin 10μM (FCCP/Oligo). Groups included Control (C), and neurons from the 4th lumbar (L4) and L5 dorsal root ganglia after spinal nerve ligation. n is the number of neurons. The median and interquartile range (IQR) are shown for the fold change of the measured parameter produced by FCCP/Oligo application, calculated as the ratio of the second depolarization (following FCCP/Oligo) divided by the first depolarization (baseline) for each neuron. 1-sample Wilcoxon is the P of an effect of vehicle or FCCP/Oligo in that group using a one-sample Wilcoxon test (compared to a value of 1). Vehicle vs. FCCP/Oligo is the P for the comparison of vehicle vs. FCCP/Oligo in control neurons using Mann-Whitney test. Group Main Effect P is the probability of a main effect comparing the three groups (C, L4, L5) during FCCP/Oligo application, using Kruskal-Wallis. Paired Comparisons gives the P for significant paired comparisons between these groups (blank when none) using Dunn’s test.
P < 0.05.
Since the application of FCCP/Oligo did not fully eliminate the shoulder of the depolarization-induced transient in some neurons, we examined whether FCCP/Oligo treatment affected the level of the shoulder, which represents the set-point of mitochondrial Ca2+ uptake and release. While the separate 0.5s and 1.3s data for the low number of neurons with shoulders both before and after FCCP/Oligo showed no significant effect (Table 2), combining these (using only a single averaged value for neurons that had shoulders for both 0.5 transients and 1.3 transients) showed a 1.51-fold elevation of the shoulder which was not evident in vehicle controls, which suggests that incomplete blockade of mitochondrial buffering results in an elevation of the shoulder.
Together, these effects of FCCP/Oligo show that mitochondrial function modulates the transient peak, area, and shoulder, and that injury affects the extent of FCCP/Oligo-sensitive mitochondrial activity.
2.3 Effect of neuronal injury on transient features regulated by mitochondrial Ca2+ buffering
We next asked which of these features are affected by injury in a fashion indicative of over or underperformance of mitochondrial Ca2+ buffering. We first examined the occurrence of a shoulder as an indicator of mitochondrial involvement in handling cytoplasmic Ca2+ loads induced by neuronal activity. Since injury may influence Ca2+ influx during depolarization of sensory neurons (Hogan et al., 2000; McCallum et al., 2006), we analyzed shoulder occurrence in a manner designed to account for varying Ca2+ loads. Specifically, we exposed each neuron to increasing K+ durations of 0.3s, 0.5s, 0.8s, 1.3s, and 2.5s to test the occurrence of a shoulder across a range of Ca2+ loads (Fig. 3A), and identified the lowest transient peak that was adequate to generate a shoulder in each neuron as an indication of the threshold for mitochondrial control of the transient (Fig. 3B). This revealed that SNL L4 neurons developed shoulders associated with lower peak [Ca2+]c compared to Control and SNL L5 neurons, which suggests that mitochondrial sensitivity to cytoplasmic Ca2+ is enhanced in the SNL L4 population.
Figure 3.

Repeated depolarization protocol and determination of Ca2+ load adequate to produce a shoulder. (A) Cytoplasmic Ca2+ concentration trace (top) and its differentiation trace (bottom) in a control neuron, showing the protocol for sequential depolarizations, and the identification of a shoulder (indicated by dashed arrows) during the recovery phase of transients with larger Ca2+ loads. (B) Determination for each neuron of the lowest peak associated with the appearance of a shoulder shows that neurons from the fourth lumbar dorsal root ganglion after spinal nerve ligation (SNL L4, n=24) developed shoulders associated with lower transient peaks than control (C, n=50) or SNL L5 (n=71) groups. This and other boxplots show median and first and third quartiles (Q1, Q3) with whiskers denoting the lowest datum still within 1.5 times the interquartile range of Q1 and the highest datum still within 1.5 times the interquartile range of Q3*P < 0.05.
We next determined whether injury affects the peak during 1.3s transients, which was shown to reflect mitochondrial function in the FCCP/Oligo experiments above. Neurons from SNL L4 ganglia developed lower transient peaks (Fig. 4A), representing a change opposite that produced by FCCP/Oligo. Since our FCCP/Oligo data also showed that transient area for 1.3s transients is regulated by mitochondrial Ca2+ buffering, we compared that measure between injury groups (Fig. 4B), but this was not affected by injury. Finally, comparison of the level of the shoulder between groups showed that 1.3s transients had higher shoulders in the SNL L5 neurons compared to Control and SNL L4 neurons (Fig. 4C). This shows release of Ca2+ from mitochondria at a higher [Ca2+]c in axotomized L5 neurons than in Control neurons, which duplicates the effect of FCCP/Oligo.
Figure 4.
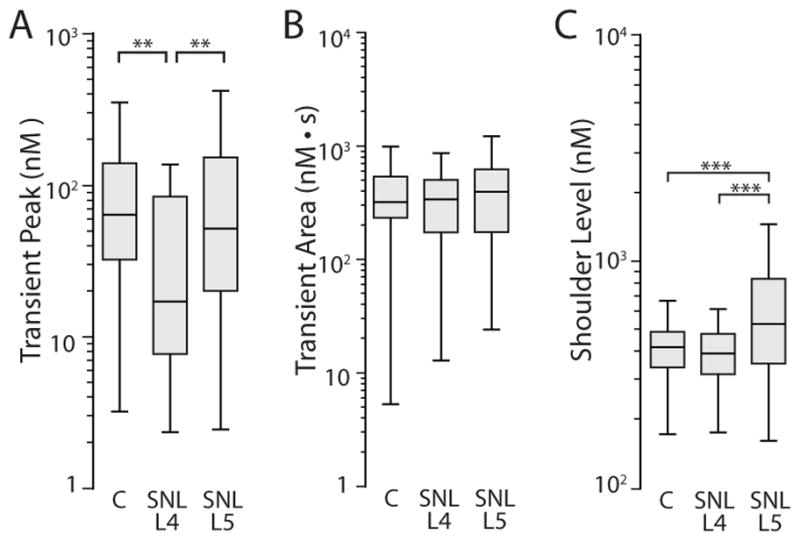
Comparison of FCCP/Oligo-sensitive dimensions of transients induced by 1.3s K+ depolarization in Control neurons compared to neurons from the fourth lumbar dorsal root ganglion after spinal nerve ligation (SNL L4) and SNL L5. (A) Determination of the transient peak. (B) Determination of the transient area. (C) Determination of the level of the shoulder during transient recovery. C, n=56; SNL L4, n=28; SNL L5, n=77. *P < 0.05, **P < 0.01, ***P < 0.001.
2.4 Mitochondrial regulation of resting [Ca2+]c and the effect of injury
In the present data, peripheral nerve injury (SNL) depressed the resting [Ca2+]c in the SNL L5 population (Fig. 5A), as we have noted before (Fuchs et al., 2005). It is unlikely that an injury-induced shift in mitochondrial buffering alone accounts for this effect as we have previously found that injury also alters performance of PMCA and SOCE (Duncan et al., 2013; Gemes et al., 2011), which contribute to setting the resting [Ca2+]c. Therefore, to directly test the role of mitochondrial Ca2+ buffering in controlling resting [Ca2+]c, we examined resting [Ca2+]c before and after FCCP/Oligo administration to resting neurons (Fig. 5B). FCCP/Oligo elevated [Ca2+]c of Control neurons significantly greater than the effect of vehicle (Fig. 5C), indicating a role of mitochondria in regulating resting [Ca2+]c. Comparison of injury groups (Fig. 5C) showed a heightened effect of FCCP/Oligo on resting [Ca2+]c in SNL L4 neurons, suggesting enhanced [Ca2+]c buffering in resting neurons of the SNL L4 population.
Figure 5.

Effect of nerve injury on mitochondrial regulation of resting [Ca2+]c. (A) Resting [Ca2+]c in Control neurons (C, n=279), neurons from the fourth lumbar dorsal root ganglion after spinal nerve ligation (SNL L4, n=76), and SNL L5 (n=113). (B) Sample traces showing the effect of co-administration of FCCP (1μM) and oligomycin (10μM, FCCP/Oligo) after application of Tyrode’s solution (Tyr). (C) Summary data comparing the response of resting [Ca2+]c in neurons from Control animals to vehicle (Veh, n=63), and the response to FCCP/Oligo of neurons from Control (C, n=94), SNL L4 (n=38) and SNL L5 (n=25) ganglia. **P < 0.01, ***P < 0.001.
2.5 Effect of nerve injury on mitochondrial Ca2+ stores
Calcium buffering by mitochondria is most strongly engaged during periods of high [Ca2+]c, such as those that follow intense neuronal activation. To test the effect of injury on mitochondrial Ca2+ sequestration under these peak conditions, we applied FCCP/Oligo 2s after the termination of a 5s K+ application to neurons of the various injury groups (Fig. 6A). To confirm that this triggered release of Ca2+ stored in mitochondria and was independent of Ca2+ influx into the neuron, we compared the response to FCCP/Oligo in neurons bathed in normal Tyrode’s solution (2mM Ca2+) to neurons bathed in Ca2+-free solution, which showed that the rate of measurable Ca2+ release (Fig. 6B, left panel) and the amplitude of the Ca2+ transient triggered by the release (Fig. 6B, right panel) were the same. Whereas most of the Control and SNL L4 neurons showed measureable FCCP/Oligo-induced Ca2+ release, axotomized SNL L5 neurons typically lacked this response (Fig. 6C, left panel). Analysis of the neurons with a measurable release showed that the amplitude of the response in the SNL L5 group was diminished compared to the other groups (Fig. 6C, right panel). However, it is known that axotomy results in reduced Ca2+ influx through voltage-gated Ca2+ channels in sensory neurons (Hogan et al., 2000; McCallum et al., 2006), which was again demonstrated in the present data by lower depolarization-induced transient peaks (Fig. 6D). Since reduced Ca2+ influx may have contributed to reduced mitochondrial Ca2+ uptake in SNL L5 neurons, we separately examined neurons with a restricted range of K+-transient peaks (200–800nM), but this did not change the reduced incidence or amplitude of FCCP/Oligo Ca2+ release (data not shown). We additionally examined the relationship between each neuron’s depolarization-induced transient peak, which reflects the Ca2+ influx ((Duncan et al., 2013), also Fig. 3C) and the amount of releasable Ca2+ in the mitochondrial stores for the entire population of neurons. Comparison of the regression relationship between cytoplasmic Ca2+ load (represented by the peak of the K+-induced transient) and mitochondrial Ca2+ release (represented by the FCCP/Oligo transient amplitude) in the various injury groups (Fig. 6E) revealed that, under comparable loading conditions, mitochondria in SNL L4 neurons accumulate more releasable Ca2+ while mitochondria in SNL L5 neurons accumulate less. Together, these findings indicate that mitochondria of axotomized SNL L5 neurons are functionally deficient and often fail to sequester Ca2+, while mitochondria of adjacent SNL L4 neurons show hyperactive mitochondrial Ca2+ buffering.
Figure 6.

Effect of nerve injury on mitochondrial Ca2+ stores following neuronal activation. (A) Sample traces from control neurons, neurons from the fourth lumbar dorsal root ganglion after spinal nerve ligation (SNL L4), and SNL L5, showing response of [Ca2+]c to K+ depolarization (5s) and to application of FCCP (1μM) and oligomycin (10μM, FCCP/Oligo) following bath superfusion of Tyrode’s solution. (B) The left panel shows the frequency of measurable Ca2+ release triggered by application of FCCP/Oligo to uninjured neurons bathed either in normal Tyrode’s solution (n=46) or to matched neurons in which the bath was switched to Ca2+-free Tyrode’s solution 5s before the application of FCCP/Oligo (in Ca2+-free vehicle) (n=36). The right panel shows amplitude of the [Ca2+]c transient induced by the application of FCCP/Oligo, excluding data from neurons in with no measurable Ca2+ release. In both cases, there was no difference between groups normal Ca2+ bath (n=34) and Ca2+-free conditions (n=27). (C) The left panel shows summary data for the frequency of measurable Ca2+ release triggered by application of FCCP/Oligo to control neurons (C, n=135), SNL L4 (n=52), and SNL L5 (n=52). The right panel shows amplitude of the [Ca2+]c transient induced by the application of FCCP/Oligo, excluding data from neurons in with no measurable Ca2+ release. Groups include control neurons (C, n=95), neurons from the fourth lumbar dorsal root ganglion after spinal nerve ligation (SNL L4, n=45), and SNL L5 (n=14). ***P < 0.001. (D) Peak of the [Ca2+]c transient induced by application of K+ (5s). Groups include control neurons (C, n=305), neurons from the fourth lumbar dorsal root ganglion after spinal nerve ligation (SNL L4, n=52), and SNL L5 (n=52). ***P < 0.001. (E) Relationships between the peak of the K+-induced transient and the FCCP/Oligo-induced transient for each neuron, separately considered by group. Groups include control neurons (C, n=135), neurons from the fourth lumbar dorsal root ganglion after spinal nerve ligation (SNL L4, n=52), and SNL L5 (n=52). Data points for neurons with no measurable Ca2+ release (0 value for Y-axis) are displayed as a group below the plot, separated vertically for clarity. The regression lines represent a linear fit for the log transform of all data in that group, including non-responding neurons, for which the value of 5 (half of the lowest non-zero value in the data set) was substituted for the purpose of log transform and fitting the regression line. Comparison of the regression lines showed that the slope for SNL L4 was greater than for SNL L5 (P < 0.05), the Y-intercept for SNL L4 was greater than for Control (P < 0.01), and the Y-intercept for Control was greater than for SNL L5 (P <0.001).
3. Discussion
The cytoplasmic Ca2+ signal is a fundamental regulator of most neuronal functions. The ability of mitochondria to transiently sequester massive amounts of Ca2+ indicates a pivotal involvement in normal neuronal function and in the management of potentially toxic Ca2+ loads. Therefore, we sought evidence of a possible role of mitochondria in contributing to disrupted Ca2+ signaling of sensory neurons after peripheral nerve injury. We chose to examine mitochondrial function in the setting of depolarization-induced Ca2+ loading since Ca2+ influx is a critical link between membrane activation and processes that control neuronal excitability (Lirk et al., 2008; Tang et al., 2012) and synaptic transmission (Kann and Kovacs, 2007). Our initial examination with FCCP/Oligo identified mitochondrial participation in generating a shoulder during transient recovery and limiting transient peak and area after entry of an adequate Ca2+ load (1.3s depolarization), as well as regulating the shoulder level. Using these markers, heightened mitochondrial Ca2+ buffering in L4 neurons after SNL was suggested by several lines of evidence, including development of a shoulder after exposure of the mitochondria to lower cytoplasmic Ca2+ concentrations, lower transient peaks, an accentuated effect of FCCP/Oligo (on transient peak, shoulder level, and resting [Ca2+]c), and greater activity-induced loading of Ca2+ into the mitochondria. In contrast, axotomized SNL L5 neurons showed changes that suggest diminished mitochondrial Ca2+ buffering, including elevated shoulder level, reduced sensitivity of peak and shoulder level to FCCP/Oligo, and diminished activity-induced mitochondrial Ca2+ loading (Fig. 7).
Figure 7.

A summary of possible underlying functional changes that could explain nerve injury effects on mitochondrial function. Cycling of Ca2+ through the mitochondria is illustrated as uptake by the Ca2+ uniporter and release by the Na+/Ca2+ exchanger. Axotomized neurons accumulate less Ca2+ in the mitochondrial matrix and retain Ca2+ less well. In contrast, the adjacent neurons that are intact but share peripheral nerve fascicles with degenerating segments of the axotomized neurons develop exaggerated Ca2+ accumulation without evidence of an altered release process.
Despite retaining axonal continuity, L4 neurons in the SNL model are exposed to inflammation from Wallerian degeneration of axotomized neurons (Shamash et al., 2002; Uceyler and Sommer, 2006), which results in functional alterations such as elevated TRPV1 expression (Hudson et al., 2001) and increased sensitivity to tumor necrosis factor-alpha (Schafers et al., 2003). Although our prior investigations of Ca2+ signaling in the SNL model have showed that injury effects are preferentially expressed in the axotomized SNL L5 population (e.g. depressed resting [Ca2+]c (Fuchs et al., 2005), loss of ER Ca2+ stores (Rigaud et al., 2009), elevated SOCE (Gemes et al., 2011), increased PMCA (Gemes et al., 2012b), and depressed SERCA function (Duncan et al., 2013)), our present study offers evidence that injury also alters Ca2+ handling in the intact L4 population, in this case via amplified mitochondrial buffering. The underlying mechanism for this functional shift could involve greater Ca2+ affinity of the Ca2+ uniporter, elevated electrochemical drive for Ca2+ uptake through the mitochondrial inner membrane, or a higher buffering capacity within the mitochondrial matrix. Even the resting [Ca2+]c of SNL L4 neurons was found to be more sensitive to FCCP/Oligo than Control or SNL L5 neurons. This finding might be considered unexpected since mitochondrial Ca2+ buffering has often been viewed as operating only at high [Ca2+]c, but recent reports demonstrate constitutive Ca2+ cycling at low [Ca2+]c in resting neurons (Colegrove et al., 2000; Kang et al., 2008).
In contrast to L4 neurons, L5 neurons after SNL are subject to axonal transection and loss of target-derived neurotrophins. In parallel with differing pathophysiology, L5 neurons showed effects on mitochondrial Ca2+ buffering that were distinct from the L4 population. Specifically, L5 neurons lacked the features indicative of accelerated Ca2+ buffering acquired by L4 neurons after SNL, but instead developed a deficient ability to accumulate Ca2+, which was evident as reduced or absent releasable mitochondrial Ca2+ despite large cytoplasmic Ca2+ loads. This is possibly due to reduced Ca2+ uniporter function, inadequate ΔΨm due to uncoupling or defective electron transport chain, a loss of Ca2+ buffering capacity in the mitochondrial matrix, or a more general structural defect. Additionally, mitochondria of SNL L5 neurons show early Ca2+ release during the transient recovery phase and have a higher shoulder level as a result. This indicates a shifted balance of mitochondrial Ca2+ uptake and release, and implicates disordered control of the Na+/Ca2+ exchanger release pathway. Since this process is coupled to Na+/H+ exchange across the inner mitochondrial membrane, diminished respiratory chain function and the resulting deficit in H+ gradient and ΔΨm would be expected to delay release of intra-mitochondrial Ca2+ during the shoulder phase in injured neurons (Montero et al., 2001) in contrast to our findings. Replication of this finding with FCCP/Oligo application nonetheless suggests loss of ΔΨm as a contributing factor. Additionally, the mitochondrial Ca2+ release mechanism is regulated by [Ca2+]c (Bathori et al., 2006; Montero et al., 2001), so a shift in this sensitivity could cause a higher shoulder after injury.
Upstream triggers for mitochondrial dysfunction after axotomy are undefined. However, several possibilities can be considered on the basis of recent observations on the sensory neuronal response to injury. It is increasingly recognized that mitochondria reside at the hub of a complex system of reciprocal interactions that requires close structural proximity (Szabadkai and Duchen, 2008) near regions of high local [Ca2+]c such as plasmalemmal Ca2+ channels (Pivovarova et al., 1999) and ER Ca2+ release channels (Hayashi et al., 2009). This allows mitochondrial participation in Ca2+ buffering despite relatively low Ca2+ affinity of the uniporter intake path. We have observed extensively disordered ER ultrastructure after axotomy (Gemes et al., 2009), so a possible consequence is the loss of the particular physical interactions that underlie mitochondrial colocalization and the coordinated physiological function of these ultrastructural elements. In support of this, we have noted a decrease in ER profile lengths in SNL L5 neurons, but an increase in the L4 population (Gemes et al., 2009). Close mitochondrial association with ER provides for bidirectional exchange of Ca2+. On the one hand, since mitochondria provide an important source of Ca2+ for refilling ER Ca2+ stores (Arnaudeau et al., 2001; Malli et al., 2005), deficient mitochondrial Ca2+ uptake in axotomized neurons may in part explain our prior finding of depleted ER Ca2+stores (Rigaud et al., 2009). On the other hand, mitochondria also receive Ca2+ from ER (Montero et al., 2001). Structural association of ER and mitochondria fails when [Ca2+]c is low (Wang et al., 2000), so the reduced resting [Ca2+]c in axotomized sensory neurons may interrupt subcellular interactions of mitochondrial with ER and contribute to loss of mitochondrial Ca2+ buffering. However, evidence that Ca2+ released from ER is not buffered by mitochondria in sensory neurons (Scheff et al., 2013; Svichar et al., 1997) raises a question whether mitochondria receive Ca2+ from closely associated ER in the same fashion as adrenal chromaffin cells (Montero et al., 2001).
The functional implications of disordered mitochondrial Ca2+ handling could be extensive. Mitochondrial energy production is sensitive to matrix Ca2+ levels via regulation of rate limiting enzymes of the citric acid cycle, so neuronal ATP/ADP ratios may be affected. Inability of mitochondria to prevent excessive [Ca2+]c could contribute to excitotoxicity and neuronal apoptosis after injury. Mitochondrial Ca2+ release in presynaptic terminals regulates synaptic function, although the evidence from various tissues is divergent, showing both mitochondrial support for presynaptic [Ca2+]c (Tang et al., 2012) or suppression of presynaptic [Ca2+]c (Shutov et al., 2013; Wan et al., 2012). The role of mitochondria in dorsal horn neurotransmission of nociceptive traffic will be an important area of investigation. Depolarized ΔΨm develops in sensory neurons of a rat model of diabetes (Huang et al., 2003) and in sensory neurons exposed to cisplatin or paclitaxel (Melli et al., 2008), all of which produce clinical neuropathic pain. Combined with our findings, these reports support our suspicion of a mitochondrial role in pain pathophysiology. Since both axotomized neurons and adjacent inflamed neurons may contribute to generating pain after nerve injury (Gold, 2000), we cannot speculate about the relative importance of the changes we have found in L4 and L5 neurons after SNL, although a disease phenotype due to excessive mitochondrial Ca2+ buffering has not been described before now.
Several important limitations of our data should be mentioned. Mitochondrial Ca2+ cycling is temperature sensitive. Whereas our recordings were performed at 25°C, greater buffering may be expected at higher temperatures (David and Barrett, 2000; Kang et al., 2008). It is unknown whether this would alter our intergroup comparisons. Additionally, while our recording of Ca2+ provides a view of averaged cytoplasmic Ca2+ levels, it nonetheless neglects important microdomains where mitochondrial effects may differ. Also, we examined only small to medium sized sensory neurons. However, the observations by Lu et al. (Lu et al., 2006) that elimination of mitochondrial Ca2+ buffering affected all size groups of sensory neurons and effects were least in small sensory neurons suggest that our findings may be representative of the full population of sensory neurons. Finally, our approach of tracking FCCP/Oligo-sensitive features after SNL suffers from a lack of specificity since we did not evaluate mitochondrial Ca2+ buffering in isolation from other Ca2+ sequestration and extrusion processes. For instance, compensatory increase of PMCA and SERCA function driven by relatively higher [Ca2+]c during mitochondrial blockade may have resulted in underestimating the role of mitochondrial Ca2+ buffering in the present experiments. However, functional examination of isolated mitochondria would not reflect their operation in the natural context of the intact neuron, whereas our approach reveals the integrated physiological consequences of altered mitochondrial Ca2+ cycling at the cellular level.
We conclude that peripheral nerve trauma substantially affects mitochondrial performance in both directly injured neurons as well as neurons with intact fibers. These populations show a divergent response in which mitochondria in axotomized neurons have a deficient ability to buffer Ca2+ following intense neuronal activity, while the adjacent inflamed neurons show enhanced buffering of cytoplasmic Ca2+. Since mitochondrial Ca2+ cycling is functionally linked with numerous other organelles and membrane pumps, these shifts in Ca2+ management support a view that mitochondrial dysfunction contributes to neuropathic pain.
4. Experimental Procedures
4.1 Animals
All methods and use of animals were approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee. Male Sprague-Dawley rats (Taconic Farms Inc., Hudson, NY) were housed in pairs in a room maintained at 22 ± 0.5°C and constant humidity (60 ± 15%) with an alternating 12hr light-dark cycle. Food and water were freely available throughout the experiments.
4.2 Injury model
Rats weighing 150 to 180g were subjected to SNL modified from the original technique (Kim and Chung, 1992). Specifically, rats were anesthetized with 2% isoflurane in oxygen and the right paravertebral region was exposed. The L6 transverse process was removed, after which the L5 and L6 spinal nerves were ligated with 6–0 silk suture and transected distal to the ligature. To minimize non-neural injury, no muscle was removed, muscles and intertransverse fascia were incised only at the site of the two ligations, and articular processes were not removed. The muscular fascia was closed with 4–0 resorbable polyglactin sutures and the skin closed with staples. Control animals received skin incision and closure only.
4.3 Sensory testing
We measured the incidence of a pattern of hyperalgesic behavior that we have previously documented to be associated with conditioned place avoidance (Hogan et al., 2004; Wu et al., 2010). Briefly, on 3 different days between 10d and 17d after surgery, right plantar skin was touched (10 stimuli/test) with a 22G spinal needle with adequate pressure to indent but not penetrate the skin. Whereas control animals respond with only a brief reflexive withdrawal, rats following SNL may display a complex hyperalgesia response that includes licking, chewing, grooming and sustained elevation of the paw. The average frequency of hyperalgesia responses over the 3 testing days was tabulated for each rat. After SNL, only rats that displayed a hyperalgesia-type response after at least 20% of stimuli were used further in this study.
4.4 Neuron isolation and plating
Neurons were rapidly harvested from L4 and L5 DRGs during isoflurane anesthesia and decapitation 21 to 28 days after SNL or skin sham surgery. This interval was chosen since hyperalgesia is fully developed by this time (Hogan et al., 2004). Ganglia were incubated in 0.5mg/ml Liberase TM (Roche, Indianapolis, IN) in DMEM/F12 with glutaMAX (Life Technologies) for 30min at 37 °C, followed with 1 mg/ml trypsin (Sigma-Aldrich, St. Louis, MO) and 150 Kunitz units/ml DNase (Sigma-Aldrich) for another 10min. After addition of 0.1% trypsin inhibitor (Type II, Sigma-Aldrich), tissues were centrifuged, lightly triturated in neural basal media (1X) (Life Technologies) containing 2% (v:v) B27 supplement (50x) (Life Technologies), 0.5mM glutamine (Sigma-Aldrich), 0.05mg/ml gentamicin (Life Technologies) and 10 ng/ml nerve growth factor 7S (Alomone Labs Ltd., Jerusalem, Israel). Cells were then plated onto poly-L-lysine (70–150kDa, Sigma-Aldrich) coated glass cover slips (Deutsches Spiegelglas, Carolina Biological Supply, Burlington, NC) and incubated at 37°C in humidified 95% air and 5% CO2 for 2hr and were studied 3–8hr after dissociation.
4.5 Solutions and agents
Unless otherwise specified, the bath contained Tyrode’s solution (in mM): NaCl 140, KCl 4, CaCl2 2, Glucose 10, MgCl2 2, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) 10, with an osmolarity of 297–300mOsm and pH 7.40. Fura-2-AM was obtained from Invitrogen (Carlsbad, CA), tetramethylrhodamine methyl ester (TMRM) was from Molecular Probes (Waltham, MA), antimycin, oligomycin, and the SERCA blocker thapsigargin (TG) were from Sigma Aldrich (St. Louis, MO), and carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) was from Tocris (Minneapolis, MN). Stock solutions of TG, FCCP, Fura-2-AM, and TMRM were dissolved in DMSO. Subsequently dilution in the relevant bath solution produced final bath concentrations of DMSO of 0.1% for TG-containing solution, 0.002% for TMRM, and 0.01% for FCCP-containing solutions. Oligomycin stock was dissolved in ethanol, and subsequent dilution produced bath concentrations containing 0.05% ethanol, which does not effect [Ca2+]c (data not shown, and (Avery and Johnston, 1997)). The 500μl recording chamber was superfused by gravity-driven flow at a rate of 3ml/min. Agents were delivered by directed microperfusion controlled by a computerized valve system (ALA Scientific Instruments, Farmingdale, NY) through a 500μm diameter hollow quartz fiber 300μm upstream from the neurons. This flow completely displaced the bath solution (demonstrated by imaging dye flow, data not shown) such that neurons in the imaging field were exposed to undiluted microperfusion fluid, and constant flow was maintained through this microperfusion pathway by delivery of bath solution when specific agents were not being administered. Dye imaging shows that solution changes were achieved within 200ms. Neurons were activated by depolarization produced by microperfusion application of 50mM K+ for various durations as specified below. Repeat depolarizations were initiated only after 60s had elapsed following recovery of the prior transient.
4.6 Fluorimetric imaging
To measure [Ca2+]c, neurons plated on cover slips were exposed to Fura-2-AM (5μM) for 30min at room temperature in a solution that contained 2% bovine albumin to aid dispersion of the fluorophore, washed 3 times with regular Tyrode’s solution, and given 30min for de-esterification. Neurons showing signs of lysis, crenulation or superimposed glial cells under brightfield illumination were excluded. For [Ca2+]c recording, the fluorophore was excited alternately with 340nm and 380nm wavelength illumination (150W Xenon, Lambda DG-4, Sutter, Novato, CA), and images were acquired at 510nm using a cooled 12-bit digital camera (Coolsnap fx, Photometrics, Tucson, AZ) and inverted microscope (Diaphot 200, Nikon Instruments, Melville, NY) through a 20x objective. Recordings from each neuron were obtained as separate regions (MetaFluor, Molecular Devices, Downingtown, PA) at a rate of 1Hz. After background subtraction, the fluorescence ratio R for individual neurons was determined as the intensity of emission during 340nm excitation (I340) divided by I380, on a pixel-by-pixel basis. The Ca2+ concentration was then estimated by the formula where β = (I380max)/(I380min). Values of Rmin, Rmax and β were determined by in-situ calibration (Fuchs et al., 2005; Gemes et al., 2011) and were 0.85, 11.10 and 5.28, and Kd was 224nM (Grynkiewicz et al., 1985).
Data were derived only from neurons with stable resting R traces, which were analyzed using Axograph X 1.1 (Axograph Scientific, Sydney, Australia). Transient amplitude was determined as the difference between the peak [Ca2+]c and the resting level just before depolarization (Fig. 1A). To characterize transient duration, it is conventional to chose a level, such as 20% of the transient amplitude above resting [Ca2+]c (i.e. 80% recovery, T80), at which to measure the duration. However, mitochondrial blockade may have divergent influences on transient duration when measured below versus above the level of the plateau (e.g. eliminating the plateau but prolonging the phase above the plateau), which combined with variance in the level of the plateau makes this type of measurement highly arbitrary. We therefore additionally evaluated the area of the transient, which incorporates both amplitude and duration without the need to record duration at a specific level. Presence of a shoulder was determined by differentiation of the trace of [Ca2+]c vs. time. A shoulder was considered present when the differentiated trace showed two negative maxima (Fig. 1B). When the transient had a shoulder, its level was measured at the time specified by the inflection point, i.e. when the slope of the [Ca2+]c vs. time trace during the recovery phase of the transient shifted from becoming less negative to becoming more negative (Fig. 1B). Recovery of transients that lacked a shoulder was fit by a mono-exponential curve from which the time constant (τ) was derived as a measure of the pace of recovery.
To indicate trends in ΔΨm, TMRM was used in a non-quenched mode, in which falling ΔΨm is accompanied by loss of mitochondrial fluorophore to the cytoplasm and hence out of the neuron, resulting in diminished total neuron fluorescence (Ward, 2010). The fluorophore was applied in a similar fashion as FURA-2 but at a concentration of 20nM (i.e. a concentration too low to permit auto-quenching within the mitochondria (Ward, 2010)), and loading the dye was performed at 37°C for 60min. TMRM was present in the bath solution during imaging at 25°C, using 550nm wavelength excitation and image acquisition at 570nm.
4.7 Statistical analysis
Sensory neuron somatic diameter is broadly associated with specific sensory modalities (Ma et al., 2003). In this study, we examined small to medium sized neurons (diameter ≤ 34μm), which represent predominantly nociceptive neurons, and are the population for which Ca2+ signaling is preferentially affected by injury (Duncan et al., 2013; Rigaud et al., 2009). Neurons were grouped separately as Control neurons from sham-operated animals, and axotomized L5 neurons and neighboring L4 neurons from SNL animals.
Statistical analysis was performed in collaboration with the Medical College of Wisconsin Statistical Consultation Service. Prism (version 4.1, GraphPad Software, Inc., San Diego, CA) and Statistica (StatSoft Inc, Tulsa, OK) were used to perform statistical evaluations. Incidence of features was evaluated by Fisher’s exact test for two groups or Chi-square for 3 groups. When a significant influence of group was shown by Chi-square, paired comparisons were performed by Fischer’s exact test and corrected by the Bonferroni method. Influence of blockade of mitochondrial Ca2+ buffering upon Ca2+ transient parameters was evaluated by nonparametric analysis since the data distribution was frequently non-Gaussian. Either one-sample Wilcoxon test for Control neurons alone, Mann-Whitney for comparing two groups, or Kruskal-Wallis test followed by Dunn’s Multiple Comparison Test for comparing the effect of mitochondrial blockade in three groups. A P value less than 0.05 was considered significant. Data are reported as Median and interquartile range (IQR) represented as first and third quartiles (as [Q1, Q3]), and plots of data show median and Q1, Q3 (box plot) with whiskers denoting the lowest datum still within 1.5 IQR of Q1 and the highest datum still within 1.5 IQR of Q3 (i.e. Tukey boxplot). Log plots were used for data that was highly skewed toward lower values.
Highlights.
Mitochondrial Ca2+ buffering was examined in injured sensory neurons.
Mitochondria regulate resting [Ca2+]c and K+-induced transient shoulder and peak.
Neurons axotomized show evidence of deficient Ca2+ buffering.
Adjacent neurons exposed to inflammation show enhanced cytoplasmic Ca2+ buffering.
Acknowledgments
We are grateful for the statistical consultation provided by Dr. Aniko Sazbo PhD, supported in part by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number 8UL1TR000055. This study was funded by NIH grants NS042150 to QH and DA-K01-024751 to H-EW.
Abbreviations
- ΔΨm
inner mitochondrial membrane potential
- [Ca2+]c
cytoplasmic Ca2+ concentration
- DRG
dorsal root ganglion
- FCCP
carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone
- L4
4th lumbar
- PMCA
plasma membrane Ca2+-ATPase
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+-ATPase
- SOCE
store-operated Ca2+ entry
- SNL
spinal nerve ligation
- TMRM
tetramethylrhodamine methyl ester
Footnotes
Conflict of Interest
The authors have no conflicts of interest relevant to the work in this report.
Authors’ Contributions
Participated in research design: Hogan, Bienengraeber, Pan, Wu.
Conducted experiments: Sprick, Guo, Mueller, Wu.
Performed data analysis: Hogan, Wu.
Wrote or contributed to the writing of the manuscript: Hogan, Wu.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnaudeau S, Kelley WL, Walsh JV, Jr, Demaurex N. Mitochondria recycle Ca(2+) to the endoplasmic reticulum and prevent the depletion of neighboring endoplasmic reticulum regions. J Biol Chem. 2001;276:29430–9. doi: 10.1074/jbc.M103274200. [DOI] [PubMed] [Google Scholar]
- Avery RB, Johnston D. Ca2+ channel antagonist U-92032 inhibits both T-type Ca2+ channels and Na+ channels in hippocampal CA1 pyramidal neurons. J Neurophysiol. 1997;77:1023–8. doi: 10.1152/jn.1997.77.2.1023. [DOI] [PubMed] [Google Scholar]
- Bathori G, Csordas G, Garcia-Perez C, Davies E, Hajnoczky G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC) J Biol Chem. 2006;281:17347–58. doi: 10.1074/jbc.M600906200. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J Neurochem. 1996;66:403–11. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- Chu C, Levine E, Gear RW, Bogen O, Levine JD. Mitochondrial dependence of nerve growth factor-induced mechanical hyperalgesia. Pain. 2011;152:1832–7. doi: 10.1016/j.pain.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrove SL, Albrecht MA, Friel DD. Dissection of mitochondrial Ca2+ uptake and release fluxes in situ after depolarization-evoked [Ca2+](i) elevations in sympathetic neurons. J Gen Physiol. 2000;115:351–70. doi: 10.1085/jgp.115.3.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett EF. Stimulation-evoked increases in cytosolic [Ca(2+)] in mouse motor nerve terminals are limited by mitochondrial uptake and are temperature-dependent. J Neurosci. 2000;20:7290–6. doi: 10.1523/JNEUROSCI.20-19-07290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. J Physiol. 1999;516(Pt 1):1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan C, Mueller S, Simon E, Renger JJ, Uebele VN, Hogan QH, Wu HE. Painful nerve injury decreases sarco-endoplasmic reticulum Ca(2)(+)-ATPase activity in axotomized sensory neurons. Neuroscience. 2013;231:247–57. doi: 10.1016/j.neuroscience.2012.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatters SJ, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain. 2006;122:245–57. doi: 10.1016/j.pain.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Lirk P, Stucky C, Abram SE, Hogan QH. Painful nerve injury decreases resting cytosolic calcium concentrations in sensory neurons of rats. Anesthesiology. 2005;102:1217–25. doi: 10.1097/00000542-200506000-00023. [DOI] [PubMed] [Google Scholar]
- Gemes G, Rigaud M, Weyker PD, Abram SE, Weihrauch D, Poroli M, Zoga V, Hogan QH. Depletion of calcium stores in injured sensory neurons: anatomic and functional correlates. Anesthesiology. 2009;111:393–405. doi: 10.1097/ALN.0b013e3181ae63b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G, Bangaru ML, Wu HE, Tang Q, Weihrauch D, Koopmeiners AS, Cruikshank JM, Kwok WM, Hogan QH. Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J Neurosci. 2011;31:3536–49. doi: 10.1523/JNEUROSCI.5053-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G, Koopmeiners A, Rigaud M, Lirk P, Sapunar D, Bangaru ML, Vilceanu D, Garrison SR, Ljubkovic M, Mueller SJ, Stucky CL, Hogan QH. Failure of Action Potential Propagation in Sensory Neurons: Mechanisms and Loss of Afferent Filtering in C-type Units after Painful Nerve Injury. J Physiol. 2012a doi: 10.1113/jphysiol.2012.242750. published ahead of print November 12, 2012, jphysiol.2012.242750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G, Oyster KD, Pan B, Wu HE, Bangaru ML, Tang Q, Hogan QH. Painful nerve injury increases plasma membrane Ca2+-ATPase activity in axotomized sensory neurons. Mol Pain. 2012b;8:46. doi: 10.1186/1744-8069-8-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS. Spinal nerve ligation: what to blame for the pain and why. [Review] [22 refs] Pain. 2000;84:117–20. doi: 10.1016/s0304-3959(99)00309-7. [DOI] [PubMed] [Google Scholar]
- Gover TD, Moreira TH, Kao JP, Weinreich D. Calcium homeostasis in trigeminal ganglion cell bodies. Cell Calcium. 2007;41:389–96. doi: 10.1016/j.ceca.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–50. [PubMed] [Google Scholar]
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19:81–8. doi: 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–28. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- Hogan Q, Sapunar D, Modric-Jednacak K, McCallum JB. Detection of neuropathic pain in a rat model of peripheral nerve injury. Anesthesiology. 2004;101:476–87. doi: 10.1097/00000542-200408000-00030. [DOI] [PubMed] [Google Scholar]
- Hogan Q, Lirk P, Poroli M, Rigaud M, Fuchs A, Fillip P, Ljubkovic M, Gemes G, Sapunar D. Restoration of calcium influx corrects membrane hyperexcitability in injured rat dorsal root ganglion neurons. Anesth Analg. 2008;107:1045–51. doi: 10.1213/ane.0b013e31817bd1f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan QH, McCallum JB, Sarantopoulos C, Aason M, Mynlieff M, Kwok WM, Bosnjak ZJ. Painful neuropathy decreases membrane calcium current in mammalian primary afferent neurons. Pain. 2000;86:43–53. doi: 10.1016/s0304-3959(99)00313-9. [DOI] [PubMed] [Google Scholar]
- Huang TJ, Price SA, Chilton L, Calcutt NA, Tomlinson DR, Verkhratsky A, Fernyhough P. Insulin prevents depolarization of the mitochondrial inner membrane in sensory neurons of type 1 diabetic rats in the presence of sustained hyperglycemia. Diabetes. 2003;52:2129–36. doi: 10.2337/diabetes.52.8.2129. [DOI] [PubMed] [Google Scholar]
- Hudson LJ, Bevan S, Wotherspoon G, Gentry C, Fox A, Winter J. VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. Eur J Neurosci. 2001;13:2105–14. doi: 10.1046/j.0953-816x.2001.01591.x. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Mitochondrial electron transport in models of neuropathic and inflammatory pain. Pain. 2006;121:105–14. doi: 10.1016/j.pain.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Kang SH, Carl A, McHugh JM, Goff HR, Kenyon JL. Roles of mitochondria and temperature in the control of intracellular calcium in adult rat sensory neurons. Cell Calcium. 2008;43:388–404. doi: 10.1016/j.ceca.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann O, Kovacs R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol. 2007;292:C641–57. doi: 10.1152/ajpcell.00222.2006. [DOI] [PubMed] [Google Scholar]
- Kawano T, Zoga V, Gemes G, McCallum JB, Wu HE, Pravdic D, Liang MY, Kwok WM, Hogan Q, Sarantopoulos C. Suppressed Ca2+/CaM/CaMKII-dependent K(ATP) channel activity in primary afferent neurons mediates hyperalgesia after axotomy. Proc Natl Acad Sci U S A. 2009;106:8725–30. doi: 10.1073/pnas.0901815106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Kojundzic SL, Puljak L, Hogan Q, Sapunar D. Depression of Ca(2+)/calmodulin-dependent protein kinase II in dorsal root ganglion neurons after spinal nerve ligation. J Comp Neurol. 2010;518:64–74. doi: 10.1002/cne.22209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lirk P, Poroli M, Rigaud M, Fuchs A, Fillip P, Huang CY, Ljubkovic M, Sapunar D, Hogan Q. Modulators of calcium influx regulate membrane excitability in rat dorsal root ganglion neurons. Anesth Analg. 2008;107:673–85. doi: 10.1213/ane.0b013e31817b7a73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SG, Zhang X, Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. J Physiol. 2006;577:169–90. doi: 10.1113/jphysiol.2006.116418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Shu Y, Zheng Z, Chen Y, Yao H, Greenquist KW, White FA, LaMotte RH. Similar electrophysiological changes in axotomized and neighboring intact dorsal root ganglion neurons. J Neurophysiol. 2003;89:1588–602. doi: 10.1152/jn.00855.2002. [DOI] [PubMed] [Google Scholar]
- Malli R, Frieden M, Trenker M, Graier WF. The role of mitochondria for Ca2+ refilling of the endoplasmic reticulum. J Biol Chem. 2005;280:12114–22. doi: 10.1074/jbc.M409353200. [DOI] [PubMed] [Google Scholar]
- McCallum JB, Kwok WM, Sapunar D, Fuchs A, Hogan QH. Painful peripheral nerve injury decreases calcium current in axotomized sensory neurons. Anesthesiology. 2006;105:160–8. doi: 10.1097/00000542-200607000-00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melli G, Taiana M, Camozzi F, Triolo D, Podini P, Quattrini A, Taroni F, Lauria G. Alpha-lipoic acid prevents mitochondrial damage and neurotoxicity in experimental chemotherapy neuropathy. Exp Neurol. 2008;214:276–84. doi: 10.1016/j.expneurol.2008.08.013. [DOI] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Albillos A, Garcia-Sancho J, Alvarez J. Mitochondrial Ca(2+)-induced Ca(2+) release mediated by the Ca(2+) uniporter. Mol Biol Cell. 2001;12:63–71. doi: 10.1091/mbc.12.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondria and calcium signaling. Cell Calcium. 2005;38:311–7. doi: 10.1016/j.ceca.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Pivovarova NB, Hongpaisan J, Andrews SB, Friel DD. Depolarization-induced mitochondrial Ca accumulation in sympathetic neurons: spatial and temporal characteristics. J Neurosci. 1999;19:6372–84. doi: 10.1523/JNEUROSCI.19-15-06372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaud M, Gemes G, Weyker PD, Cruikshank JM, Kawano T, Wu HE, Hogan QH. Axotomy depletes intracellular calcium stores in primary sensory neurons. Anesthesiology. 2009;111:381–92. doi: 10.1097/ALN.0b013e3181ae6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapunar D, Ljubkovic M, Lirk P, McCallum JB, Hogan QH. Distinct membrane effects of spinal nerve ligation on injured and adjacent dorsal root ganglion neurons in rats. Anesthesiology. 2005;103:360–76. doi: 10.1097/00000542-200508000-00020. [DOI] [PubMed] [Google Scholar]
- Schafers M, Lee DH, Brors D, Yaksh TL, Sorkin LS. Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-alpha after spinal nerve ligation. J Neurosci. 2003;23:3028–38. doi: 10.1523/JNEUROSCI.23-07-03028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatzmann HJ, Vincenzi FF. Calcium movements across the membrane of human red cells. J Physiol. 1969;201:369–95. doi: 10.1113/jphysiol.1969.sp008761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff NN, Lu SG, Gold MS. Contribution of endoplasmic reticulum Ca2+ regulatory mechanisms to the inflammation-induced increase in the evoked Ca2+ transient in rat cutaneous dorsal root ganglion neurons. Cell Calcium. 2013;54:46–56. doi: 10.1016/j.ceca.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamash S, Reichert F, Rotshenker S. The cytokine network of Wallerian degeneration: tumor necrosis factor-alpha, interleukin-1alpha, and interleukin-1beta. J Neurosci. 2002;22:3052–60. doi: 10.1523/JNEUROSCI.22-08-03052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutov LP, Kim MS, Houlihan PR, Medvedeva YV, Usachev YM. Mitochondria and plasma membrane Ca2+-ATPase control presynaptic Ca2+ clearance in capsaicin-sensitive rat sensory neurons. J Physiol. 2013;591:2443–62. doi: 10.1113/jphysiol.2012.249219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svichar N, Kostyuk P, Verkhratsky A. Mitochondria buffer Ca2+ entry but not intracellular Ca2+ release in mouse DRG neurones. Neuroreport. 1997;8:3929–32. doi: 10.1097/00001756-199712220-00017. [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 2008;23:84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- Tang Q, Bangaru ML, Kostic S, Pan B, Wu HE, Koopmeiners AS, Yu H, Fischer GJ, McCallum JB, Kwok WM, Hudmon A, Hogan QH. Ca(2)(+)-dependent regulation of Ca(2)(+) currents in rat primary afferent neurons: role of CaMKII and the effect of injury. J Neurosci. 2012;32:11737–49. doi: 10.1523/JNEUROSCI.0983-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer SA, Miller RJ. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. Journal of Physiology. 1990;425:85–115. doi: 10.1113/jphysiol.1990.sp018094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uceyler N, Sommer C. Wallerian degeneration and neuropathic pain. Drug Discovery Today: Disease Mechanisms. 2006;3:351–56. [Google Scholar]
- Wan QF, Nixon E, Heidelberger R. Regulation of presynaptic calcium in a mammalian synaptic terminal. J Neurophysiol. 2012;108:3059–67. doi: 10.1152/jn.00213.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HJ, Guay G, Pogan L, Sauve R, Nabi IR. Calcium regulates the association between mitochondria and a smooth subdomain of the endoplasmic reticulum. J Cell Biol. 2000;150:1489–98. doi: 10.1083/jcb.150.6.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward MW. Quantitative analysis of membrane potentials. Methods Mol Biol. 2010;591:335–51. doi: 10.1007/978-1-60761-404-3_20. [DOI] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci. 1994;14:348–56. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HE, Gemes G, Zoga V, Kawano T, Hogan QH. Learned avoidance from noxious mechanical simulation but not threshold semmes weinstein filament stimulation after nerve injury in rats. J Pain. 2010;11:280–6. doi: 10.1016/j.jpain.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]