

Abstract
The etiology of colorectal cancer (CRC) is multifactorial with genetic, molecular, inflammatory, and environmental risk factors. Recently, the gut microbiota is recognized as a new environmental contributor to CRC in both animal models and human studies. An additional interplay of the gut microbiome with inflammation is also evident in studies that have demonstrated that inflammation alone or the presence of bacteria/bacterial metabolites alone is not enough to promote tumorigenesis. Rather, complex interrelationships with the gut microbiome, inflammation, genetics, and other environmental factors are evident in colorectal tumor progression.
Keywords: Gut Microbiome, Colorectal Cancer, Inflammation, Carcinogenesis
Introduction
The last decade has brought a revolution in the understanding of microorganisms viz-a-viz their environment/mammalian hosts. These radical changes in thought not only challenge ideas that dominated biological and medical sciences for over a hundred years, but at a visceral level call into question the very definition of the human identity. The emergence of the germ theory of disease in the late 19th century, highlighted by the propagation of Robert Koch’s famous postulates, and the ensuing discovery of antibiotics some decades later, exemplify the view of microorganism as a foreign ‘other’ with disease-causing potential (pathogens) that often need to be treated via medical eradication. In the common parlance germs are bad and not to be spread. Although there was a movement recognizing the potential for bacteria to benefit their host (probiotics) during the 20th century, it was only in the last decade or so that the true extent, complexity and intimacy of this relationship have taken form.
It is now generally accepted that bacteria are (critical to their ecosystems) ubiquitous and colonizers of all exposed human body surfaces including the entire alimentary tract. In fact, bacterial organisms living in/on a human host outnumber that host’s native cells by a factor of 10. These bacterial communities (microbiota) become a part of us from birth and participate in what is now regarded as a relationship of symbiotic mutualism, whereby the human provides a nutrient-enriched tailored living environment. In return, bacteria play a critical role for the health and development of the human species. There is evidence, for example, that the presence of the bacterial microbiome is integral for modulation of the human immune system, digestion of dietary nutrients otherwise impervious to human enzymes, and prevention of pathogenic bacterial disease. Given the above, some go as far as to characterize the human and his corresponding microbiota as parts of a vastly greater super-organism. At a minimum, it is clear that mammals and microorganisms have coevolved to produce an intricate and vital symbiotic relationship.
Reminiscent of the inextricable linkage between the invention/popularization of the microscope and the discovery of microorganisms, both attributed to Van Leeuwenhoek (late 17th century), the recent charge to characterize whole populations of bacteria and viruses was permitted by advances in experimental techniques and laboratory sciences. These include advancements in bioinformatics, biological analytics and DNA/RNA collection and sequencing techniques that allow for high throughput approaches to specify and quantitate myriads of different bacteria. While a single strain of bacteria may be held accountable as an etiologically specific cause for diseases, such as Clostridium difficile for pseudomembranous colitis, perhaps the more pertinent question is: what changes in the usually protective microbiome (dysbiosis) allowed for such infection? In the above example the answer would be antibiotic-induced dysbiosis. Moreover, the state of microbiota has been associated with conditions such as diabetes, skin disease, obesity, inflammatory bowel disease and even cancer, all of which are commonly regarded as non-infectious processes.
Although inflammatory, infectious and neoplastic diseases are often considered categorically distinct processes, evidence has shown significant overlap between them. In fact, it is estimated that 15% of worldwide cancer is of infectious nature, with human papillomavirus, hepatitis B virus, hepatitis C virus, human herpesvirus-8, and Helicobacter pylori recognized as the definitive cause of cervical cancer, liver cancer, Kaposi’s sarcoma and stomach cancer/lymphoma, respectively. Furthermore, direct causation of cancer by chronic inflammatory conditions is very well documented. The association of inflammatory bowel disease (IBD) with increased risk of colon cancer is a case in point. Thus, it should come as no surprise that alterations of the microbiome may lead to infectious, inflammatory and ultimately cancerous disease. It is the focus of this review to detail the interrelationship between colorectal cancer (CRC) and the gut microbiome.
Background
CRC is the second leading type of cancer in females and the third in males worldwide with over 1.2 million new cases and over 600,000 estimated deaths in 2008 [1]. In the United States, an estimate of 142,820 new cases of CRC with over 50,000 deaths occur annually [2]. However, both incidence and mortality rates of CRC in the United States have steadily declined, and this decrease may be attributed to prevention, early screening, detection, and treatment of CRC [3].
Major risk factors of CRC have also been established. In sporadic CRC, age is a risk factor with increased incidence between the ages of 40–50 with 90% of cases occurring after the age of 50 [4]. In the United States, men have a 25% higher incidence of CRC than women, and African Americans have a 20% higher incidence than Caucasians.
Genetic risk factors are evident in hereditary CRC syndromes such as familial adenomatous polyposis (FAP) and hereditary nonpolyposis colorectal cancer (HNPCC). In FAP, the adenomatous polyposis coli (APC) gene located on chromosome 5 is mutated and accounts for less than 1% of CRCs [5]. HNPCC accounts for 3–5% of CRCs and has a germline mutation in one allele of a mismatch repair gene including hMLH1, hMSH2, hMSH6, or PMS2, with inactivation of the second allele by loss of heterozygosity, somatic mutation, or promoter hypermethylation [5,6]. HNPCC-related CRCs present with KRAS mutations and do not have BRAF mutations [7]. Additional risk factors include personal or family history of CRC or adenomatous colon polyps [8, 9]. (Figure 1)
Figure 1.
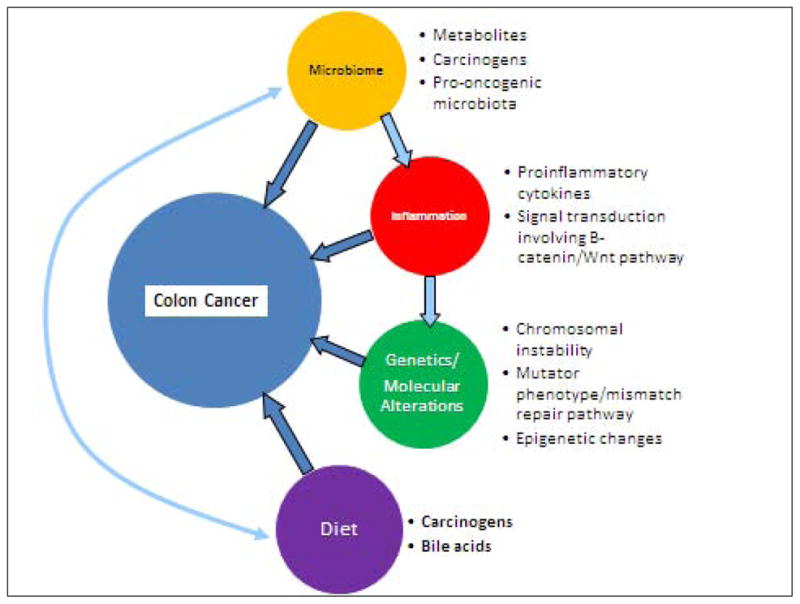
Overview of factors leading to colorectal carcinogenesis. The microbiome interacts with inflammatory mechanisms as well as dietary factors in progression of tumorigenesis.
The majority of CRCs are sporadic with tumorigenesis that involves mutations in APC (5q), DNA hypomethylation, and acquisition of multiple additional alterations, especially in KRAS2 (12p), DCC (18q), and p53 (17p) [10, 11]. BRAF mutations are especially prevalent in sporadic CRC of smokers [12].
At least three molecular pathways have been outlined in colorectal tumorigenesis. The chromosomal instability (CIN) pathway is seen in FAP as well as in sporadic CRC and is characterized by chromosomal abnormalities including deletions, insertions, and loss of heterozygosity [13]. The mutator phenotype/mismatch repair pathway is represented by HNPCC as outlined above. The third hypermethylation phenotype, hyperplastic/serrated polyp pathway, includes epigenetic changes including hypermethylation of some CpG islands. This alteration may result in hypermethylation of the promoter region of mismatch repair enzymes such as MLH1 [14].
IBD including ulcerative colitis (UC) and Crohn disease also predispose to CRC. Although the pathogenesis of CRC in the setting of IBD is poorly understood, studies suggest that there are differences from sporadic CRC. In contrast to sporadic CRC, mutations in the ras protooncogene are less frequently observed in CRC associated with UC and occur as a late event [15–17]. In CRC associated with IBD, loss of heterozygosity for p53 and SRC activation occur earlier [15, 17].
In addition to genetic factors, environmental factors also predispose individuals to CRC. In particular, diet has been linked to CRC. Some studies have shown that high intake of red and processed meats, highly-refined grains and starches, sugars, fat, and alcohol are associated with an increased risk of CRC [18, 19].
Lastly, microorganisms such as bacteria have been suggested as links to CRC. One example is the association of Streptococcus bovis with CRC that has been previously recognized [20]. However, the etiological nature of this association is unclear. The gut microbiota is an emerging environmental contributor to CRC that has led to various investigations in both animal and human models, which will be further discussed.
Normal Bacterial Microbiota
The colon contains an estimated load of 1013–1014 microorganisms that is composed of more than 1,000 different bacterial species [21, 22]. These microorganisms collectively comprise the microbiota [23, 24]. The normal colonic microbiota includes anaerobes such as Bacteroides, Eubacterium, Bifidobacterium, Fusobacterium, Peptostreptococcus, and Atopobium [24, 25]. Facultative anaerobes include Lactobacilli, Enterococci, Streptococci, and Enterobacteriaceae and are present at approximately 1,000-fold lower levels [24]. However, the exact number and variability of bacterial species among individuals remain to be characterized [24, 26].
Microbiota and Carcinogenesis in Colon
The population of microbiota in healthy adults is relatively stable over time with fluctuations occurring in response to environmental and pathological events [24, 27]. An association between colon cancer and specific bacterial species including Streptococcus bovis, Bacteroides, and Clostridia has been established [24, 28–30]. Interestingly, other bacterial strains such as Lactobacillus acidophilus and Bifidobacterium longum have been shown to inhibit tumorigenesis [24, 31, 32]. Hence, the microbiota appears to be a balance between beneficial and “harmful” bacteria with shifts influencing carcinogenesis.
Adherent/invasive Escherichia coli strains were abundant in the colonic mucosa of patients with CRC and adenoma but not in those with normal colonic mucosa [33]. Other studies have reported microbiome maps generated from late-stage CRC tissue. A relative abundance of Bacteroidaceae, Streptococcaceae, Fusobacteriaceae, Peptostreptococcaceae, Veillonellaceae, and Pasteurellaceae and a significantly lower level of Lachnospiraceae, Ruminococcaceae, and Lactobacillaceae have been found in cancerous tissues compared to the intestinal lumen [24, 34]. Chen et al. examined 16S rRNA genes to profile the microbiota present in patients with CRC compared to healthy controls and found considerable differences between the two groups. The mucosa-adherent microbiota, Bifidobacterium, Faecalibacterium, and Blautia, were reduced in CRC patients, whereas Porphyromonas and Mogibacterium were enriched [24, 34]. In the lumen, Erysipelotrichaceae, Prevotellaceae, and Coriobacteriaceae were also increased in CRC patients and suggest that intestinal lumen microflora increases CRC risk through direct interactions with the host [24, 34].
In a study by Gueimonde et al., quantitative reverse transcriptase polymerase chain reaction was used to analyze colonic mucosa samples obtained from 21 patients with CRC, 9 patients with diverticulitis, and 4 patients with inflammatory bowel disease. The CRC patients had significantly lower levels of B. longum and Bifidobacterium bifidum when compared to the other patients [35]. In a study by Shen et al., sequencing of 335 clones for phylogenetic and taxonomic analyses of adherent bacteria present in 21adenoma and 23 non-adenoma subjects showed higher numbers of Proteobacteria and lower numbers of Bacteroidetes in adenoma subjects [36]. Sobhani et al. analyzed stool bacterial DNA using pyrosequencing and subsequent PCA to detect shifts in the composition of the microbiota of CRC patients and found Bacteroides/Prevotella species to be more abundant in cancer patients than in control subjects [37]. These studies show a complex association of gut microbiota with CRC development.
Fusobacterium, a Gram-negative anaerobe that is often associated with periodontal disease [38], has received heightened attention recently after several groups demonstrated its link to CRC. Early studies revealed that Fusobacterium is more abundant in human CRC tissue than in adjacent normal tissue [39, 40] but a late study found that even in normal rectal mucosa, Fusobacterium is enriched in CRC case subjects compared with control subjects [34]. The difference is also evident in stool samples [41,42]. The enrichment of Fusobacterium can also be identified in colorectal adenoma, the precursor of CRC [42, 43]. CRC patients with high Fusobacterial levels had a significantly longer overall survival time than patients with low and moderate levels of the bacterium [42]. CRC with high level of Fusobacterium is associated with CpG island methylator phenotype, TP53 wild-type, hMLH1 methylation positivity, microsatellite instability, and CHD7/8 mutation [44]. FadA adhesin of Fusobacterium can mediate bacterial adherence and invasion and induce oncogenic and inflammatory responses to stimulate growth of CRC cells by activation of β-catenin signaling via FadA binding to E-cadherin [45].
Several animal studies have shown bacterial strains to be implicated in colorectal carcinogenesis. The first reported link between the gut microbiota and CRC development was in 1975 by Reddy et al. [46] where 93% of conventionally maintained rats developed chemically-induced CRC and only 20% of germ-free rats developed CRC. In carcinogen-treated rats, Streptococcus bovis and its antigens extracted from the bacterial cell wall led to increased expression of proliferation markers and formation of aberrant, hyperproliferative colonic crypts [24, 47].
Animal studies have also shown a link between the effects of the microbiota on metabolites and progression to CRC. Many carcinogenic compounds are metabolized in the liver and then conjugated to glucuronic acid before being excreted via the bile into the small intestine [24]. In the colon, bacterial β-glucuronidase hydrolyzes the conjugates and releases the parent compound and activated metabolite [48]. One example is seen with the colon carcinogen, dimethylhydrazine (DMH), which is metabolized in the liver. Small amounts of the procarcinogenic conjugate of the activated metabolite, methylazoxymethanol (MAM), are excreted in the bile and released in the colon through hydrolysis by bacteria [49]. Germ-free animals treated with DMH had fewer colon tumors when compared to conventional animals, and microflora-derived β-glucuronidase played an important role in the etiology of CRC [24, 49].
Intestinal microbiota also plays an important role in the metabolism of bile acids. The process of 7α-dehydroxylation involves the conversion of cholic to deoxycholic acid (DCA) and chenodeoxycholic to lithocholic acid (LCA) [24, 50]. In an animal model, infusion of DCA caused intestinal mucosal damage and led to increase in cell proliferation [51], and DCA-induced DNA damage also triggered calcium ion-dependent apoptosis independent of p53 [24, 52]. In a rat model, the capacity for DCA to enhance colon tumor development was shown to be attenuated by all-trans retinoic acid [53]. Secondary bile acids may also lead to progression of CRC by supporting apoptosis-resistant cells or by mediating interactions with important secondary messenger signaling systems known to be activated in CRC [24, 54].
Enterotoxigenic Bacteroides fragilis (ETBF) belongs to a group of bacterial drivers of CRC that are defined as intestinal bacteria with procarcinogenic features that may initiate CRC development [24]. One mechanism for this process involves the production of DNA-damaging compounds [24, 55]. For example, certain E. coli strains that harbor a polyketide synthetase island, which encodes a genotoxin called colibactin, can induce single-strand DNA breaks and lead to tumorigenesis [24, 55].
Furthermore, gut microbiota also appears to induce chronic inflammation and generate reactive metabolites and carcinogens leading to development of CRC [56].
Several experimental models have been used to study the role of microorganisms during the development of inflammation and CRC. ETBF can secrete a B. fragilis toxin (BFT) which can cause human inflammatory diarrhea and stimulate cleavage of the tumor suppressor protein, E-cadherin [57]. Loss of membrane-associated E-cadherin in HT29/C1 cells triggers the nuclear localization of β-catenin, which then binds with T cell factor-dependent transcriptional activators to induce expression of c-Myc and cyclin D1, which results in persistent cell proliferation [24, 58].
A mouse model of ETBF-induced colitis and carcinogenesis demonstrated enhanced tumorigenesis through induction of infiltration of the lamina propria by IL-17-producing CD4+ T cells (Th17) and γδ-T cells via STAT3 signaling [24, 59]. IL-17 can also promote tumor growth in vitro and in vivo via the production of IL-6 by IL-17 receptor-bearing tumor cell lines [60]. Both NF-κB [61] and STAT-3 [62] are key mediators of inflammation-driven carcinogenesis via their putative antiapoptotic and cell cycle activity in colonic epithelial cells and their promotion of procarcinogenic mediators by immune cells [24].
Microbiota and Inflammation in Colon
An inflammatory microenvironment has long been associated with contributing to and increasing the risk of colorectal cancer (CRC). The prototypical inflammatory bowel diseases, Cohn’s disease and ulcerative colitis, both carry an increased risk of malignancy [63]. The interplay between the gut microbiome and inflammation is complex namely because there are several microorganisms implicated acting either singly, concurrently, or synergistically with each other. These microorganisms interact with a complex immune system of which several cytokines, chemokines, factors, proteins and cells are implicated in contributing to setting the stage for tumorigenesis.
Chronic inflammation alters the microenvironment in several ways. It sustains an environment that promotes DNA damage of epithelial cells by introducing and maintaining the presence of nitric oxide and other reactive oxygen species. The cytokines and chemokines produced by inflammatory cells, in response to a microorganism or a by-product of its metabolism, act to eliminate the threat but at the same time suppress the immune response against cells undergoing transformation. These include nitric oxide, TNF-α, IL-1, IL-8, prostaglandin-2 derivatives as well as several molecules triggered in the inflammatory signaling pathway. Cytokines and chemokines also enhance tumor survival by promoting angiogenesis. Chief factors include TNF-α, IL-6, and IL-1. The transformation of epithelial cells is modulated by both groups of factors.
Inflammation alone or the presence of bacteria/bacterial metabolites alone is not enough to promote tumorigenesis. As an example, a series of experiments by Joshua et al in which IL-10−/− mice exposed to pro-carcinogenic compound azoxymethane (AOM) developed colitis and colorectal carcinoma (CRC) whereas wild type mice were colitis free and developed only low grade dysplasia [64]. Intestinal bacteria are needed to metabolically activate AOM [65] and trigger IL-10 production [66, 67]. These experiments support a mechanism of tumorigenesis in which induced chronic inflammation (by the microbiota) and disruption of the balance between pro-inflammatory and tolerogenic mediators promote disease.
In sporadic cases of CRC and experimental models of colitis associated cancer (CAC) the inducible mediator of prostaglandin synthesis, COX2, is upregulated [68, 69]. The pro-tumorogenic effects of COX2 upregulation lies in the synthesis of prostaglandin E2 (PGE2) [70, 71, 72, 73]. PGE2 also promotes tumor survival and proliferation via β-catenin dependent signaling. COX2 inhibits apoptosis by increasing Bcl-2 expression via MAP-kinase or PI3K/AKT signaling pathways [74, 75, 76]. It also enhances tumor survival by inducing production of the pro-angiogenic factors VEGF and b-FGF [77].
Probiotic Effects on Carcinogenesis
The use of diet to alter the intestinal microbiota can be seen in the example of probiotics. Probiotics are defined as live microorganisms which confer a health benefit to the host when administered in adequate amounts [78]. Species include Lactobacillus rhamnosus, Lactobacillus reuteri, and Lactobacillus acidophilus [24]. Probiotic mechanisms include immunological modulation, providing bioactive metabolites, binding mutagens, inhibition of intestinal bacterial enzymes, competition for limited nutrients, inhibition of harmful bacterial mucosa adherence, and inhibition of epithelial cell invasion [79, 80]. Molecular mechanisms involve macrophage activation, blocking of cytochrome P450, a reduction in carcinogen generation, downregulation of Ras-p21 expression, promotion of cell differentiation, inhibition of COX-2 upregulation, inhibition of nitric oxide synthase, an increase in short-chain fatty acid production, and a reduction in intestinal pH due to a decrease in the number of putrefactive bacteria [24, 79, 81].
Corthesy et al. summarized several studies that revealed that ingested probiotic strains may persist for relatively short periods and do not become permanent members of the normal microbiota [82]. However benefits are seen as various studies have shown antiproliferative effects of probiotic species in certain cancer cell lines [83, 84, 85]. In particular, Kim et al. assessed the anticancer activity and bacterial enzyme inhibition of Bifidobacterium adolescentis SPM0212 in human colon cancer cell lines [85]. This strain also was found to inhibit harmful fecal enzymes, including β-glucuronidase, β-glucosidase, tryptophanase, and urease [85]. There is, however, a need for more well-controlled clinical studies to elucidate therapeutic and preventive effects of probiotics in various diseases. Even so, the beneficial effects of certain probiotics have been documented in treatment of pouchitis, traveler’s, and antibiotic-associated diarrhea, irritable bowel syndrome, and rotavirus enteritis [24, 86].
Key Points.
The etiology of colorectal cancer (CRC) is multifactorial with genetic, molecular, inflammatory, and environmental risk factors. Recently, the gut microbiota is recognized as a new environmental contributor to CRC in both animal models and human studies.
An additional interplay of the gut microbiome with inflammation is also evident in studies that have demonstrated that inflammation alone or the presence of bacteria/bacterial metabolites alone is not enough to promote tumorigenesis.
Complex interrelationships with the gut microbiome, inflammation, genetics, and other environmental factors are evident in colorectal tumor progression.
Acknowledgments
This work was supported in part by grants U01CA18237, UH3CA140233, R01CA159036, and R03CA159414 from the National Cancer Institute and NIH Human Microbiome Project, and by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development.
Footnotes
Disclosure:
The Authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013 Jan;63(1):11–30. doi: 10.3322/caac.21166. Epub 2013 Jan 17. [DOI] [PubMed] [Google Scholar]
- 3.Kohler BA, Ward E, McCarthy BJ, Schymura MJ, Ries LA, Eheman C, Jemal A, Anderson RN, Ajani UA, Edwards BK. Annual report to the nation on the status of cancer, 1975–2007, featuring tumors of the brain and other nervous system. J Natl Cancer Inst. 2011;103(9):714–36. doi: 10.1093/jnci/djr077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eddy DM. Screening for colorectal cancer. Ann Intern Med. 1990;113(5):373–84. doi: 10.7326/0003-4819-113-5-373. [DOI] [PubMed] [Google Scholar]
- 5.Burt RW, DiSario JA, Cannon-Albright L. Genetics of colon cancer: impact of inheritance on colon cancer risk. Annu Rev Med. 1995;46:371–9. doi: 10.1146/annurev.med.46.1.371. [DOI] [PubMed] [Google Scholar]
- 6.Lynch HT, Smyrk TC, Watson P, Lanspa SJ, Lynch JF, Lynch PM, Cavalieri RJ, Boland CR. Genetics, natural history, tumor spectrum, and pathology of hereditary nonpolyposis colorectal cancer: an updated review. Gastroenterology. 1993;104(5):1535–49. doi: 10.1016/0016-5085(93)90368-m. [DOI] [PubMed] [Google Scholar]
- 7.Domingo E, Niessen RC, Oliveira C, Alhopuro P, Moutinho C, Espín E, Armengol M, Sijmons RH, Kleibeuker JH, Seruca R, Aaltonen LA, Imai K, Yamamoto H, Schwartz S, Jr, Hofstra RM. BRAF-V600E is not involved in the colorectal tumorigenesis of HNPCC in patients with functional MLH1 and MSH2 genes. Oncogene. 2005;24(24):3995–8. doi: 10.1038/sj.onc.1208569. [DOI] [PubMed] [Google Scholar]
- 8.Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med. 1992;326(10):658–62. doi: 10.1056/NEJM199203053261002. [DOI] [PubMed] [Google Scholar]
- 9.Imperiale TF, Ransohoff DF. Risk for colorectal cancer in persons with a family history of adenomatous polyps: a systematic review. Ann Intern Med. 2012;156(10):703–9. doi: 10.7326/0003-4819-156-10-201205150-00006. [DOI] [PubMed] [Google Scholar]
- 10.Potter JD. Colorectal cancer: molecules and populations. J Natl Cancer Inst. 1999;91(11):916–932. doi: 10.1093/jnci/91.11.916. [DOI] [PubMed] [Google Scholar]
- 11.Glei M, Latunde-Dada GO, Klinder A, Becker TW, Hermann U, Voigt K, Pool-Zobel BL. Iron-overload induces oxidative DNA damage in the human colon carcinoma cell line HT29 clone 19A. Mutation Research. 2002;519(1–2):151–161. doi: 10.1016/s1383-5718(02)00135-3. [DOI] [PubMed] [Google Scholar]
- 12.Samowitz WS, Albertsen H, Sweeney C, Herrick J, Caan BJ, Anderson KE, Wolff RK, Slattery ML. Association of smoking, CpG island methylator phenotype, and V600E BRAF mutations in colon cancer. J Natl Cancer Inst. 2006;98(23):1731–8. doi: 10.1093/jnci/djj468. [DOI] [PubMed] [Google Scholar]
- 13.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 14.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, Leggett B, Levine J, Kim M, French AJ, Thibodeau SN, Jass J, Haile R, Laird PW. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–93. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 15.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319(9):525–32. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 16.Burmer GC, Levine DS, Kulander BG, Haggitt RC, Rubin CE, Rabinovitch PS. c-Ki-ras mutations in chronic ulcerative colitis and sporadic colon carcinoma. Gastroenterology. 1990;99(2):416–20. doi: 10.1016/0016-5085(90)91024-z. [DOI] [PubMed] [Google Scholar]
- 17.Itzkowitz SH. Inflammatory bowel disease and cancer. Gastroenterol Clin North Am. 1997;26(1):129–39. doi: 10.1016/s0889-8553(05)70287-9. [DOI] [PubMed] [Google Scholar]
- 18.Chan AT, Giovannucci EL. Primary prevention of colorectal cancer. Gastroenterology. 2010;138(6):2029–2043. doi: 10.1053/j.gastro.2010.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bingham SA. Epidemiology and mechanisms relating diet to risk of colorectal cancer. Nutr Res Rev. 1996;9(1):197–239. doi: 10.1079/NRR19960012. [DOI] [PubMed] [Google Scholar]
- 20.McIIImurray MB, Langman MJ. Large bowel cancer: causation and management. Gut. 1975;16(10):815–820. doi: 10.1136/gut.16.10.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arthur JC, Jobin C. The complex interplay between inflammation, the microbiota and colorectal cancer. Gut Microbes. 2013;4(3):253–258. doi: 10.4161/gmic.24220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sobhani I, Amiot A, Le Baleur Y, Levy M, Auriault ML, Van Nhieu JT, Delchier JC. Microbial dysbiosis and colon carcinogenesis: could colon cancer be considered a bacteria-related disease? Ther Adv Gastroenterol. 2013;6(3):215–229. doi: 10.1177/1756283X12473674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–33. doi: 10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- 24.Zhu Q, Gao R, Wu W, Qin H. The role of gut microbiota in the pathogenesis of colorectal cancer. Tumor Biol. 2013;34(3):1285–1300. doi: 10.1007/s13277-013-0684-4. [DOI] [PubMed] [Google Scholar]
- 25.Tlaskalová-Hogenová H, Stepánková R, Hudcovic T, Tucková L, Cukrowska B, Lodinová-Zádníková R, et al. Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunol Lett. 2004;93(2–3):97–108. doi: 10.1016/j.imlet.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 26.Mai V. Dietary modification of the intestinal microbiota. Nutr Rev. 2004;62(6 Pt 1):235–42. doi: 10.1301/nr2004.jun235-242. [DOI] [PubMed] [Google Scholar]
- 27.Stanghellini V, Barbara G, Cremon C, Cogliandro R, Antonucci A, Gabusi V, et al. Gut microbiota and related diseases: clinical features. Intern Emerg Med. 2010;5 (Suppl 1):S57–63. doi: 10.1007/s11739-010-0451-0. [DOI] [PubMed] [Google Scholar]
- 28.Gold JS, Bayar S, Salem RR. Association of Streptococcus bovis bacteremia with colonic neoplasia and extracolonic malignancy. Arch Surg. 2004;139(7):760–5. doi: 10.1001/archsurg.139.7.760. [DOI] [PubMed] [Google Scholar]
- 29.Moore WE, Moore LH. Intestinal floras of populations that have a high risk of colon cancer. Appl Environ Microbiol. 1995;61 (9):3202–7. doi: 10.1128/aem.61.9.3202-3207.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura J, Kubota Y, Miyaoka M, Saitoh T, Mizuno F, Benno Y. Comparison of four microbial enzymes in Clostridia and Bacteroides isolated from human feces. Microbiol Immunol. 2002;46(7):487–90. doi: 10.1111/j.1348-0421.2002.tb02723.x. [DOI] [PubMed] [Google Scholar]
- 31.McIntosh GH, Royle PJ, Playne MJ. A probiotic strain of L. acidophilus reduces DMH-induced large intestinal tumors in male Sprague-Dawley rats. Nutr Cancer. 1999;35(2):153–9. doi: 10.1207/S15327914NC352_9. [DOI] [PubMed] [Google Scholar]
- 32.Rowland IR, Bearne CA, Fischer R, Pool-Zobel BL. The effect of lactulose on DNA damage induced by DMH in the colon of human flora-associated rats. Nutr Cancer. 1996;26(1):37–47. doi: 10.1080/01635589609514461. [DOI] [PubMed] [Google Scholar]
- 33.Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA. 2010;107(25):11537–42. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7(6):e39743. doi: 10.1371/journal.pone.0039743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gueimonde M, Ouwehand A, Huhtinen H, Salminen E, Salminen S. Qualitative and quantitative analyses of the bifidobacterial microbiota in the colonic mucosa of patients with colorectal cancer, diverticulitis and inflammatory bowel disease. World J Gastroenterol. 2007;13(29):3985–9. doi: 10.3748/wjg.v13.i29.3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen XJ, Rawls JF, Randall T, Burcal L, Mpande CN, Jenkins N, et al. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010;1(3):138–47. doi: 10.4161/gmic.1.3.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6(1):e16393. doi: 10.1371/journal.pone.0016393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Signat B, Roques C, Poulet P, Duffaut D. Fusobacterium nucleatum in periodontal health and disease. Curr Issues Mol Biol. 2011;13(2):25–36. [PubMed] [Google Scholar]
- 39.Kostic AD, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22(2):292–298. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castellarin M, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22(2):299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahn J, Sinha R, Pei Z, Dominianni C, Wu W, Goedert JJ, Hayes RB, Yang L. Human gut microbiome and risk for colorectal cancer. J National Cancer Institute. 2013;105(24):1907–11. doi: 10.1093/jnci/djt300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flanagan L, Schmid J, Ebert M, Soucek P, Kunicka T, Liska V, Bruha J, Neary P, Dezeeuw N, Tommasino M, Jenab M, Prehn JH, Hughes DJ. Fusobacterium nucleatum associates with stages of colorectal neoplasia development, colorectal cancer and disease outcome. Eur J Clin Microbiol Infect Dis. 2014 Aug;33(8):1381–90. doi: 10.1007/s10096-014-2081-3. [DOI] [PubMed] [Google Scholar]
- 43.McCoy AN, Araújo-Pérez F, Azcárate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium is associated with colorectal adenomas. PLoS One. 2013;8(1):e53653. doi: 10.1371/journal.pone.0053653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, Jelinek J, Yamano HO, Sugai T, An B, Shureiqi I, Toyota M, Kondo Y, Estécio MR, Issa JP. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 2014 Mar 1;74(5):1311–8. doi: 10.1158/0008-5472.CAN-13-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013 Aug 14;14(2):195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reddy BS, Mastromarino A, Wynder EL. Further leads on metabolic epidemiology of large bowel cancer. Cancer Res. 1975;35(11 Pt 2):3403–6. [PubMed] [Google Scholar]
- 47.Ellmerich S, Djouder N, Schöller M, Klein JP. Production of cytokines by monocytes, epithelial and endothelial cells activated by Streptococcus bovis. Cytokine. 2000;12(1):26–31. doi: 10.1006/cyto.1999.0521. [DOI] [PubMed] [Google Scholar]
- 48.Rowland IR. The role of the gastrointestinal microbiota in colorectal cancer. Curr Pharm Des. 2009;15(13):1524–7. doi: 10.2174/138161209788168191. [DOI] [PubMed] [Google Scholar]
- 49.Gill CI, Rowland IR. Diet and cancer: assessing the risk. Br J Nutr. 2002;88 (Suppl 1):S73–87. doi: 10.1079/BJN2002632. [DOI] [PubMed] [Google Scholar]
- 50.de Giorgio R, Blandizzi C. Targeting enteric neuroplasticity: diet and bugs as new key factors. Gastroenterology. 2010;138 (5):1663–6. doi: 10.1053/j.gastro.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 51.Rubin DC, Shaker A, Levin MS. Chronic intestinal inflammation: inflammatory bowel disease and colitis-associated colon cancer. Front Immunol. 2012;3:107. doi: 10.3389/fimmu.2012.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Powolny A, Xu J, Loo G. Deoxycholate induces DNA damage and apoptosis in human colon epithelial cells expressing either mutant or wild-type p53. Int J Biochem Cell Biol. 2001;33 (2):193–203. doi: 10.1016/s1357-2725(00)00080-7. [DOI] [PubMed] [Google Scholar]
- 53.Narahara H, Tatsuta M, Iishi H, Baba M, Uedo N, Sakai N, et al. K-ras point mutation is associated with enhancement by deoxycholic acid of colon carcinogenesis induced by azoxymethane, but not with its attenuation by all-trans-retinoic acid. Int J Cancer. 2000;88(2):157–61. doi: 10.1002/1097-0215(20001015)88:2<157::aid-ijc2>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 54.Radley S, Davis AE, Imray CH, Barker G, Morton DG, Baker PR, et al. Biliary bile acid profiles in familial adenomatous polyposis. Br J Surg. 1992;79(1):89–90. doi: 10.1002/bjs.1800790134. [DOI] [PubMed] [Google Scholar]
- 55.Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313(5788):848–51. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 56.Hope ME, Hold GL, Kain R, El-Omar EM. Sporadic colorectal cancer—role of the commensal microbiota. FEMS Microbiol Lett. 2005;244(1):1–7. doi: 10.1016/j.femsle.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 57.Rhee KJ, Wu S, Wu X, Huso DL, Karim B, Franco AA, et al. Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infect Immun. 2009;77(4):1708–18. doi: 10.1128/IAI.00814-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu S, Morin PJ, Maouyo D, Sears CL. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology. 2003;124(2):392–400. doi: 10.1053/gast.2003.50047. [DOI] [PubMed] [Google Scholar]
- 59.Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15 (9):1016–22. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206(7):1457–64. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 62.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7(1):41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 63.Moossavi S, Bishehsari F. Inflammation in sporadic colorectal cancer. Arch Iran Med. 2012;15(3):166–70. [PubMed] [Google Scholar]
- 64.Uronis JM, Mühlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C. Modulation of the intestinal microbiota alters colitis associated colorectal cancer susceptibility. PLoS One. 2009;4(6):e6026. doi: 10.1371/journal.pone.0006026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc. 2007;2:1998–2004. doi: 10.1038/nprot.2007.279. [DOI] [PubMed] [Google Scholar]
- 66.Ivanov II, de Frutos RL, Manel N, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453(7195):620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 68.Sheehan KM, Sheahan K, O’Donoghue DP, et al. The relationship between cyclooxygenase-2 expression and colorectal cancer. JAMA. 1999;282(13):1254–1257. doi: 10.1001/jama.282.13.1254. [DOI] [PubMed] [Google Scholar]
- 69.Arber N, Eagle CJ, Spicak J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355(9):885–895. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 70.Taketo MM. COX-2 and colon cancer. Inflamm Res. 1998;47(Suppl 2):S112–S116. doi: 10.1007/s000110050295. [DOI] [PubMed] [Google Scholar]
- 71.Pugh S, Thomas GA. Patients with adenomatous polyps and carcinomas have increased colonic mucosal prostaglandin E2. Gut. 1994;35(5):675–678. doi: 10.1136/gut.35.5.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang VW, Shields JM, Hamilton SR, et al. Size-dependent increase in prostanoid levels in adenomas of patients with familial adenomatous polyposis. Cancer Res. 1998;58(8):1750–1753. [PubMed] [Google Scholar]
- 73.Kawamori T, Uchiya N, Sugimura T, Wakabayashi K. Enhancement of colon carcinogenesis by prostaglandin E2 administration. Carcinogenesis. 2003;24(5):985–990. doi: 10.1093/carcin/bgg033. [DOI] [PubMed] [Google Scholar]
- 74.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310(5753):1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 75.Tessner TG, Muhale F, Riehl TE, Anant S, Stenson WF. Prostaglandin E2 reduces radiation-induced epithelial apoptosis through a mechanism involving AKT activation and bax translocation. J Clin Invest. 2004;114(11):1676–1685. doi: 10.1172/JCI22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pozzi A, Yan X, Macias-Perez I, et al. Colon carcinoma cell growth is associated with prostaglandin E2/EP4 receptor-evoked ERK activation. J Biol Chem. 2004;279(28):29797–29804. doi: 10.1074/jbc.M313989200. [DOI] [PubMed] [Google Scholar]
- 77.Tsujii M, Kawano S, DuBois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci U S A. 1997;94(7):3336–3340. doi: 10.1073/pnas.94.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu Y, Michelle Luo T, Jobin C, Young HA. Gut microbiota and probiotics in colon tumorigenesis. Cancer Lett. 2011;309(2):119–27. doi: 10.1016/j.canlet.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pala V, Sieri S, Berrino F, Vineis P, Sacerdote C, Palli D, et al. Yogurt consumption and risk of colorectal cancer in the Italian European prospective investigation into cancer and nutrition cohort. Int J Cancer. 2011;129(11):2712–9. doi: 10.1002/ijc.26193. [DOI] [PubMed] [Google Scholar]
- 80.de Vrese M, Schrezenmeir J. Probiotics, prebiotics, and synbiotics. Adv Biochem Eng Biotechnol. 2008;111:1–66. doi: 10.1007/10_2008_097. [DOI] [PubMed] [Google Scholar]
- 81.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–80. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Corthésy B, Gaskins HR, Mercenier A. Cross-talk between probiotic bacteria and the host immune system. J Nutr. 2007;137(3 Suppl 2):781S–90S. doi: 10.1093/jn/137.3.781S. [DOI] [PubMed] [Google Scholar]
- 83.Orlando A, Messa C, Linsalata M, Cavallini A, Russo F. Effects of Lactobacillus rhamnosus GG on proliferation and polyamine metabolism in HGC-27 human gastric and DLD-1 colonic cancer cell lines. Immunopharmacol Immunotoxicol. 2009;31(1):108–16. doi: 10.1080/08923970802443631. [DOI] [PubMed] [Google Scholar]
- 84.Lee NK, Park JS, Park E, Paik HD. Adherence and anticarcinogenic effects of Bacillus polyfermenticus SCD in the large intestine. Lett Appl Microbiol. 2007;44(3):274–8. doi: 10.1111/j.1472-765X.2006.02078.x. [DOI] [PubMed] [Google Scholar]
- 85.Kim Y, Lee D, Kim D, Cho J, Yang J, Chung M, et al. Inhibition of proliferation in colon cancer cell lines and harmful enzyme activity of colon bacteria by Bifidobacterium adolescentis SPM0212. Arch Pharm Res. 2008;31(4):468–73. doi: 10.1007/s12272-001-1180-y. [DOI] [PubMed] [Google Scholar]
- 86.Tlaskalová-Hogenová H, Stìpánková R, Kozáková H, Hudcovic T, Vannucci L, Tuèková L, et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol. 2011;8(2):110–20. doi: 10.1038/cmi.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]