

Abstract
The comparison of DNA methylation patterns across cancer types (pan-cancer methylome analyses) has revealed distinct subgroups of tumors that share similar methylation patterns. Integration of these data with the wealth of information derived from cancer genome profiling studies performed by large international consortia has provided novel insights into the cellular aberrations that contribute to cancer development. There is evidence that genetic mutations in epigenetic regulators (such as DNMT3, IDH1/2 or H3.3) mediate or contribute to these patterns, although a unifying molecular mechanism underlying the global alterations of DNA methylation has largely been elusive. Knowledge gained from pan-cancer methylome analyses will aid the development of diagnostic and prognostic biomarkers, improve patient stratification and the discovery of novel druggable targets for therapy, and will generate hypotheses for innovative clinical trial designs based on methylation subgroups rather than on cancer subtypes. In this review, we discuss recent advances in the global profiling of tumor genomes for aberrant DNA methylation and the integration of these data with cancer genome profiling data, highlight potential mechanisms leading to different methylation subgroups, and show how this information can be used in basic research and for translational applications. A remaining challenge is to experimentally prove the functional link between observed pan-cancer methylation patterns, the associated genetic aberrations, and their relevance for the development of cancer.
Introduction
Ongoing molecular characterizations of large cohorts of cancer patients using tumor samples from all major organs have made available a wealth of genomic, epigenomic, transcriptomic and proteomic data, enabling integrated analysis across different tumor types - so called pan-cancer analyses. These studies aim to identify genomic and epigenomic similarities and differences among distinct cancer types, independent of their tissue of origin [1]. The large number of available tumor sample datasets increases statistical power, allowing researchers to detect molecular aberrations that otherwise would have been missed. From these integrated analyses, mutational landscapes are emerging that have revealed novel oncogenic signatures and cancer driver mutations [2-4].
Cancer is no longer seen as solely a genetic disease; epigenetic alterations are now being taken into account as additional layers in the regulation of gene expression. Epigenetic modifications, including DNA methylation, non-coding RNAs, histone modifications and nucleosome positioning, modify chromatin structure and hence gene transcription. These mechanisms act coordinately to form an epigenetic landscape regulated by various enzymes, either establishing (writers), interpreting (readers), modifying (editors) or removing (erasers) epigenetic marks (reviewed in [5]).
DNA methylation is by far the best characterized epigenetic modification and is involved in the regulation of gene expression, genome stability and developmental processes (reviewed in [6]). High-throughput techniques, including array and sequencing-based technologies, now provide genome-scale DNA methylation maps (also called methylomes), which have confirmed aberrant methylation as a hallmark of all cancer types and are used to identify novel methylation-based cancer biomarkers.
Multidisciplinary international consortia such as The Cancer Genome Atlas (TCGA) or the International Cancer Genome Consortium (ICGC) have produced methylomes for thousands of samples from at least 15 cancer types (Box 1). Integrative data analyses have revealed that methylomes in subgroups within one tumor type might differ more than between distinct cancer types. Even within the same tumor, regional differences in DNA methylation alterations have been identified, associated with intrinsic tumor heterogeneity [7].
The TCGA Pan-Cancer project was launched in 2012 with the goal of collecting, analyzing and interpreting data across distinct tumor types and of making these resources publically available [2]. One of the aims of this project is to define pan-cancer methylation patterns and to integrate them with genomic, transcriptomic and proteomic data. A remarkable initial finding was that tumor samples cluster largely according to their tissue of origin [1]. Analyses of single tumor entities revealed that colorectal, gastric and endometrial cancers have similar highly methylated subgroups that are associated with tumors with microsatellite instability and hypermethylation of the MLH1 promoter. Subtypes of breast, serous endometrial, high-grade serous ovarian, colorectal and gastric carcinomas are associated with high chromosomal instability as well as with recurrent TP53 mutations and share patterns of low methylation. Moreover, emerging evidence shows that cancer genomes exhibit frequent mutations in epigenetic regulators, suggesting a close interplay between epigenomic and genomic events (reviewed in [8]). Identifying commonalities between tumor entities might help to identify therapeutic regimens that are in place for one tumor type as being of use for another, less well characterized one, and will allow better patient stratification [1]. Deciphering the mechanisms underlying methylation patterns will facilitate the identification of novel therapeutic targets.
In this review, we aim to highlight recent findings from genome-wide DNA methylation profiling studies. We describe DNA methylation subgroups in 11 distinct tumor entities and analyses across cancer types, and discuss the potential mechanisms underlying the different methylation subgroups. We also explore the potential use of DNA methylation as a biomarker for diagnostic, prognostic and treatment response, and as a target for epigenetic therapy.
Definition and function of DNA methylation
DNA methylation usually occurs at cytosine-guanine (CpG) dinucleotides, where DNA methyltransferases (DNMTs) catalyze the transfer of a methyl group to position 5 of a cytosine, generating 5-methylcytosine (5mC). DNMT1 maintains the patterns of DNA methylation after cell division using hemi-methylated DNA as a template [9], while the de novo methyltransferases DNMT3A and DNMT3B establish cytosine methylation during early development [10]. For a long time, it was believed that methyl groups are only removed passively after cell replication. However, active mechanisms of DNA demethylation were recently identified. For instance, DNA repair pathways have an essential role in the active removal of 5mC, involving proteins such as GADD45 (reviewed in [11]). Another mechanism implicates the ten-eleven translocation (TET) family of proteins, which catalyze the hydroxylation of 5mC to 5-hydroxymethylcytosine (5hmC) [12]. Subsequent studies showed that 5hmC can be further converted to 5-formylcytosine and/or 5-carboxylcytosine, which can then be excised by thymine-DNA glycosylase [13].
The location and distribution of 5mCs across the genome have important implications for understanding the roles of DNA methylation [6]. In mammalian genomes CpGs are unevenly distributed: they are depleted on a global scale but enriched at short CpG-rich DNA stretches known as CpG islands (CGIs), which are preferentially located at transcription start sites of gene promoters (reviewed in [14]). In normal cells, cytosines within CGIs are generally protected from DNA methylation, in contrast to the vast majority of CpGs, which are usually methylated (that is, at non-coding regions and repetitive elements) [15]. Flanking regions of CGIs (±2 kilobases), referred to as CGI shores, show tissue-specific DNA methylation and are associated with gene silencing [16].
The patterns of DNA methylation observed in normal genomes change during tumorigenesis. The first epigenetic alteration reported in cancer cells was a widespread loss of 5mC [17], which has been recently confirmed in single-base-resolution methylomes of colorectal cancer, chronic lymphocytic leukemia (CLL) and medulloblastoma [18-20]. Loss of DNA methylation occurs mainly at repetitive sequences, centromeric DNA and gene bodies, leading to genomic instability, reactivation of transposable elements or loss of imprinting, which ultimately contribute to tumor initiation or progression [21]. Hypomethylation can also lead to transcriptional activation of normally silenced genes such as oncogenes (reviewed in [22]). Additionally, whole-genome bisulfite sequencing (WGBS) analyses have shown that global hypomethylation usually coincides with large partially methylated domains (PMDs) that are associated with late replication lamina-associated domains and might lead to long-range epigenetic silencing through repressive chromatin domain formation [23,24]. Recent studies have also revealed that hypomethylation occurs at more localized regions, termed DNA methylation valleys (DMVs), which are enriched for developmental genes and may regulate tissue-specific expression [20,25]. Global or localized DNA hypomethylation in cancer cells is often accompanied by focal hypermethylation of CGIs (Figure 1), which contributes to carcinogenesis by transcriptional silencing of genes including tumor suppressor genes (TSGs) [26].
Figure 1.
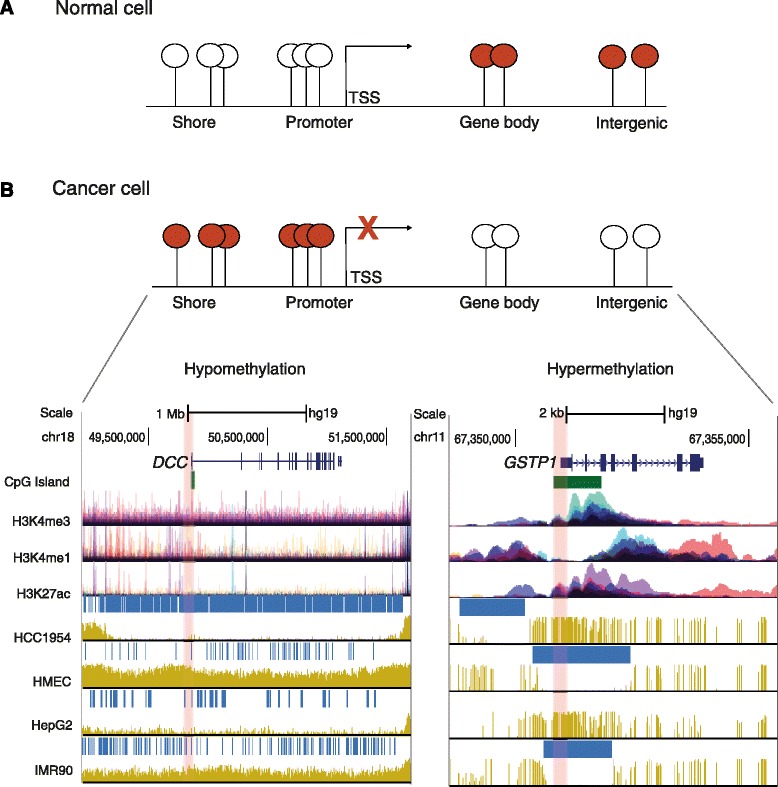
DNA methylation patterns in normal and cancer cells. (A) In normal cells, most CpGs located outside of promoters in gene bodies and intergenic regions are methylated (red circles), whereas promoter-associated CpG islands are protected from DNA methylation (white circles). (B) In cancer cells, a global or localized loss of 5-methylcytosine occurs at gene bodies and intergenic regions, whereas CpG-rich regions like promoters are usually heavily methylated, which might lead to transcriptional repression. Regions of intermediate CpG levels such as shores are associated with tissue-specific methylation. Global loss (left plot) and focal gain (right plot) of DNA methylation are depicted as tracks of the University of California Santa Cruz genome browser [118] using whole-genome bisulfite sequencing data for normal and cancer cell lines. Tracks for CpG islands and selected histone modifications, including H3K4me3, which is associated with transcriptionally active promoters, and H3K4me1 and H3K27ac as markers for enhancers, are illustrated below the gene track. Each color of the histone tracks represents an individual ENCODE cell line. The deleted in colon cancer gene (DCC) was taken as an exemplary locus for which long-range hypomethylation regions (horizontal blue bars) are observed in the breast cancer cell line HCC1954 and in the liver carcinoma cell line HepG2, but not in normal mammary epithelial cells (HMEC) or the myofibroblast cell line IMR90. The glutathione S-transferase P1 gene (GTSP1) represents an example of promoter hypermethylation (highlighted in red) in cancer cell lines compared to normal cells. TSS, transcription start site.
DNA methylation subgroups according to tumor types
It has long been thought that each tumor type has a characteristic DNA methylation pattern. For example, a specific pattern of high methylation at CGIs, defined as the CpG island methylator phenotype (CIMP), was first discovered in colorectal cancer [27], even before the omics era. Now, genome-wide sequencing projects have confirmed the existence of this and additional DNA methylation subgroups in multiple cancer types. The question remains as to what extent these DNA methylation patterns are unique for a specific tumor type or comparable across different types of cancers. The comprehensive molecular catalogs generated by the TCGA might help to shed light on this (summarized in Table 2).
Table 2.
Pan-cancer patterns of DNA methylation
| Tumor type (number of methylation groups) | Methylation subgroup | Genomic aberrations | Methylation pattern* | Comments | References |
|---|---|---|---|---|---|
| AML | High | IDH1/2 or TET2 mutations | A | Associated with patients presenting with an intermediate-risk karyotype | [43,51,107] |
| Co-occurrence of IDH1/2 and NPM1 mutations is associated with good clinical outcome | |||||
| Bladder urothelial (3) |
High | RB1 mutations | Smoking-pack years as predictor of CIMP phenotype Frequent mutations in chromatin regulators such as MLL2, ARID1A, KDM6A, and EP300 † Mutations in chromatin regulators were more frequent than in any other TCGA tumor |
[35] | |
| Low | ↑ TP53 mutations | B | |||
| Breast (5) |
B-CIMP | ↓ mutation rate | Luminal ER/PR-positive tumors | [31,32] | |
| Low metastatic risk and better clinical outcome | |||||
| Enriched for genes targeted by the PRC2 (e.g. SUZ12 and EZH2) | |||||
| B-CIMP-negative | ↑ TP53 mutations | B | Basal-like tumors (ER/PR-negative) | ||
| High metastatic risk and poor clinical outcome | |||||
| Cholangiocarcinoma | High | IDH1 and/or IDH2 mutations | A | Longer survival | [47] |
| Chondrosarcoma | High | IDH1 and/or IDH2 mutations | A | [46,64] | |
| Colorectal (4) |
CIMP-H | MLH1 hypermethylation | C | MSI | [29,108] |
| Right/ascending colonic region | |||||
| ↑ mutation rate | |||||
| ↑ BRAF V600E mutation | |||||
| Good prognosis | |||||
| ↑ BRAF V600E mutation | |||||
| CIMP-L | KRAS mutations | CIN (non-MSI) | |||
| Poor prognosis | |||||
| Two non-CIMP | ↑ TP53 mutations | B | Anatomic origins distinct from CIMP groups | ||
| ↑ SCNAs | |||||
| Endometrial (4) |
High | MLH1 hypermethylation | C | MSI | [33] |
| ARID5B mutations | |||||
| ↑ mutation rate | |||||
| Low | ↑ TP53 mutations | B | Serous-like tumors | ||
| ↑ SCNAs | Poor prognosis | ||||
| Two non-methylated | ↑ POLE mutations | Endometrioid tumors | |||
| ↑ SCNAs | ARID1A and PTEN mutations were present in all groups without high TP53 mutations | ||||
| Gastric (4) |
EBV-CIMP | ↑ PIK3CA, ARID1A and BCOR mutations | EBV-positive tumors Highest frequency of hypermethylation events among TCGA tumors |
[30] | |
| CDKN2A hypermethylation | |||||
| Amplifications of JAK2, CD274 and PDCD1LG2 | |||||
| Gastric CIMP | MLH1 silencing | C | MSI | ||
| ↑ mutation rate | |||||
| Cluster 3 – low | RHOA and CDH1 mutations | Enriched for the diffuse histological variant | |||
| Genomically stable | Also fusions involving RHO-family GTPase-activating proteins | ||||
| Cluster 4 – low | ↑ TP53 mutation | B | CIN | ||
| Focal amplifications of receptor tyrosine kinases | |||||
| Glioblastoma (6) |
G-CIMP | IDH1 mutations | A | Secondary tumors with proneural expression | [41,42,48] |
| ATRX mutations | |||||
| MYC mutations and amplifications | |||||
| Younger age at diagnosis | |||||
| Better survival rates | |||||
| G-CIMP negative proneural | No IDH1 mutations | Relative hypomethylation | |||
| PDGFRA amplifications | Proneural subtype cases without IDH1 mutations | ||||
| Pediatric glioblastoma (6) |
Global loss of methylation at non-promoter regions | H3F3A mutations | H3F3A mutations are mutually exclusive with IDH1 mutations and are associated with TP53 mutations and alternative lengthening of telomeres (ALT) | [49,109] | |
| Renal cell carcinoma | Global loss of methylation | SETD2 mutations | VHL hypermethylation in about 7 % of the tumors† | [36] | |
| Loss of methylation at non-promoter regions | |||||
| One of the tumor types with the lowest frequency of DNA methylation events | |||||
| Lung ADCA (3) |
CIMP-high | CDKN2A hypermethylation | Associated either with ↑ ploidy, ↑ mutation and the PI subtype or with ↓ ploidy, ↓ mutation rate and the TRU subtype | [39] | |
| MYC overexpression | |||||
| Mutations in chromatin modifiers such as SETD2, ARID1A, SMARCA4 † | |||||
| Lung SQCC (4) |
High | CDKN2A inactivation | Classical expression subtype | [38] | |
| NFE2L2, KEAP1, PTEN mutations | |||||
| Chromosomal instability | |||||
| ↑ SCNAs | |||||
| Low | Primitive expression subtype | ||||
| Serous ovarian (4) |
High | Germline and somatic BRCA1 mutations | More differentiated tumors | [34] | |
| Better survival | |||||
| Low | ↑ TP53 mutation | B | TP53 mutations occur in 90 % of the tumors and are not exclusive for the low methylation group | ||
| ↑ SCNAs | |||||
| BRAC1 hypermethylation | |||||
| Poor clinical outcome |
*Methylation patterns A, B and C indicate common genetic and epigenetic aberrations across different tumors. †These molecular aberrations were not necessarily associated with a specific methylation subgroup. ADCA, adenocarcinoma; AML, acute myeloid leukemia; CIMP, CpG island methylator phenotype; CIN, chromosomal instability; EBV, Epstein-Barr virus; ER, estrogen receptor; MSI, microsatellite instability; PI, proximal inflammatory; PR, progesterone receptor; PRC, polycomb repressor complex; SCNAs, somatic copy-number alterations; SQCC, squamous cell carcinoma; TCGA, The Cancer Genome Atlas; TRU, terminal respiratory unit.
However, a caveat should be noted: the methylation data underlying these reports were derived from 27 k and 450 k Illumina platforms. Only CpG sites covered on both platforms were considered and filtered for sites overlapping with single-nucleotide polymorphisms, resulting in around 10,000 eligible CpGs. From these, the most variable CpG sites were used for cluster analyses. The interpretation of these datasets is to a certain extent biased, as 27 k arrays mainly cover sites located within CGIs, while information on additional regulatory regions (for example, shores, intra- and intergenic enhancers) is missing. Also, information on larger genomic domains such as PMDs and DMVs cannot be determined from these datasets.
Colorectal and gastric cancer
According to the degree of methylation, colorectal cancer is currently divided into four DNA-methylation subgroups with specific genetic and clinical features [28,29]; that is, CIMP high (CIMP-H), CIMP low (CIMP-L) and two non-CIMP subgroups. CIMP-H is associated with hypermethylation of the repair gene MLH1, the activating BRAFV600E mutation and microsatellite instability (MSI). Tumors in this subgroup are often derived from the right/ascending colon, show high mutation rates (hypermutation) and low somatic copy-number alterations (SCNAs). The molecular mechanisms underlying these relationships need more investigation. CIMP-L is associated with tumors enriched for KRAS mutations and chromosomal instability (non-MSI). The non-CIMP subgroups, corresponding to the majority of colorectal tumors, do not show specific mutations, but are enriched for SCNAs and originate from distinct anatomical sites compared with the CIMP groups.
Epstein-Barr virus (EBV)-positive gastric tumors display an extreme EBV-CIMP profile [30], with hypermethylation of CDKN2A but not of MLH1. This phenotype has the highest frequency of DNA hypermethylation when compared with other cancer types reported by TCGA [30]. In contrast, gastric CIMP tumors showed hypermutation, MSI and epigenetic silencing of MLH1.
Breast, endometrial and ovarian carcinomas
A breast CpG island methylator phenotype (B-CIMP) was first reported in 2011 [31]. B-CIMP is enriched in estrogen and progesterone receptor (ER/PR)-positive tumors and is associated with good survival rates and low metastatic risk. It is characterized by high methylation of genes targeted by the polycomb repressor complex 2 (PRC2), including SUZ12 and EZH2 [31]. In contrast, the B-CIMP-negative group shows high metastatic risk and poor clinical outcome. TCGA analyses confirmed these findings, although they defined five distinct DNA methylation subgroups. The high methylation group overlapped with luminal B tumors (ER/PR-positive) and had a low rate of mutations. Conversely, the methylation-low group had a high TP53 mutation rate and was enriched in basal-like tumors (ER/PR-negative) [32].
In endometrial carcinomas, TCGA identified four DNA methylation subtypes. Similar to colorectal cancer, the high methylator phenotype was mainly composed of hypermutated MSI tumors showing extensive MLH1 promoter hypermethylation and an under-representation of TP53 mutations [33].
Four DNA methylation clusters were defined for serous ovarian cancer. This cancer type has a 90 % prevalence of TP53 mutations. TCGA identified a methylation-high group enriched for highly differentiated tumors with germline BRCA1 mutations. BRCA1 mutations were mutually exclusive with BRCA1 hypermethylation, which is characteristic of methylation-low tumors with high SCNAs. Survival analysis showed that cases with hypermethylated BRCA1 had a poorer clinical outcome compared to tumors with BRCA1/2 mutations [34].
Bladder urothelial and kidney renal clear cell carcinomas
Bladder urothelial carcinomas were divided into three DNA methylation subgroups; one of these groups had a CIMP-like hypermethylation profile and was enriched for tumors with RB1 mutations. Similar to the low methylation groups in breast, endometrial, gastric and colorectal tumors, the methylation-low group had the highest percentage of TP53 mutations, suggesting a common molecular mechanism of epigenetic regulation. Interestingly, chromatin regulators such as the histone methyltransferase MLL2, the chromatin remodeling gene ARID1A, the histone demethylase KDM6A and the histone acetyltransferase EP300 were frequently mutated in this cancer type [35].
For renal clear cell carcinoma, the most common type of kidney cancer, TCGA identified epigenetic silencing of the tumor suppressor VHL in about 7 % of the tumors, which was mutually exclusive with VHL mutations. Increased promoter methylation was linked to tumors with a higher grade and stage. Tumors with a widespread loss of DNA methylation were associated with mutations of the H3K36 methyltransferase SETD2, in contrast to methylation-low subgroups in other cancer types [36].
Lung adenocarcinoma and squamous cell carcinoma
Non-small-cell lung carcinoma (NSCLC), the most common type of lung cancer, is divided into three subtypes: adenocarcinoma, squamous cell carcinoma (SQCC), and large cell carcinoma [37]. Methylation analysis of SQCC identified four groups with distinct DNA methylation patterns. The methylation-high group overlapped with tumors from the so-called classical subtype, which are characterized by chromosomal instability. Moreover, the TSG CDKN2A was inactivated in 72 % of cases, 21 % of which were due to epigenetic silencing [38].
Recent results for adenocarcinoma revealed three different methylation subgroups: CIMP-H, a subgroup with intermediate methylation levels, and CIMP-L. Remarkably, these methylation subgroups were not specifically related to genomic, transcriptomic or histopathological subtypes. CIMP-H subtypes were either associated with tumors with high ploidy and a high mutation rate and were classified as proximal inflammatory (previously known as squamoid), or were associated with tumors presenting with low ploidy and a low mutation rate and were classified as terminal respiratory unit (formerly bronchioid). Moreover, an association between tumors enriched for SETD2 and CDKN2A methylation was found, suggesting an interaction between SETD2 mutations and altered chromatin structure for these tumors [39].
Glioblastoma
Aberrant DNA methylation has been widely described for glioblastoma multiforme (GBM) – the most common adult brain tumor. In 2008, TCGA chose GBM as the first cancer to be comprehensively characterized, revealing an important association between MGMT methylation, mutations in mismatch repair genes and response to therapy [40]. Subsequently, TCGA identified three DNA methylation groups, one of which showed hypermethylation at a large number of loci and was termed G-CIMP [41]. This group was enriched in secondary tumors with proneural expression and somatic mutations of the isocitrate dehydrogenase 1 (IDH1) gene [42]. This gain-of-function mutation results in increased catalysis of α-ketoglutarate to d-2-hydroxyglutarate (2-HG), which inhibits the activity of TET and KDM proteins, affecting chromatin remodeling and leading to an increase in DNA methylation. IDH1/2 mutations are also common in hematopoietic malignancies, including acute myeloid leukemia (AML) [43], myelodysplastic syndromes (MDS), myeloproliferative neoplasms [44] and T-cell lymphomas [45], as well as in solid tumors such as chondrosarcoma [46] and cholangiocarcinoma [47].
The G-CIMP group is associated with better survival compared with G-CIMP-negative tumors. The survival advantage of G-CIMP tumors was confirmed by a follow-up TCGA study characterizing more than 500 GBM tumors [48]. In this study, six DNA methylation clusters, including the G-CIMP subgroup, were identified. Additionally, the G-CIMP phenotype was associated with a younger age at diagnosis, enrichment for mutations in the chromatin remodeling gene ATRX, and MYC alterations.
The landscape of DNA methylation and genomic aberrations in pediatric GBM varies. Instead of having a hypermethylator phenotype, these tumors show a global loss of 5mC, which is mainly associated with extensive changes in histone modifications caused by mutations in H3F3A (reviewed in [8]). This was defined by Sturm et al., who found six epigenetic subgroups harboring specific mutations, SCNAs and transcriptome patterns [49]. Two methylation subgroups specifically correlated with hotspot mutations in H3F3A, namely at K27 and G34, and were associated with a younger age at diagnosis. Strikingly, the G34 tumors showed a global loss of methylation occurring mainly at chromosome ends. The presence of IDH1 mutations was mutually exclusive with H3F3A mutations.
Acute myeloid leukemia
AML is a highly heterogeneous myeloid disorder and the most common acute leukemia in adults. AML patients from the normal or intermediate cytogenetic risk category frequently have mutations in epigenetic regulators such as IDH1/2, DNMT3 and TET enzymes (reviewed in [50]). Similar to GBM, AML with a DNA hypermethylation phenotype is associated with IDH1/2 mutations [43]. These mutations are mutually exclusive with mutations in the demethylating enzyme TET2, suggesting a complementary role. It might be that DNA methylation is a consequence of mutant IDH expression and that this phenotype contributes to AML development. The association of IDH1/2 mutations with the hypermethylation phenotype in AML was confirmed by a recent TCGA study. Gain of DNA methylation was mainly observed at CpG-sparse regions of the genome. Other subtypes of tumors were associated with a substantial loss of DNA methylation and with the presence of MLL fusion genes or co-occurring mutations in NPM1, DNMT3A or FLT3 [51].
Potential mechanisms leading to DNA methylation subgroups
The observation that many tumor types carry numerous mutations in enzymes regulating epigenetic patterns suggests that these defects contribute to the global alterations seen in cancer genomes [5,8]. However, despite this expected molecular link, there are currently only reports associating methylome subgroups with gene mutations [29,49], rather than detailed molecular studies. Exceptions are studies on the histone H3.3 mutation H3F3A(K27M), which inactivates EZH2 in the PRC2 complex [52-54]. In addition, introduction of an IDH1 mutant, R132H, into astrocytes induces a specific methylome pattern [55]. Mutations in IDH1/2 cause accumulation of the oncometabolite 2-HG, which disturbs the DNA demethylation process, causing hypermethylation [43].
Epigenetic subgroups might also represent preexisting epigenetic states. For example, PRC2 target genes are commonly hypermethylated in cancer, and EZH2 is up-regulated in various cancer subtypes. These changes were associated with gene amplifications, and alterations in the regulation of gene expression by noncoding RNAs and mutations (reviewed in [56]). Apart from mutations affecting epigenetic modifiers, other genes are certainly also affected. Colorectal CIMP is tightly associated with BRAF mutations, although it appears that these mutations do not drive the hypermethylation phenotype [28]. Methylation subgroups might reflect the survival advantage of cell populations that have acquired early defects in DNA repair genes (for example, MLH1, MGMT and BRCA1). Distinct methylation clusters might also represent a common cell type of origin. As an example, the basal breast cancer subgroup shares characteristics of low methylation, high TP53 mutations and high chromosomal instability with serous endometrial and serous ovarian cancer subgroups [33]. Different epigenetic subgroups have been suggested to represent differences in tumor etiology induced by environmental factors, such as recently shown for EBV in gastric cancer [30].
Again, the question of whether there is a causal relationship between epigenetic changes and cancer or whether these associations represent changes in the methylome that are non-functional events and thus do not contribute to the carcinogenic process (passengers) rather than methylation events that drive the carcinogenic process (drivers) remains open. However, there are some general observations that extend across studies. First, mutations in epigenetic enzymes such as IDH1/2 are causally linked to the pathogenesis of subtypes of GBM and AML, as well as to the formation of CIMP. Second, mutations in the gene H3F3A encoding the histone variant H3.3 are associated with global loss of methylation, especially in sub-telomeric regions, and with the alternative lengthening of telomeres phenotype that is characteristic of a fraction of cancer cells, for example in pediatric GBM. Third, mutations in chromatin regulatory factors such as SETD2, ARID1, SMARCA4, KDM6A, EP300 and MLL are emerging in various cancer types [57] but, so far, only a few have been linked to altered methylome patterns. Many of these factors act in protein complexes, indicating that mutations in any of these could disrupt the function of the complex. Fourth, current cancer epigenome research points to the fact that methylation of polycomb group targets (PCGTs) is detectable even in pre-neoplastic lesions and could represent a risk factor for neoplastic transformation [58]. Fifth, recent reports have described particular methylation patterns related to infectious agents such as EBV or human papilloma virus (HPV), which can initiate carcinogenesis [30,59]; whether these methylation alterations are primarily useful biomarkers for patient stratification or whether there is a causal relationship to carcinogenesis has yet to be demonstrated. Last, similarities in methylation patterns across tumor types could indicate the accumulation of as yet unidentified, low frequency molecular aberrations that lead to a common phenotype and contribute to cancer development. Future research will have to address these points to draw clear conclusions.
Methylome analyses across different cancer types
The genome-wide methylation profiles generated by TCGA and others have shown that aberrant methylomes are a hallmark of cancer, and are useful for classifying tumor subgroups as well as for identifying novel clinical biomarkers. Currently, efforts are being made to integrate different methylomes and to determine common and tissue-specific DNA methylation patterns across multiple tumor entities (pan-cancer). These integrative analyses might also help to distinguish the driver methylation events (that contribute to the carcinogenic process) from the passenger methylation events (which do not contribute to the carcinogenic process).
In 2013, TCGA published the first integrative analysis of genomic data across 12 cancer types. In this study, SCNAs, somatic mutations and DNA methylation were integrated, although methylation changes were limited to a selection of 13 epigenetically silenced genes. From these genes, MGMT, GSTP1, MLH1 and CDKN2A were found to be aberrantly methylated in a large number of samples in different types of tumors. Hypermethylation of MLH1 was associated with the so-called ‘M class’, characterized by recurrent mutations, whereas BRCA1 hypermethylation correlated with the ‘C class’ of tumors enriched for SCNAs [3]. These findings confirm the previous TCGA reports for single tumor entities. However, by using this selected panel of genes, the results of this investigation might not reflect the actual similarities and differences in DNA methylation patterns across distinct tumor types, as for example shown in Figure 2.
Figure 2.

Pan-cancer methylome representation for ten cancer cohorts from The Cancer Genome Atlas. The Cancer Genome Atlas PANCAN12 DNA methylation data, representing 24,980 CpG sites acquired from the 27 k Illumina platform and corresponding to 2,224 tumor samples, were downloaded from the University of California Santa Cruz Cancer Genomics Browser [119]. CpG sites located on chromosome X and Y were removed, as well as the ones associated with single-nucleotide polymorphisms (n = 2,750). DNA methylation data for ten tumor entities - OV (n = 600), UCEC (n = 117), BRCA (n = 315), LUAD (n = 126), LUSC (n = 133), READ (n = 67), COAD (n = 166), GBM (n = 287), KIRC (n = 219) and AML (n = 194) - are included in the PANCAN12 dataset. For each of the tumor entities, color-coded on the top of the graph, the 500 most variable CpGs of the remaining 21,844 data points were selected. From the overlap, Qlucore Omics Explorer 3.0 software was used to select the 1,430 most variable CpGs, which were then hierarchically clustered as a heat map. Beta values are offset by −0.5 to shift the whole dataset to values between −0.5 (in dark blue) and 0.5 (in yellow) for improved graphical display [119]. DNA methylation patterns show relatively high homogeneity within tumor entities. We do not observe a common CpG island methylator phenotype-like group across several tumor types, suggesting that the ‘tissue of origin’ methylation signature is a strong decisive factor for the pattern. Colorectal cancer shows the highest overall methylation, whereas kidney cancer is characterized by low variance of methylation. The methylation patterns of ovarian, endometrial and breast cancer display a similar distribution of high and low methylation. CpG sites fall into high and intermediate DNA methylation clusters, covering all tumors entities, and a low methylation cluster with genes methylated in glioblastoma multiforme (GBM) or colorectal tumors and unmethylated in ovarian cancer. Unexpectedly, the high methylation cluster shows enrichment for membrane-associated genes including claudins (CLDN) and cadherins (CDH), while polycomb repressor complex PRC2 target genes are highly enriched in the intermediate and low methylation clusters. Some of these genes, as well as a selection of differentially methylated genes mentioned in the text such as MLH1, APC, BRCA1/2 and VHL, are indicated on the right side of the graph. For abbreviations of the tumor entities see Table 1.
By combining the methylomes of ten distinct tumor entities, Kim et al. found that aberrant DNA methylation affects similar biological pathways across the cancer types analyzed [60]. Over 50 % of the hypermethylation events were involved in early development and morphogenesis, including neurogenesis and embryonic development, whereas the remaining hypermethylation changes were related to transcription factor activity. A significant overlap between those pathways and PCGT genes was observed. Among the pan-cancer hypermethylated genes targeted by PRC2 were several members of the HOX family as well as the TSG CDKN2A. This finding is in agreement with previous studies reporting that methylation of PCGT genes is frequent in distinct cancer types (reviewed in [61]).
The integration of genome-wide DNA methylation data across four different gynecological tumors, namely breast, ovarian, endometrial and cervical carcinomas, revealed similar results [62]. This study additionally investigated the dynamics of DNA methylation through different stages of cervical carcinogenesis (that is, normal, invasive and metastatic stages). Hypermethylation at stem-cell PCGT genes was found to occur in cytologically normal cervical cells 3 years before the appearance of the first neoplastic alterations. Moreover, a loss of DNA methylation in CpGs termed ‘methylated embryonic stem-cell loci’ was predominantly observed in invasive tissues, suggesting that hypomethylation at these CpG sites might constitute a poor prognostic signature for these four gynecological tumor entities.
In contrast to these findings, a comparative analysis of methylomes from seven different tissue types revealed that hypermethylated genes tend to be already repressed in precancerous tissues and that aberrant methylation does not contribute to cancer progression under the classical model of epigenetic silencing [63]. It was suggested that pan-cancer patterns of hypermethylation occur owing to the variable gene expression profiles in the corresponding normal tissues. Hypermethylation of specific genes might then account for passenger methylation events rather than for driver events.
Apart from analyzing pan-cancer methylomes, integrative analyses of different tumors harboring mutations in common epigenetic regulators might provide clues about the molecular mechanisms affecting DNA methylation. Guilhamon et al. performed an exemplary meta-analysis of the DNA methylation profiles of tumors with IDH mutations and intrinsic high methylator phenotypes – namely AML, low-grade GBM, cholangiocarcinomas and chondrosarcomas [64]. The retinoic acid receptor pathway, which is usually dysregulated in the early steps of tumorigenesis, was enriched in the four tumor types. The early B-cell factor 1 (EBF1) was identified as a novel interaction partner of the dioxygenase TET2, suggesting that TET-mediated demethylation is regulated in a tissue-specific manner through EBF1 acting at the transcriptional or post-transcriptional level.
Clinical applications of DNA methylation in oncology
The identification of a wide number of genes that are affected by aberrant DNA methylation in cancer has highlighted the potential use of this epigenetic modification as a biomarker for cancer risk diagnosis, prognosis and prediction of therapy response. Moreover, the stable nature of DNA compared with RNA and the availability of high-throughput techniques for measurement of DNA methylation in large sample sets add advantages for its clinical application. The most prominent DNA methylation biomarkers are summarized in Table 3.
Table 3.
DNA methylation biomarkers and their potential clinical applications
| Biomarker name | Cancer type | Tissue detected |
|---|---|---|
| Risk | ||
| BRCA1 DNAm signature (1,829 CpGs) | Breast | Whole blood DNA [65] |
| 140 variable CpGs | Cervical | Normal uterine cervix cells [58] |
| Diagnosis | ||
| GSTP1 | Prostate | Serum, urine, ejaculate [70] |
| APC, EDNRB, GSTP1 | Prostate | Urine [71] |
| CDKN2A, ARF, MGMT, GSTP1 | Prostate | Urine [72] |
| GSTP1, APC, PTGS2 | Prostate | Paraffin-embedded tissues [110] |
| SETP9 | Colorectal | Blood plasma [77] |
| APC, MGMT, RASSF2A, WIF1 | Colorectal | Blood plasma [78] |
| SHOX2 | NSCLC | Bronchial fluid aspirates/ blood plasma [76] |
| CDKN2A, MGMT | NSCLC | Sputum [74] |
| CCND2, RASSF1A, APC, HIN1 | Breast | Fine needle aspiration biopsy [111] |
| ZNF154, HOXA9, POU4F2, EOMES | Bladder | Urine [112] |
| Prognosis | ||
| 20-gene signature | ALL | Leukemic cells from bone marrow and peripheral blood [88] |
| 15-gene classifier | AML | |
| RASSF1A, APC | Breast | Serum [82] |
| ZAP70 | CLL | CD19 sorted mononuclear cells [80] |
| CDKN2A | CCR | Blood plasma [81] |
| DAPK1 | Head and neck | Tumor samples [84] |
| DAPK1 | NSCLC | Tumor samples [83] |
| CDKN2A, RASSF1A, CDH13, APC | NSCLC | Primary tumors and lymph nodes [85] |
| HIST1H4F, PCDHGB6, NPBWR1, ALX1, HOX9 | NSCLC | Tumor samples [89] |
| ALDH1A, OSR2, GATA4, GRIA4, IRX4 | OPSCC | Tumor samples [59] |
| GSTP1, APC, PTGS2 | Prostate | Tumor samples [110] |
| Response to therapy | ||
| BRCA1 | Breast | Tumor samples [92,93] |
| BCL2 | Breast | Tumor samples [113] |
| PITX2 | Breast | Tumor samples [114] |
| TFAP2E | Colon | Tumor samples [115] |
| MGMT | Glioma | Tumor samples [90,91] |
| APAF1 | Melanoma | Tumor samples/cell lines [116] |
| IGFBP3 | NSCLC | Tumor samples/cell lines [117] |
| BRCA1 | Ovary | Tumor samples [94] |
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CCR, colorectal cancer; CLL, chronic lymphocytic leukemia; DNAm, DNA methylation; NSCLC, non-small-cell lung cancer; OPSCC, oropharyngeal squamous cell carcinoma.
DNA methylation for risk prediction and as a diagnostic biomarker
Recently, it has been proposed that the inherent epigenetic variability of normal cells can be used to predict the risk of neoplastic transformation. DNA methylation is being implemented as a molecular biomarker for early cancer detection that is able to distinguish early precancerous lesions from non-cancerous ones. Moreover, the analysis of DNA methylation offers the possibility of non-invasively detecting disease at early stages using biological fluids such as blood, saliva, urine and semen.
For instance, alterations in DNA methylation in healthy cervical tissues collected 3 years before detectable cytological and morphological transformations could predict the risk of acquiring cancer [58]. Differentially variable CpGs showed increased variance in normal cells from people predisposed to cervical neoplasia; the differentially variable CpGs were also enriched for developmental genes and PCGTs. Age-associated variation in DNA methylation was also correlated with the risk of neoplastic transformation.
A study analyzing whole blood from BRCA1 mutation carriers identified a methylation signature that predicted sporadic breast cancer risk and death years in advance of diagnosis [65]. Hypermethylated CpGs in BRCA1 mutation carriers were enriched for stem cell PCGTs, demonstrating that alterations of PCGTs occur early in tumorigenesis, as previously described [62,66]. Another study using whole blood samples identified a PCGT methylation signature present in preneoplastic conditions that was prone to become methylated with age, suggesting that age might predispose to tumorigenesis by irreversibly maintaining stem-cell properties [67]. Although attractive as a surrogate tissue, analyses in whole blood should be cautiously interpreted and stringently validated owing to its cellular heterogeneity [68].
Aberrant DNA methylation is also emerging as a potential tool for cancer detection. The list of methylation-based diagnostic biomarkers for different tumor types is enormous. For some of these biomarkers commercially kits are available. Hypermethylation of GSTP1, one of the first epigenetic biomarkers to be implemented in the clinic, is used for early diagnosis of prostate cancer [69]. The promoter of this gene is highly methylated in about 90 % of prostate cancers and can be detected in serum, urine and semen [70]. By combining GSTP1 hypermethylation with (1) the DNA methylation levels of the TSGs APC and EDNRB [71], (2) the DNA methylation levels of CDKN2A, ARF and MGMT [72], or (3) the levels of the prostate-specific antigen, prostate cancer diagnosis sensitivity is improved [73]. In NSCLC, aberrant DNA methylation of CDKN2A and MGMT were used to detect malignant lung carcinoma 3 years before its diagnosis using samples from a small cohort of patients [74]. Hypermethylation of the homeobox gene SHOX2 in bronchial fluid aspirates of more than 500 patient samples allowed the differentiation of benign lung lesions from carcinogenic lesions [75]. A subsequent study analyzing blood plasma from 411 individuals confirmed the specificity and sensitivity of SHOX2 hypermethylation [76], identifying it as a potential clinical biomarker for early non-invasive lung cancer diagnosis.
Another exemplary diagnostic biomarker is the hypermethylation of SET pseudogene 9 (SETP9) in colorectal cancer, which can be sensitively and specifically detected in blood plasma and is able to differentiate between all the stages of the disease [77]. Tumor-specific methylation of APC, MGMT, RASSF2A and WIF1 have also been suggested as potential biomarkers for early detection of colorectal cancer [78]. Moreover, a recent genome-wide screen using DNA methylation data from more than 700 colorectal cancer samples identified hypermethylation of the thrombin receptor THBD and of C9orf50 as novel blood-based biomarkers for colorectal cancer detection [79].
DNA methylation as a prognosis biomarker
In addition to its diagnostic applications, aberrant DNA methylation could help to predict and stratify patients with risks of distinct clinical outcomes. Studies using DNA methylation as a prognostic biomarker have identified more aggressive tumors and predicted overall survival and risk of disease progression and/or recurrence. Initially, studies combined clinical characteristics with aberrant DNA methylation at single or multiple genes, but genome-wide DNA methylation profiling of thousands of CpG sites is now leading to the identification of prognostic signatures.
In CLL, DNA methylation of a single CpG within the zeta-chain-associated protein kinase 70 (ZAP70) gene promoter predicted disease outcome better than current genetic approaches [80]. Examples of other hypermethylated genes used to predict poor clinical prognosis include CDKN2A in colorectal cancer [81], RASSF1A and APC in breast cancer [82], the apoptosis-associated gene DAPK1 in lung and head and neck cancers [83,84], and CDKN2A, RASSF1A, cadherin 13 (CDH13) and APC in stage I NSCLC [85].
The first studies characterizing DNA methylation at a genome-wide scale and using large cohorts of patients to investigate prognostic signatures were performed on hematopoietic malignancies. In AML, the methylomes of 344 patients were used to classify 16 distinct AML subgroups. From these, 5 subgroups defined new AML subtypes without any reported cytogenetic, molecular or clinical features. This study also revealed a 15-gene methylation classifier that predicted overall survival [86]. A recent investigation that focused on cytogenetically normal AML patients identified a seven-gene score which combined DNA methylation and gene expression and was associated with patient outcome [87]. In childhood acute lymphoblastic leukemia (ALL), distinct biological ALL subtypes were identified, as well as a group of genes whose DNA methylation levels correlated with a higher risk of relapse [88]. Another study in HPV-driven oropharyngeal squamous cell carcinoma defined a DNA methylation score of five genes (ALDH1A2, OSR2, GATA4, GRIA4 and IRX4), which was associated with clinical outcome [59]. Moreover, DNA hypermethylation of five genes (HIST1H4F, PCDHGB6, NPBWR1, ALX1 and HOXA9) was used to classify high- and low-risk stage I NSCLC and patients with shorter relapse-free survival [89]. Apart from these studies, the efforts of TCGA have shown that methylomes could be used to stratify tumors with distinct biological and clinical characteristics, as mentioned earlier.
DNA methylation as a biomarker to predict treatment response
The individual response of each patient to chemotherapeutic drugs is quite heterogeneous and, hence, biomarkers that predict response to therapy as well as the development of drug resistance are urgently required. DNA methylation has proven to be a suitable biomarker to predict treatment outcome in various types of tumors. Such a marker was identified in GBM, where hypermethylation of the DNA repair gene MGMT predicted treatment response. Silencing of MGMT diminishes DNA repair activity and removal of alkyl lesions, and thus predicts responsiveness to chemotherapeutic agents such as temozolomide and carmustine [90,91]. TCGA confirmed these findings and further identified that MGMT hypermethylation in GBM patients might predict responders from non-responders more accurately than the classical expression subgroups [48].
Hypermethylation of the DNA repair gene BRCA1 in sporadic triple-negative breast tumors has also been proposed as a biomarker to predict sensitivity of breast cancers to the cross-linking agent cisplatin [92] and to the poly(ADP)-ribose polymerase inhibitor olaparib [93]. Similar results were observed in ovarian tumors with BRCA1/2 mutations, where BRCA1 hypermethylation predicted better response to poly(ADP)-ribose polymerase inhibitor treatment [94].
Therapeutic use
Owing to its reversible nature in comparison to genetic alterations, aberrant DNA methylation can also be therapeutically targeted. Epigenetic drugs such as the histone deacetylase (HDAC) inhibitors, DNA demethylating agents or small molecule inhibitors of the BET family of bromodomain proteins have been shown to modify chromatin structure and modify DNA methylation patterns across the genome [95,96]. DNMT inhibitors can be incorporated into the DNA or RNA of replicating cells, blocking the catalytic domain of DNMTs and thus inhibiting the maintenance of DNA methylation after cell division. The DNMT inhibitors azacitidine (5-azacytidine) and decitabine (5-aza-2'-deoxycytidine) have been tested in clinical trials for hematopoietic malignancies and were approved by the US Food and Drug Administration for the treatment of MDS and AML [97,98]. Moreover, azacitidine in combination with an HDAC inhibitor has been used as a treatment regimen in a phase II clinical trial for solid tumors including NSCLC, breast cancer and colorectal cancer [95,99]. The results obtained for NSCLC showed durable responses and better patient survival, suggesting that combined epigenetic therapy may have clinical benefits for the treatment of this and other solid tumor types.
Conclusions and future perspectives
The integration of genome-wide DNA methylation profiles with genomic and other omic profiles is just emerging, and further efforts are needed to complete cross-tumor analyses, which will then help us to understand the molecular mechanisms responsible for the epigenetic defects that can result from aberrant DNA methylation. Several interesting findings have been revealed. Subgroups of cancers with high methylation (including CIMP), are associated with individual genomic aberrations underlying these patterns, and have been identified in various cancer entities. At present, however, there is no evidence for a unifying mechanism leading to these high methylation phenotypes.
Moreover, several tumor types, such as basal breast, high-grade serous ovarian and subtypes of serous endometrial, gastric and colorectal carcinomas, related to frequent TP53 mutations and high levels of SCNAs, share a pattern of low methylation in CGIs. Apparently, in these tumor subtypes, CGIs retain the low methylation patterns observed in normal tissues and are protected from methylation or are subjected to active demethylation. Again, the molecular mechanism underlying these observations is not known. We hypothesize that in this case structural genomic alterations are sufficient to drive carcinogenesis.
Although still in its infancy, pan-cancer methylome analyses have provided some interesting insights into the mechanisms of cancer development. First, it is becoming more apparent that multiple cancer types are affected by mutations in genes encoding epigenetic regulatory enzymes, histone variants and chromatin regulatory factors. Some of these have been experimentally shown to contribute to alterations in methylation patterns. Comparing methylomes across cancer types might now help to identify novel non-recurrent mutations converging on common biological pathways that might lead to the development of altered methylation phenotypes in specific subgroups of cancers. Second, hypermethylation of PCGTs is apparent in basically every tumor type and can even be observed in preneoplastic tissues. Third, the influence of environmental factors on DNA methylomes might have been underestimated until now. For example, infectious agents have been recently linked to specific methylation patterns.
However, pan-cancer methylome analyses still need to overcome some challenges. First, in the past, DNA methylation data were generated on two different platforms for some tumor types. Integration of these data restricts the output to overlapping CpG sites, mostly representing CGIs, and strongly reduces the genome-wide coverage. With the generation of larger datasets derived from the 450 k platform, these limitations will be overcome in the future. Second, comparing datasets derived from different platforms, and from samples provided by various centers, is intrinsically prone to systematic batch effects that need to be carefully monitored. Third, some tumor types are characterized by high tumor heterogeneity that is difficult to control and might lead to false positive results. Also, high tumor purity is an important prerequisite for correct data interpretation, but is often difficult to achieve. Enrichment of certain cell types by sorting or laser capture microdissection prior to analysis might be desirable. Fourth, for the development of clinical predictive, diagnostic or prognostic biomarkers and stratification of patient subgroups, the availability of well documented clinical data is essential. Last, integrative and comparative analyses of multi-platform datasets require powerful bioinformatic and biostatistical algorithms. Dedicated computational centers have to develop and rigorously test and validate these tools.
The epigenetic field is rapidly evolving, and in the near future more single-base resolution methylomes for a large number of tumors will be available. The generation of such methylomes is now affordable due to a considerable reduction in next-generation sequencing costs, improved computational expertise and emerging technologies that use lower DNA input, such as tagmentation-based WGBS. This method is used for WGBS library preparation, and is based on the enzymatic activity of a transposase to simultaneously fragment and tag DNA with adapters [100]. High-resolution methylation maps will provide additional information to the current methylomes, especially regarding cytosine methylation in a non-CpG context, long-range methylation interactions, and better assessment of allele-specific DNA methylation (reviewed in [101]). In addition, high sequencing coverage will accurately quantify DNA methylation in genomic regions such as enhancers, insulators, intergenic regions and repetitive elements, which are currently not included in pan-cancer methylome analyses.
In the longer term, novel technologies will also allow genomic and epigenomic analyses of single cells. These analyses will generate more precise datasets by avoiding the problems associated with tissue impurities or heterogeneity, and will allow a direct link between the methylome and the transcriptome [102]. However, the broad application of single-cell analyses still requires methodological development to reduce technical artefacts. To fully understand the interplay between the genome, epigenome and transcriptome, existing datasets need to be integrated with information about additional mechanisms of epigenomic regulation, including the emerging non-coding transcriptome and higher-order chromatin organization. Importantly, hypotheses generated from these combined efforts need to be experimentally tested to prove their functional relevance.
Finally, in terms of translation to the clinic, an essential aspect is to use the knowledge generated by methylome analyses as well as from the integration of methylation data with other omic data to identify novel clinical markers that should be able to stratify patients better and to define molecular signatures across different tumor types. On the basis of these molecular markers, novel epigenetic therapies could be developed, setting the stage for better clinical trial strategies across cancer types as well as for personalized medicine based on next-generation sequencing data. Already, pan-cancer analyses have revealed molecular similarities that will allow existing therapies to be applied to different cancer types.
Box 1 The International Cancer Genome Consortium: characterizing cancer genomes in different tumor types
Cancer genomes are complex. The integration of comprehensive catalogues of genomic, transcriptomic, epigenomic and proteomic data is a promising strategy for tackling this complexity. Institutions from across the globe have joined forces to achieve this ambitious goal. In 2006, The Cancer Genome Atlas (TCGA) Research Network was launched in the USA with the aim of generating molecular profiles of thousands of samples from more than 25 distinct tumor types [2]. A year later, the International Cancer Genome Consortium (ICGC) was created, with the goal of characterizing genomes from 50 different cancer types and subtypes worldwide [103]. By 2013, TCGA – now an ICGC member – produced comprehensive molecular profiles of more than 7,000 samples from 27 types of cancer [2]. All the data generated by these research networks are publicly available via the ICGC [104], TCGA [105] and the cancer genomics hub [106] data portals.
To make these data comparable, the ICGC aims to standardize the collection, processing and analysis of samples across multiple institutions. Infinium HumanMethylation27 and HumanMethylation450 BeadChips have been used by ICGC to produce genome-wide DNA methylation profiles. From at least 15 cancer methylomes generated so far, the breast cancer methylome comprises the largest number of samples, followed by serous ovarian and kidney renal clear cell carcinoma (Table 1). Moreover, whole-genome bisulfite sequencing (WGBS) will be applied for some tumors and has already been used to generate the methylomes of pediatric brain tumors and chronic lymphocytic leukemia (CLL).
Table 1.
International Cancer Genome Consortium projects with methylomes generated by Infinium BeadChips
| Tumor type | Project and country identification | Number of methylomes |
|---|---|---|
| Breast | BRCA-US | 971 |
| Ovary | OV-US | 572 |
| Kidney | KIRC-US | 491 |
| Head and neck | THCA-US | 488 |
| Uterus | UCEC-US | 481 |
| Lung | LUAD-US | 460 |
| Colorectal | COAD-US | 414 |
| Lung | LUSC-US | 410 |
| Head and neck | HNSC-US | 407 |
| Brain | GBM-US | 393 |
| Skin | SKCM-US | 338 |
| Stomach | STAD-US | 328 |
| Brain | LGG-US | 293 |
| Bladder | BLCA-US | 198 |
| Prostate | PRAD-US | 196 |
| Blood | LAML-US | 194 |
| Pancreas | PACA-AU | 167 |
| Blood | CLLE-ES | 159 |
| Colorectal | READ-US | 150 |
| Liver | LIHC-US | 149 |
| Kidney | KIRP-US | 142 |
| Cervix | CESC-US | 127 |
| Brain | PBCA-DE | 115 |
| Ovary | OV-AU | 93 |
| Pancreas | PAAD-US | 72 |
| Pancreas | PAEN-AU | 23 |
Modified from the International Cancer Genome Consortium data portal [104]. AU, Australia; DE, Germany; ES, Spain; US, United States.
Acknowledgements
This work was supported in part by the German Federal Ministry of Education and Science through the program for medical genome research (FKZ: 01KU1001A) to CP and CG and by the German Funding Agency (DFG SPP1463) to TW and CP. TW is supported by a PhD scholarship provided by the Helmholtz International Graduate School for Cancer Research at the German Cancer Research Center.
Abbreviations
- 2-HG
d-2-hydroxyglutarate
- 5hmC
5-hydroxymethylcytosine
- 5mC
5-methylcytosine
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- CGI
CpG island
- CIMP
CpG island methylator phenotype
- CLL
chronic lymphocytic leukemia
- DMV
DNA methylation valley
- DNMT
DNA methyltransferase
- EBF1
early B-cell factor 1
- EBV
Epstein-Barr virus
- ER
estrogen receptor
- GBM
glioblastoma multiforme
- HDCA
histone deacetylase
- HPV
human papilloma virus
- ICGC
International Cancer Genome Consortium
- MDS
myelodysplastic syndrome
- MSI
microsatellite instability
- NSCLC
non-small-cell lung carcinoma
- PCGT
polycomb group target
- PMD
partially methylated domain
- PR
progesterone receptor
- PRC
polycomb repressor complex
- SCNA
somatic copy-number alteration
- SQCC
squamous cell carcinoma
- TCGA
The Cancer Genome Atlas
- TET
ten-eleven translocation
- TSG
tumor suppressor gene
- WGBS
whole-genome bisulfite sequencing
Footnotes
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Tania Witte, Email: t.witte@dkfz.de.
Christoph Plass, Email: c.plass@dkfz.def.
Clarissa Gerhauser, Email: c.gerhauser@dkfz.de.
References
- 1.Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D, Zhu J. Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Sci Rep. 2013;3:2652. doi: 10.1038/srep02652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DT, Jones D, Knappskog S, Kool M, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 7.Brocks D, Assenov Y, Minner S, Bogatyrova O, Simon R, Koop C, Oakes C, Zucknick M, Lipka DB, Weischenfeldt J, Feuerbach L, Cowper-Sal Lari R, Lupien M, Brors B, Korbel J, Schlomm T, Tanay A, Sauter G, Gerhauser C, Plass C, ICGC Early Onset Prostate Cancer Project Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep. 2014;8:798–806. doi: 10.1016/j.celrep.2014.06.053. [DOI] [PubMed] [Google Scholar]
- 8.Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet. 2013;14:765–780. doi: 10.1038/nrg3554. [DOI] [PubMed] [Google Scholar]
- 9.Hermann A, Goyal R, Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem. 2004;279:48350–48359. doi: 10.1074/jbc.M403427200. [DOI] [PubMed] [Google Scholar]
- 10.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 11.Niehrs C, Schafer A. Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol. 2012;22:220–227. doi: 10.1016/j.tcb.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 16.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, Ehrlich M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983;11:6883–6894. doi: 10.1093/nar/11.19.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kulis M, Heath S, Bibikova M, Queiros AC, Navarro A, Clot G, Martinez-Trillos A, Castellano G, Brun-Heath I, Pinyol M, Barberan-Soler S, Papasaikas P, Jares P, Bea S, Rico D, Ecker S, Rubio M, Royo R, Ho V, Klotzle B, Hernandez L, Conde L, Lopez-Guerra M, Colomer D, Villamor N, Aymerich M, Rozman M, Bayes M, Gut M, Gelpi JL, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44:1236–1242. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- 19.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, Briem E, Zhang K, Irizarry RA, Feinberg AP. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hovestadt V, Jones DT, Picelli S, Wang W, Kool M, Northcott PA, Sultan M, Stachurski K, Ryzhova M, Warnatz HJ, Ralser M, Brun S, Bunt J, Jager N, Kleinheinz K, Erkek S, Weber UD, Bartholomae CC, von Kalle C, Lawerenz C, Eils J, Koster J, Versteeg R, Milde T, Witt O, Schmidt S, Wolf S, Pietsch T, Rutkowski S, Scheurlen W, et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 2014;510:537–541. doi: 10.1038/nature13268. [DOI] [PubMed] [Google Scholar]
- 21.Hoffmann MJ, Schulz WA. Causes and consequences of DNA hypomethylation in human cancer. Biochem Cell Biol. 2005;83:296–321. doi: 10.1139/o05-036. [DOI] [PubMed] [Google Scholar]
- 22.Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev. 2012;22:50–55. doi: 10.1016/j.gde.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 23.Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CP, van Dijk CM, Tollenaar RA, Van Den Berg D, Laird PW. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet. 2012;44:40–46. doi: 10.1038/ng.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE, Camargo AA, Stevenson BJ, Ecker JR, Bafna V, Strausberg RL, Simpson AJ, Ren B. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012;22:246–258. doi: 10.1101/gr.125872.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeong M, Sun D, Luo M, Huang Y, Challen GA, Rodriguez B, Zhang X, Chavez L, Wang H, Hannah R, Kim SB, Yang L, Ko M, Chen R, Gottgens B, Lee JS, Gunaratne P, Godley LA, Darlington GJ, Rao A, Li W, Goodell MA. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat Genet. 2014;46:17–23. doi: 10.1038/ng.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 27.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, Malik S, Pan F, Noushmehr H, van Dijk CM, Tollenaar RA, Laird PW. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012;22:271–282. doi: 10.1101/gr.117523.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The Cancer Genome Atlas Research Network: Comprehensive molecular characterization of gastric adenocarcinoma.Nature 2014, doi:10.1038/nature13480. [DOI] [PMC free article] [PubMed]
- 31.Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, Baylin SB, Ahuja N, Viale A, Massague J, Norton L, Vahdat LT, Moynahan ME, Chan TA. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011;3:75ra25. doi: 10.1126/scitranslmed.3001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.The Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The Cancer Genome Atlas Research Network Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.The Cancer Genome Atlas Research Network Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hensing T, Chawla A, Batra R, Salgia R. A personalized treatment for lung cancer: molecular pathways, targeted therapies, and genomic characterization. Adv Exp Med Biol. 2014;799:85–117. doi: 10.1007/978-1-4614-8778-4_5. [DOI] [PubMed] [Google Scholar]
- 38.The Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.The Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.The Cancer Genome Atlas Research Netowrk Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K, The Cancer Genome Atlas Research Network Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B, Hunault-Berger M, Slama B, Vey N, Lacombe C, Solary E, Birnbaum D, Bernard OA, Fontenay M. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia. 2010;24:1094–1096. doi: 10.1038/leu.2010.52. [DOI] [PubMed] [Google Scholar]
- 45.Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset P, Chan LC, Chan WC, Gaulard P, Mak TW. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119:1901–1903. doi: 10.1182/blood-2011-11-391748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O'Donnell P, Grigoriadis A, Diss T, Eskandarpour M, Presneau N, Hogendoorn PC, Futreal A, Tirabosco R, Flanagan AM. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224:334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 47.Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, Andersen JB, Jiang W, Savich GL, Tan TX, Auman JT, Hoskins JM, Misher AD, Moser CD, Yourstone SM, Kim JW, Cibulskis K, Getz G, Hunt HV, Thorgeirsson SS, Roberts LR, Ye D, Guan KL, Xiong Y, Qin LX, Chiang DY. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32:3091–3100. doi: 10.1038/onc.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, Zucknick M, Hielscher T, Liu XY, Fontebasso AM, Ryzhova M, Albrecht S, Jacob K, Wolter M, Ebinger M, Schuhmann MU, van Meter T, Fruhwald MC, Hauch H, Pekrun A, Radlwimmer B, Niehues T, von Komorowski G, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 50.Im AP, Sehgal AR, Carroll MP, Smith BD, Tefferi A, Johnson DE, Boyiadzis M: DNMT3AandIDHmutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies.Leukemia 2014. doi:10.1038/leu.2014.124. [DOI] [PMC free article] [PubMed]
- 51.The Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, Radlwimmer B, Hojfeldt JW, Truffaux N, Castel D, Schubert S, Ryzhova M, Seker-Cin H, Gronych J, Johann PD, Stark S, Meyer J, Milde T, Schuhmann M, Ebinger M, Monoranu CM, Ponnuswami A, Chen S, Jones C, Witt O, Collins VP, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013;24:660–672. doi: 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 53.Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, Bax DA, Carvalho D, Taylor KR, Vinci M, Bajrami I, McGonnell IM, Lord CJ, Reis RM, Hargrave D, Ashworth A, Workman P, Jones C. Histone H3.3 mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 2013;3:512. doi: 10.1158/2159-8290.CD-12-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7:299–313. doi: 10.1016/j.stem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gonzalez-Perez A, Jene-Sanz A, Lopez-Bigas N. The mutational landscape of chromatin regulatory factors across 4,623 tumor samples. Genome Biol. 2013;14:r106. doi: 10.1186/gb-2013-14-9-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teschendorff AE, Jones A, Fiegl H, Sargent A, Zhuang JJ, Kitchener HC, Widschwendter M. Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med. 2012;4:24. doi: 10.1186/gm323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kostareli E, Holzinger D, Bogatyrova O, Hielscher T, Wichmann G, Keck M, Lahrmann B, Grabe N, Flechtenmacher C, Schmidt CR, Seiwert T, Dyckhoff G, Dietz A, Hofler D, Pawlita M, Benner A, Bosch FX, Plinkert P, Plass C, Weichenhan D, Hess J. HPV-related methylation signature predicts survival in oropharyngeal squamous cell carcinomas. J Clin Invest. 2013;123:2488–2501. doi: 10.1172/JCI67010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim JH, Karnovsky A, Mahavisno V, Weymouth T, Pande M, Dolinoy DC, Rozek LS, Sartor MA. LRpath analysis reveals common pathways dysregulated via DNA methylation across cancer types. BMC Genomics. 2012;13:526. doi: 10.1186/1471-2164-13-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kalari S, Pfeifer GP. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv Genet. 2010;70:277–308. doi: 10.1016/B978-0-12-380866-0.60010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhuang J, Jones A, Lee SH, Ng E, Fiegl H, Zikan M, Cibula D, Sargent A, Salvesen HB, Jacobs IJ, Kitchener HC, Teschendorff AE, Widschwendter M. The dynamics and prognostic potential of DNA methylation changes at stem cell gene loci in women's cancer. PLoS Genet. 2012;8:e1002517. doi: 10.1371/journal.pgen.1002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sproul D, Kitchen RR, Nestor CE, Dixon JM, Sims AH, Harrison DJ, Ramsahoye BH, Meehan RR. Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns. Genome Biol. 2012;13:R84. doi: 10.1186/gb-2012-13-10-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guilhamon P, Eskandarpour M, Halai D, Wilson GA, Feber A, Teschendorff AE, Gomez V, Hergovich A, Tirabosco R, Fernanda Amary M, Baumhoer D, Jundt G, Ross MT, Flanagan AM, Beck S. Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat Commun. 2013;4:2166. doi: 10.1038/ncomms3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anjum S, Fourkala EO, Zikan M, Wong A, Gentry-Maharaj A, Jones A, Hardy R, Cibula D, Kuh D, Jacobs IJ, Teschendorff AE, Menon U, Widschwendter M. A BRCA1-mutation associated DNA methylation signature in blood cells predicts sporadic breast cancer incidence and survival. Genome Med. 2014;6:47. doi: 10.1186/gm567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, Issa JP. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20:332–340. doi: 10.1101/gr.096826.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, Savage DA, Mueller-Holzner E, Marth C, Kocjan G, Gayther SA, Jones A, Beck S, Wagner W, Laird PW, Jacobs IJ, Widschwendter M. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–446. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Michels KB, Binder AM, Dedeurwaerder S, Epstein CB, Greally JM, Gut I, Houseman EA, Izzi B, Kelsey KT, Meissner A, Milosavljevic A, Siegmund KD, Bock C, Irizarry RA. Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods. 2013;10:949–955. doi: 10.1038/nmeth.2632. [DOI] [PubMed] [Google Scholar]
- 69.Esteller M, Corn PG, Urena JM, Gabrielson E, Baylin SB, Herman JG. Inactivation of glutathione S-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res. 1998;58:4515–4518. [PubMed] [Google Scholar]
- 70.Goessl C, Krause H, Muller M, Heicappell R, Schrader M, Sachsinger J, Miller K. Fluorescent methylation-specific polymerase chain reaction for DNA-based detection of prostate cancer in bodily fluids. Cancer Res. 2000;60:5941–5945. [PubMed] [Google Scholar]
- 71.Rogers CG, Gonzalgo ML, Yan G, Bastian PJ, Chan DY, Nelson WG, Pavlovich CP. High concordance of gene methylation in post-digital rectal examination and post-biopsy urine samples for prostate cancer detection. J Urol. 2006;176:2280–2284. doi: 10.1016/j.juro.2006.07.047. [DOI] [PubMed] [Google Scholar]
- 72.Hoque MO, Topaloglu O, Begum S, Henrique R, Rosenbaum E, Van Criekinge W, Westra WH, Sidransky D. Quantitative methylation-specific polymerase chain reaction gene patterns in urine sediment distinguish prostate cancer patients from control subjects. J Clin Oncol. 2005;23:6569–6575. doi: 10.1200/JCO.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 73.Sunami E, Shinozaki M, Higano CS, Wollman R, Dorff TB, Tucker SJ, Martinez SR, Mizuno R, Singer FR, Hoon DS. Multimarker circulating DNA assay for assessing blood of prostate cancer patients. Clin Chem. 2009;55:559–567. doi: 10.1373/clinchem.2008.108498. [DOI] [PubMed] [Google Scholar]
- 74.Palmisano WA, Divine KK, Saccomanno G, Gilliland FD, Baylin SB, Herman JG, Belinsky SA. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res. 2000;60:5954–5958. [PubMed] [Google Scholar]
- 75.Schmidt B, Liebenberg V, Dietrich D, Schlegel T, Kneip C, Seegebarth A, Flemming N, Seemann S, Distler J, Lewin J, Tetzner R, Weickmann S, Wille U, Liloglou T, Raji O, Walshaw M, Fleischhacker M, Witt C, Field JK. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer. 2010;10:600. doi: 10.1186/1471-2407-10-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kneip C, Schmidt B, Seegebarth A, Weickmann S, Fleischhacker M, Liebenberg V, Field JK, Dietrich D. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. J Thorac Oncol. 2011;6:1632–1638. doi: 10.1097/JTO.0b013e318220ef9a. [DOI] [PubMed] [Google Scholar]
- 77.Warren JD, Xiong W, Bunker AM, Vaughn CP, Furtado LV, Roberts WL, Fang JC, Samowitz WS, Heichman KA. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011;9:133. doi: 10.1186/1741-7015-9-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee BB, Lee EJ, Jung EH, Chun HK, Chang DK, Song SY, Park J, Kim DH. Aberrant methylation of APC, MGMT, RASSF2A, and Wif-1 genes in plasma as a biomarker for early detection of colorectal cancer. Clin Cancer Res. 2009;15:6185–6191. doi: 10.1158/1078-0432.CCR-09-0111. [DOI] [PubMed] [Google Scholar]
- 79.Lange CP, Campan M, Hinoue T, Schmitz RF, van der Meulen-de Jong AE, Slingerland H, Kok PJ, van Dijk CM, Weisenberger DJ, Shen H, Tollenaar RA, Laird PW. Genome-scale discovery of DNA-methylation biomarkers for blood-based detection of colorectal cancer. PLoS ONE. 2012;7:e50266. doi: 10.1371/journal.pone.0050266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Claus R, Lucas DM, Stilgenbauer S, Ruppert AS, Yu L, Zucknick M, Mertens D, Buhler A, Oakes CC, Larson RA, Kay NE, Jelinek DF, Kipps TJ, Rassenti LZ, Gribben JG, Dohner H, Heerema NA, Marcucci G, Plass C, Byrd JC. Quantitative DNA methylation analysis identifies a single CpG dinucleotide important for ZAP-70 expression and predictive of prognosis in chronic lymphocytic leukemia. J Clin Oncol. 2012;30:2483–2491. doi: 10.1200/JCO.2011.39.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lecomte T, Berger A, Zinzindohoue F, Micard S, Landi B, Blons H, Beaune P, Cugnenc PH, Laurent-Puig P. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int J Cancer. 2002;100:542–548. doi: 10.1002/ijc.10526. [DOI] [PubMed] [Google Scholar]
- 82.Muller HM, Widschwendter A, Fiegl H, Ivarsson L, Goebel G, Perkmann E, Marth C, Widschwendter M. DNA methylation in serum of breast cancer patients: an independent prognostic marker. Cancer Res. 2003;63:7641–7645. [PubMed] [Google Scholar]
- 83.Tang X, Khuri FR, Lee JJ, Kemp BL, Liu D, Hong WK, Mao L. Hypermethylation of the death-associated protein (DAP) kinase promoter and aggressiveness in stage I non-small-cell lung cancer. J Natl Cancer Inst. 2000;92:1511–1516. doi: 10.1093/jnci/92.18.1511. [DOI] [PubMed] [Google Scholar]
- 84.Sanchez-Cespedes M, Esteller M, Wu L, Nawroz-Danish H, Yoo GH, Koch WM, Jen J, Herman JG, Sidransky D. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res. 2000;60:892–895. [PubMed] [Google Scholar]
- 85.Brock MV, Hooker CM, Ota-Machida E, Han Y, Guo M, Ames S, Glockner S, Piantadosi S, Gabrielson E, Pridham G, Pelosky K, Belinsky SA, Yang SC, Baylin SB, Herman JG. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med. 2008;358:1118–1128. doi: 10.1056/NEJMoa0706550. [DOI] [PubMed] [Google Scholar]
- 86.Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, Campagne F, Mazumdar M, Greally JM, Valk PJ, Lowenberg B, Delwel R, Melnick A. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marcucci G, Yan P, Maharry K, Frankhouser D, Nicolet D, Metzeler KH, Kohlschmidt J, Mrozek K, Wu YZ, Bucci D, Curfman JP, Whitman SP, Eisfeld AK, Mendler JH, Schwind S, Becker H, Bar C, Carroll AJ, Baer MR, Wetzler M, Carter TH, Powell BL, Kolitz JE, Byrd JC, Plass C, Garzon R, Caligiuri MA, Stone RM, Volinia S, Bundschuh R, et al. Epigenetics meets genetics in acute myeloid leukemia: clinical impact of a novel seven-gene score. J Clin Oncol. 2014;32:548–556. doi: 10.1200/JCO.2013.50.6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Milani L, Lundmark A, Kiialainen A, Nordlund J, Flaegstad T, Forestier E, Heyman M, Jonmundsson G, Kanerva J, Schmiegelow K, Soderhall S, Gustafsson MG, Lonnerholm G, Syvanen AC. DNA methylation for subtype classification and prediction of treatment outcome in patients with childhood acute lymphoblastic leukemia. Blood. 2010;115:1214–1225. doi: 10.1182/blood-2009-04-214668. [DOI] [PubMed] [Google Scholar]
- 89.Sandoval J, Mendez-Gonzalez J, Nadal E, Chen G, Carmona FJ, Sayols S, Moran S, Heyn H, Vizoso M, Gomez A, Sanchez-Cespedes M, Assenov Y, Muller F, Bock C, Taron M, Mora J, Muscarella LA, Liloglou T, Davies M, Pollan M, Pajares MJ, Torre W, Montuenga LM, Brambilla E, Field JK, Roz L, Lo Iacono M, Scagliotti GV, Rosell R, Beer DG, et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J Clin Oncol. 2013;31:4140–4147. doi: 10.1200/JCO.2012.48.5516. [DOI] [PubMed] [Google Scholar]
- 90.Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350–1354. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 91.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 92.Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, Juul N, Leong CO, Calogrias D, Buraimoh A, Fatima A, Gelman RS, Ryan PD, Tung NM, De Nicolo A, Ganesan S, Miron A, Colin C, Sgroi DC, Ellisen LW, Winer EP, Garber JE. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J Clin Oncol. 2010;28:1145–1153. doi: 10.1200/JCO.2009.22.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Veeck J, Ropero S, Setien F, Gonzalez-Suarez E, Osorio A, Benitez J, Herman JG, Esteller M. BRCA1 CpG island hypermethylation predicts sensitivity to poly(adenosine diphosphate)-ribose polymerase inhibitors. J Clin Oncol. 2010;28:e563–e564. doi: 10.1200/JCO.2010.30.1010. [DOI] [PubMed] [Google Scholar]
- 94.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, A'Hern R, Tutt A, Ashworth A, Stone J, Carmichael J, Schellens JH, de Bono JS, Kaye SB. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28:2512–2519. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 95.Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours - lessons from the past. Nat Rev Clin Oncol. 2013;10:256–266. doi: 10.1038/nrclinonc.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chung CW, Coste H, White JH, Mirguet O, Wilde J, Gosmini RL, Delves C, Magny SM, Woodward R, Hughes SA, Boursier EV, Flynn H, Bouillot AM, Bamborough P, Brusq JM, Gellibert FJ, Jones EJ, Riou AM, Homes P, Martin SL, Uings IJ, Toum J, Clement CA, Boullay AB, Grimley RL, Blandel FM, Prinjha RK, Lee K, Kirilovsky J, Nicodeme E. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J Med Chem. 2011;54:3827–3838. doi: 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- 97.Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, Bayar E, Lyons J, Rosenfeld CS, Cortes J, Kantarjian HM. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2'-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103:1635–1640. doi: 10.1182/blood-2003-03-0687. [DOI] [PubMed] [Google Scholar]
- 98.Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, Stone RM, Nelson D, Powell BL, DeCastro CM, Ellerton J, Larson RA, Schiffer CA, Holland JF. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–2440. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 99.Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, Lee B, Tsai S, Delgado IE, Rudek MA, Belinsky SA, Herman JG, Baylin SB, Brock MV, Rudin CM. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1:598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang Q, Gu L, Adey A, Radlwimmer B, Wang W, Hovestadt V, Bahr M, Wolf S, Shendure J, Eils R, Plass C, Weichenhan D. Tagmentation-based whole-genome bisulfite sequencing. Nat Protoc. 2013;8:2022–2032. doi: 10.1038/nprot.2013.118. [DOI] [PubMed] [Google Scholar]
- 101.Stirzaker C, Taberlay PC, Statham AL, Clark SJ. Mining cancer methylomes: prospects and challenges. Trends Genet. 2014;30:75–84. doi: 10.1016/j.tig.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 102.Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, Andrews SR, Stegle O, Reik W, Kelsey G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods. 2014;11:817–820. doi: 10.1038/nmeth.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.The International Cancer Genome Consortium International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.International Cancer Genome Consortium. In [http://ICGC.org/]
- 105.The Cancer Genome Atlas. In [http://cancergenome.nih.gov]
- 106.Cancer Genomics Hub. In [http://cghub.ucsc.edu/]
- 107.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, Socci ND, Heguy A, Cherry A, Vance G, Higgins RR, Ketterling RP, Gallagher RE, Litzow M, van den Brink MR, Lazarus HM, Rowe JM, Luger S, Ferrando A, Paietta E, Tallman MS, Melnick A, Abdel-Wahab O, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Issa JP. Colon cancer: it's CIN or CIMP. Clin Cancer Res. 2008;14:5939–5940. doi: 10.1158/1078-0432.CCR-08-1596. [DOI] [PubMed] [Google Scholar]
- 109.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jager N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Fruhwald MC, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 110.Bastian PJ, Ellinger J, Wellmann A, Wernert N, Heukamp LC, Muller SC, von Ruecker A. Diagnostic and prognostic information in prostate cancer with the help of a small set of hypermethylated gene loci. Clin Cancer Res. 2005;11:4097–4106. doi: 10.1158/1078-0432.CCR-04-1832. [DOI] [PubMed] [Google Scholar]
- 111.Jeronimo C, Monteiro P, Henrique R, Dinis-Ribeiro M, Costa I, Costa VL, Filipe L, Carvalho AL, Hoque MO, Pais I, Leal C, Teixeira MR, Sidransky D. Quantitative hypermethylation of a small panel of genes augments the diagnostic accuracy in fine-needle aspirate washings of breast lesions. Breast Cancer Res Treat. 2008;109:27–34. doi: 10.1007/s10549-007-9620-x. [DOI] [PubMed] [Google Scholar]
- 112.Reinert T, Modin C, Castano FM, Lamy P, Wojdacz TK, Hansen LL, Wiuf C, Borre M, Dyrskjot L, Orntoft TF. Comprehensive genome methylation analysis in bladder cancer: identification and validation of novel methylated genes and application of these as urinary tumor markers. Clin Cancer Res. 2011;17:5582–5592. doi: 10.1158/1078-0432.CCR-10-2659. [DOI] [PubMed] [Google Scholar]
- 113.Stone A, Cowley MJ, Valdes-Mora F, McCloy RA, Sergio CM, Gallego-Ortega D, Caldon CE, Ormandy CJ, Biankin AV, Gee JM, Nicholson RI, Print CG, Clark SJ, Musgrove EA. BCL-2 hypermethylation is a potential biomarker of sensitivity to antimitotic chemotherapy in endocrine-resistant breast cancer. Mol Cancer Ther. 2013;12:1874–1885. doi: 10.1158/1535-7163.MCT-13-0012. [DOI] [PubMed] [Google Scholar]
- 114.Maier S, Nimmrich I, Koenig T, Eppenberger-Castori S, Bohlmann I, Paradiso A, Spyratos F, Thomssen C, Mueller V, Nahrig J, Schittulli F, Kates R, Lesche R, Schwope I, Kluth A, Marx A, Martens JW, Foekens JA, Schmitt M, Harbeck N. DNA-methylation of the homeodomain transcription factor PITX2 reliably predicts risk of distant disease recurrence in tamoxifen-treated, node-negative breast cancer patients - technical and clinical validation in a multi-centre setting in collaboration with the European Organisation for Research and Treatment of Cancer (EORTC) PathoBiology group. Eur J Cancer. 2007;43:1679–1686. doi: 10.1016/j.ejca.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 115.Ebert MP, Tanzer M, Balluff B, Burgermeister E, Kretzschmar AK, Hughes DJ, Tetzner R, Lofton-Day C, Rosenberg R, Reinacher-Schick AC, Schulmann K, Tannapfel A, Hofheinz R, Rocken C, Keller G, Langer R, Specht K, Porschen R, Stohlmacher-Williams J, Schuster T, Strobel P, Schmid RM. TFAP2E-DKK4 and chemoresistance in colorectal cancer. N Engl J Med. 2012;366:44–53. doi: 10.1056/NEJMoa1009473. [DOI] [PubMed] [Google Scholar]
- 116.Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, Cordon-Cardo C, Lowe SW. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 2001;409:207–211. doi: 10.1038/35051606. [DOI] [PubMed] [Google Scholar]
- 117.Cortes-Sempere M, Moratilla C, Machado-Pinilla R, Rodriguez-Fanjul V, Manguan-Garcia C, Cejas P, Lopez-Rios F, Paz-Ares L, de CastroCarpeno J, Nistal M, Belda-Iniesta C, Perona R. IGFBP-3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer. Oncogene. 2010;29:1681–1690. doi: 10.1038/onc.2009.454. [DOI] [PubMed] [Google Scholar]
- 118.UCSC Genome Bioinformatics. In [http://genome.ucsc.edu/]
- 119.UCSC Cancer Genomics Browser. In [https://genome-cancer.ucsc.edu/]