

Abstract
The concept of myocyte division and myocyte-mediated regeneration has re-emerged in the past five years through development of sophisticated transgenic mice and carbon-dating of cells. Although, recently, a couple of studies have been conducted as an attempt to intervene in myocyte division, the efficiency in adult animals remains discouragingly low. Re-enforcing myocyte division is a vision that has been desired for decades, leading to years of experience in myocytes resistance to pro-proliferative stimuli. Previous attempts have indeed provided a platform for basic knowledge on molecular players and signaling in myocytes. However, natural biological processes such as hypertrophy and binucleation provide layers of complexity in interpretation of previous and current findings. A major hurdle in mediating myocyte division is a lack of insight in the myocyte cell cycle. To date, no knowledge is gained on myoycte cell cycle progression and/or duration. The current review will provide an overview of previous and current literature on myocytes cell cycle and division. Furthermore, this overview will point-out the limitations of current approaches and focus on re-igniting basic questions that may be essential in understand myocardial resistance to division.
Expansion means complexity and complexity decay… Parkinson’s third Law
The complexity of myocyte cell cycle regulation is manifest. Efforts to sort out conundrums in the concept of myocyte division have suffered from the tendency to extrapolate cell cycle knowledge from other fields to a myocyte context. The term cell cycle refers to occurrence of subsequent events leading to cell duplication and generation of progeny. However, certain fundamental attributes of a cardiac myocyte do not integrate seamlessly with our general comprehension of cell cycle biology such as hypertrophy and physiologic binucleation. In addition, cell cycle terminology has migrated to the field of ‘myocyte cell cycle’ without explicit insight into the substantive meaning of those processes in a cardiac myocyte context. Cumulative experience in myocardial biology has prompted re-examination of previous assumptions, some of which may turn out to be inaccurate with regards to myocyte cell cycle control. Revisiting such long held assumptions based on newly built knowledge will redefine new directions ultimately culminating with a new appreciation for the potential for myocardial regeneration. One such common belief up for challenge today is that “terminal differentiation is an irreversible withdrawal from the myocyte cell cycle”[1–3]. Cell division is not the sole function of myocyte cell cycle, which may also involve biological processes such as hypertrophy [4–7] and DNA-repair [8–12] requiring participation of proteins and processes that are present and active during the cell cycle. The presumption of “terminal differentiation representing an irreversible withdrawal from cell cycle” implies that such cells are incapable of cell division as well as blocked from undergoing general biological processes that require cell cycle activity. Furthermore, references to withdrawal from the cell cycle and cell cycle arrest are often used interchangeably and carelessly [1]. Specifically, withdrawal from cell cycle indicates a G0-arrest caused by nutrient and mitogens deprivation, while non-G0-arrest (G1 and G2) is characterized by high levels of cyclins, CDKs and other growth stimuli that promote cellular growth [13]. Hypertrophic growth has been described as non-G0 arrest [13, 14] that is reversible, but a prolonged state of growth can push a hypertrophic cell into senescence [13, 14]. Senescence is irreversible cell cycle arrest decision point executed by a cell in response to very specific triggers. For example, DNA damage activates the DNA-Damage Response at cell cycle checkpoints intended to execute DNA-repair [15, 16]. If DNA-repair is successful then the check-point arrested cells resume cell cycle progression but if DNA-damage is not sufficiently resolved the cell becomes irreversibly arrested and is now a senescent cell [15–17]. Senescent cells do not contribute to tissue homeostasis and may eventually undergo apoptotic cell death. There are multiple types of cellular senescence; replicative senescence is caused by critical telomere shortening, premature senescence occurs as a response to exposure to reactive oxygen species and DNA-damage and hypermitogenic arrest as a protective mechanism to oncogenic stimuli [16, 17]. Senescent cells are not only irreversibly arrested in the cell cycle but also detrimentally affect the environment via their senescence-associated secretory phenotype (SASP)[16, 17]. SASP is crucial in the context of normal myocytes since neighboring senescent cells adversely impact upon regenerative and reparative potential.
Myocyte division has recently received renewed attention as a candidate for myocardial regeneration, driving the recent spate of studies that are redefining understanding of myocyte cell cycle and revisiting previous definitions. Distinctions between myocyte cell cycle processes, senescence and quiescence will have important consequences for future interventional approaches. Comprehension and interpretation of myocyte cell cycle has been extremely challenging due to technical limitations in the field. Unlike other organs, an adult heart is a difficult platform for myocyte cell cycle studies due to scarcity of the number of proliferating cells. Neonatal hearts have been studied rigorously in their proliferative potential; however, data and mechanisms studied in a young heart do not extrapolate to adult myocytes in any straightforward fashion. Culture of adult myocytes in vitro is possible, but technically challenging and accompanied by a plethora of biological changes prompted by isolation and artificial culture conditions that cannot faithfully recapitulate the in vivo contextual environment. Molecular pathways of myocyte proliferation in neonatal hearts resemble hypertrophic signaling in an adult heart. The overlap in signaling between these two processes and the ultimate phenotypic differences requires expertise in both developmental growth and pathological remodeling to decipher relationships between time-points and specific proteins that interact in these distinct juvenile versus adult processes. In most cell types, measurements of ploidy provides insight into the stage of cell cycle; however, binucleation and multinucleation of myocytes lead to ploidy ranging from 2N to 16N or more. Binucleation or endoreplication on a molecular levels shares certain aspects with cell cycle signaling and intracellular remodeling, thereby necessitating a cautious and circumspect approach to interpretation of myocyte proliferation studies.
One distinct luxury of living in 21st century for many humans is the advances in civilization and associated increases in longevity. Unfortunately, longevity does not necessarily come with more youth but rather extended old age. The worldwide cardiac disease pandemic has prompted attempts to instigate myocardial regeneration via a variety of approaches. From clinical trials using stem cells, generation of tissue engineered patches, gene therapy clinical trials, to reprogramming somatic cells in an effort to intervene in cardiac disease. Unquestionably, an ideal approach would be to manipulate innate myocyte properties for proliferation when possible and promote myocytes to provide a natural replacement mechanism for the scared ventricle with newly formed young competent myocytes. Fortunately, despite inherent technical limitations, the field has gained a considerable amount of knowledge on molecular pathways involved in myocyte cell cycle. This initiates a shift towards the second part of Parkinson’s law: complexity decay wherein elucidation of distinct processes ultimately provides sufficient understanding of the system in order to deconstruct and simplify mysterious fundamentals that governs myocyte division and myocardial regeneration. The ultimate challenge is to assimilate our collective knowledge in order to define caveats and processes that could be used as a mechanistic foundation for future interventions. This review summarizes cell cycle pathways that have been studied in myocytes, the process of terminal cell cycle arrest; senescence and one particularly neglected aspect of cell cycle in the context of myocardial regeneration: quiescence.
Myocyte Cell Cycle
Adult cardiac myocytes possess intricate almost crystalline-like myofibrillar structure required for the demanding job of providing contractile force. During fetal life myocytes undergo cell division that, soon after birth, seems to cease for all practical terms of mediating reparative processes. Although cell division is blocked, myocytes continue to increase in cell size in the form of hypertrophic growth and DNA-content in the form of bi-and multinucleation [18, 19]; two processes that require cell cycle activity but do not lead to cytokinesis. Molecular regulators involved in myocyte cell cycle progression can be subdivided in ‘cytokines and growth factors’, ’transcription factors’ and ‘cell cycle regulators’ and ‘pocket proteins’.
Growth Factors
The role of cardiomyokines is distinct from physiologic cardiac development and cardiac pathologic growth and is therefore beyond the scope of this review and is discussed elsewhere [20–24]. Fibroblast growth factors (FGFs) are proteins of ~150–300 amino acids playing multiple roles in development and metabolism. Neonatal and embryonic cardiomyocyte proliferation is stimulated by FGF in vitro [25] and inhibited by blocking FGF signaling in vivo [26]. Although FGF-2 receptor has been suggested to be cardiac specific, early genetic deletion of FGFR1 and FGFR2 leads to a hypoplastic ventricle and dilated atria implying an important role of the FGF-pathway in myocyte proliferation [25]. Similarly, ligand activation of IGF-1 stimulates myocyte proliferation in culture [27], while IGF-1 inhibition causes a decrease in growth, nuclear mitosis and DNA synthesis [27]. Overexpression of IGF-1 in transgenic mice causes an increase in body weight that is accompanied with a hyperplastic cardiac phenotype, implicating an increased myocyte division rate [28]. Although IGF-1 has been shown to play a primary role in myocyte proliferation rather than hypertrophy, downstream signaling and consequences of IGF-1 overexpression seem to play an important role in the equilibrium of myocyte division versus hypertrophy. A major downstream effector of IGF-1 signaling is Akt, which when overexpressed at non-physiologic levels increases cardiac size by hypertrophy [29]. However, targeting Akt to the nucleus of myocytes that more faithfully recapitulates the normal biological behavior of the kinase leads to a hyperplastic phenotype with an increased number of young myocytes [30]. Since inactivation of Akt during early development does not lead to lethality in mice created with genetic deletion of the kinase, a requisite role for Akt in myocyte division remains unclear. With the nascent advent of stem cell therapy, the role of nuclear targeted Akt could be revisited in the context of endogenous stem cells activation and stem cell mediated myocyte regeneration, thereby causing a hyperplastic phenotype in part via paracrine signaling to enhance cell survival and the growth milieu [31]. IGF-1 has also been associated with antagonism of senescence, increasing telomerase activity and contributing to preservation of cardiac stem cell pool, thereby implying a general beneficial effect of IGF-1 on both myocytes and stem cells [32]. That interaction, by itself, can be beneficial in mediating terminal growth arrest through senescence as discussed later in this review.
Cyclins, Cyclin-Dependent Kinases and Cyclin-Dependent Kinase Inhibitors
Cyclin dependent kinases (CDKs) are serine-threonine kinases, which become enzymatically active upon formation of a complex with the corresponding Cyclin. The regulatory role of Cyclins and CDKs are very well studied since their function has been evolutionary conserved [33]. Cell cycle progression requires accurate orchestration of Cyclins and CDKs at the corresponding checkpoints to assure specific events at very specific phases. The CDKs that are the most crucial at the G1/S-phase transition are CDK4 and CKD6, which bind and regulate the Cyclin D-family [34]. These complexes in turn lead to phosphorylation of Rb-proteins thereby manipulating E2F activity and subsequent gene expression and cell cycle progression [34]. Accelerated phosphorylation of Rb is mediated during the S-phase by the active complex of Cyclin E and CDK2 [35]. Further into the cell cycle at the G2/M transition, the main players are Cyclin E and CDC2[35]. CDK-Activating Kinase (CAK) and Cyclin-Dependent Kinases-Inhibitors (CDKIs) are positive and negative regulators of CDKs, respectively. Not surprisingly, Cyclins and CDKs are present in the embryonic heart in conjunction with other proteins involved in transcription and DNA-replication. Cardiac development has been seems to be affected the most by deletion of the Cyclin-D family. Although genetic deletion of single Cyclin-Ds does not cause a cardiac phenotype, triple deletion is lethal, partially due to cardiac defects [36]. Mutant Cyclin D mice reveal a hypoplastic ventricle and ventricle septal defects. Overexpression of Cyclin-D family members lead to increased DNA-synthesis at baseline in an adult heart [37]. Another finding is the knockout of CDK2 in conjunction with CDK4 that is similarly embryonic lethal due to cardiac defects. CDK2 and CKD4-deletion leads to hypophosphorylation of Rb that in turn affects E2F and downstream E2F targets [34]. Double-mutant mice reveal a hypoplastic ventricle, dilated atria and ventricular wall thinning. Upon cardiac growth and maturation, the expression pattern of these cell cycle regulators is altered. Whether downregulation of these regulators cause the decrease in myocyte proliferation after birth or vice versa remains a mystery. Upon birth, Cyclin D, A, B1 and E and the corresponding kinases are significantly downregulated. Temporal studies reveal that downregulation of cell cycle proteins is accompanied by an upregulation of CDKIs [35, 38, 39]. CDKIs consist of two major protein families that are structurally and functionally different; the INK4 family (p15, p16, p18 and p19) and the Cip/Kip-family (p21, p27, p57). INK4-family CDKIs are selective inhibitors of CDK4 and CDK6 thereby blocking complex formation with Cyclin D and subsequent enzymatic activity. Since, Rb is an essential downstream effector of CDK4/6, INK4-family mediated inhibition of cell cycle progression requires a responsive Rb-protein. The Cik/kip-family of CDKIs, are more effective in inhibition of CDK2, which forms a complex with cyclin E and reinforces progression through the S-phase [40]. In addition, the members of cik/kip-family of CDKIs inhibit cdc2 and Cyclin A activity, thereby playing a broader role in inhibition of cell cycle progression throughout mitosis [40]. In the heart, expression of CDKIs has been studied during development as well as upon pathologic challenge. Although the expression of p16 has been suggested to be low in young adult hearts and increase in aged hearts, p16 and p18 are predominantly present during the embryonic development of the heart [20]. Expression of the Cip/kip family of CDKIs are undetectable during embryonic development, increase in the perinatal phase and peak in adult myocytes. Both, p21 and p27 are downregulated during cardiac injury [20]. Although not very well understood, this could potentiate increased DNA-synthesis that is required to combat the metabolic need during a hypertrophic response.
Transcription Factors
An important transcription factor in cell cycle progression of a variety of cell types is E2F. E2F represents a family of eight members from which some function as transcription initiators and others transcription inhibitors. The main targets for E2F members are Cyclins, DNA-repair genes, checkpoint genes and apoptosis genes [10]. Due to the multitude of family members, studying the effects of specific E2F members is rather flawed, due to their compensatory tendencies. Specific roles of distinct E2F family members in the heart haven’t been provided as yet, and expression patterns of E2F during cardiac development yield confusing and sometimes overtly contradictory results thus far [41]. Although deletion of E2F3 is mostly embryonic lethal and leaves survivors with early congestive failure, knock-out of other members do not cause an evident phenotype, presumably due to the functional redundancy of the family members [42, 43]. Overexpression of E2F1-4 in cultured neonatal rat cardiac myocytes increases the rate of S-phase entry, while E2F1 and 3 induce apoptosis [43]. Overall, an accurate role of E2F family members in cardiac proliferation remains unresolved. Nonetheless, since E2F-family members are key regulators of cell cycle progression, extensive and comprehensive studies regarding the regulation of the G1/S-phase is mandatory in understanding and manipulation of myocyte division. Another rather well studied transcription factor of myocyte proliferation is c-myc [44–49]. The myc family of transcription factors consists of three main members, N-myc, L-myc and c-myc, which regulate transcription by forming a heterodimer with the protein Max. C-myc has a long history of understanding in the oncology field where it is predominantly associated with excessive cellular proliferation. G1 exit is mediated by c-myc by various mechanisms including upregulation of Cdk4, cyclin D1 and D2, Cdc25A, cyclin E, and cyclin A [50]. In addition, it antagonizes the action of at least one Cdk inhibitor, p27[51]. Transgenic mice lacking c-myc expression do not survive post early embryonic stage and c-myc-null mice displayed a general developmental retardation that cannot, necessarily, be attributed to the heart [52]. Overexpression of c-myc mRNA causes a hyperplastic ventricle during early postnatal development [53]. This increase in proliferation does not continue throughout the processes of maturation and aging. In an adult heart, overexpression of this proto-oncogene translates into a hypertrophic growth phenotype [53]. Although there are clues for a role of c-myc in myocyte proliferation the mechanisms for c-myc action in myocytes remain multifaceted and complex. Another, interesting, transcription factor in myocyte division is HIF1alpha. Although HIF1alpha knockout mice developed a hyperplastic phenotype, this hyperplasia caused an obstruction of outract flow and subsequent complications [54, 55]. In addition, hyperplastic growth was not complimented by increased angiogenesis due to a lack of VEGF, which is naturally induced by HIF1alpa [55]. Mechanistically, HIF1 is a known antagonist of c-myc, hereby explaining the overall phenotype of HIF1 knockout mice [56]. Future studies on myocyte transcription factors require rigorous mechanistic hypothesis on whether such manipulations should be aimed at ‘inducing proliferation’ or ‘inhibiting a checkpoint inhibition’.
Pocket Proteins
The pocket protein family consists of three proteins involved in regulation of CDKs in the G1-phase of the cell cycle; Rb, p107 and p130. During development, levels of Rb increase in myocytes, while p130 has the opposite expression pattern. Pocket proteins are well known for regulating E2F-effector genes thereby regulating cell cycle progression at the G1/S-transition [57]. In its active form, Rb is hypophosphorylated that allows binding to E2F and subsequent recruitment of transcription repressors and inhibiting cell cycle progression. Upon phosphorylation, Rb in incapable of binding to E2F, thereby enabling active transcription of genes crucial for cell cycle progression [58]. Major Rb phosphorylating kinases are CDK2 and CDK4. Rb plays an important role in cell cycle exit and myocyte differentiation [59]. Rb-deficient mice are lethal [60], however, animals deficient in Rb and p130 have an increased heart-weight-to-body weight and show enhanced BrdU-incorporation and pH3-staining, indicating persistent myocyte division [61]. Although, the role and pattern of pocket proteins require more elucidation, there are hints towards a role of these proteins in myocyte cell cycle progression.
One significant drawback of molecular studies regarding myocyte cell cycle is the necessity of using transgenic mouse models where cell cycle regulators are constitutively manipulated. The cell cycle is regulated in a tremendous dynamic fashion, where timely degradation of one protein in necessary for functionality of the next regulatory protein. This temporal oscillation is crucial for accurate regulation of cell cycle progression. Although conditional transgenic mice are available for c-myc and Cyclin D, the duration and intensity of cell cycle regulators in these model systems is anything but normal or physiologic so that insight and knowledge derived from such investigations likely only provides a jaded and biased glimpse of the functional properties of these important regulators in normal growth and proliferation. Unfortunately, to date, no knowledge is available on the temporal regulation for “molecular clocks” that time the distinct phases of a myocyte cell cycle. Thus, successful manipulation of cardiomyocyte proliferation versus hypertrophic growth remains rather complex. Recently, studies conducted on myocyte turnover in an adult heart using transgenic mice models [62] and carbon isotope-labeling [63] indicate that myocytes do possess potential for division. These studies were followed by overexpression of miRNA’s [64] and knock-out of meis1[65] in an attempt to further promote myocyte division. Although these initial probing exploratory approaches are acceptable for the current state of affairs, the efficiency of myocyte division remains poor. Ultimately, we still await new techniques and models that not only provide a system to be manipulated but will also provide insight into distinct phases of the cell cycle, the kinetics of molecular signaling in real-time, and the balancing act between stimulatory and inhibitory pathways.
Myocyte Senescence
Cellular senescence is a mechanism of protective irreversible cell cycle arrest in the face of threats stemming from DNA-damage and potential oncogenic risk. Cellular senescence can be induced by genetic and or epigenetic abnormalities. In addition, telomere shortening and/or oxidative stress-mediated metabolic changes can prompt acquisition of a senescent phenotype. A major initiator of senescence is the DNA-damage response (DDR) initiating cell cycle arrest. Persistent DDR activity presents as nuclear foci with high levels of DDR-proteins [16, 17]. Re-initiating or resuming cell cycle progression for a senescent cell is an uphill battle that would need to overcome high expression of p16 and p53, upregulation of pH2AX and increased lysosomal content. Inability of senescent cells to continue cycle progression consequently leads to lack of senescent cell contribution to maintenance of tissue homeostasis. Noteworthy is the fact that senescence is not a developmental process, but rather a degenerative process that predominantly is forced upon myocytes by the environment and demands adaptations that ultimately lead to a twilight existence typified by marginal function that can only end in death. Senescent cells can withdraw from cell cycle and hold up in G0 if cellular damage is detected during G1-phase (e.g telomere erosion). Detection of DNA-damage at a DNA-checkpoint at G2/M (where ATM/ATR is activated) does not lead to cell cycle withdrawal but a permanent cell cycle arrest at the checkpoint [13, 14, 66]. Although these concepts are used interchangeably in the literature, the interpretation of different types of cell cycle disruptions in a senescent cell is crucial for manipulation and reversal of such phenotypes.
Myocyte senescence is associated with mitochondrial dysfunction. Oxidative stress and nutrient deprivation result in excessive amount of reactive oxygen species (ROS) production in the mitochondria as a by-product of oxidative phosphorylation processes. Oxidized proteins can aggregate and interfere in biological function. Simultaneously, ROS attacks mitochondrial membranes and causes additional damage to mitochondrial DNA and oxidizing enzymes that, in turn, contributes to additional ROS production. Concurrently with these changes, mitochondrial biogenesis can be hampered due to lack of energy substrates as well as loss of intact mitochondrial DNA [67, 68]. Mitochondria carry their own telomeres [69], and telomeric attrition within the mitochondrial themselves presumably compromises their capacity for self-biogenesis. Collectively, the accrual of these aforementioned adverse events leaves a rather cytotoxic internal milieu. Mitochondrial preservation as a strategy for staving off aging has been shown by overexpression of mitochondrial catalases as well as deletion of p66shc [70]. Thus, mitochondrial functional impairment serves as a primary inciting stimulus for transition into senescence.
Another major contributor to myocyte senescence is chronic adrenergic signaling. Although a powerful compensatory system in a short run, hyper-activation of the renin-angiotensin-aldosterone signaling axis inflicts damage directly upon myocytes. In particular, angiotensin II has a detrimental effect upon myocytes through downregulation of SERCA2 and diminished calcium homeostasis [71, 72]. Mice with local cardiac overexpression of AngII developed dilated cardiomyopathy and an aged phenotype [73]. Consistent with these findings in experimental models, patients with dilated cardiomyopathy are now regularly treated with angiotensin-converting enzyme-inhibitors to blunt ongoing cardiac remodeling [72].
And finally, as described in multiple other systems, the mTORC pathway is involved in myocyte senescence and inhibition of mTORC1 has been linked to blunting of senescence phenotype acquisition [74] consistent with caloric restriction diets that prolonged lifespan in rodents [75]. Overstimulation of mTORC1 pathway through growth factors promotes cardiac senescence [76]. Typically, inhibition of mTORC1 has been performed pharmacologically with rapamycin, but side-effect toxicity of the drug limits clinical utilization. Alternatively, a molecular interventional approach to selective mTORC1 inhibition and diversion toward mTORC2 has been studied by our group using PRAS40 in the myocardial context, where overexpression of PRAS40 ameliorates hypertrophy and prevents development of diabetic cardiomyopathy in rodents.[77–79]
Any possibility for molecular interventional strategies toward senescence reversal will require understanding the underlying molecular cues responsible for prompting and maintaining the senescent state. Antagonism of replicative senescence caused by telomere attrition is a feasible target for reversal as previously performed in murine and human cardiac stem cells [80–82]. Similarly, metabolic and catecholamine-based senescence could be amenable to manipulation and subsequent rejuvenation [83]. However, a judicious approach is mandatory in reversal of senescence in systems with DNA-damage and cellular senescence due to excessive DNA-damage. Reinforcing cell cycle progression in cells that show evidence of “non-reliable” DNA is probably not worth the risk.
Myocyte Quiescence
Quiescence (Latin: quiescere = to rest) refers to a state of quietness and rest. The role of quiescence is crucial in phylogeny. Exponential growth is historically predicted to inevitably surmount the supplies required for survival of species, as would be the case for organisms and their constituent cells that may be challenged to survive under nutrient deprivation or other compromised environments. Thus, growth arrested state is biologically reasonable if not mandatory for adaptation to stress. Long-standing presumptions asserted that cells become quiescent in response to external stimuli, but recent insights suggest that cells carry an autonomous propensity for quiescence as a mechanism to preserve their fundamental biological characteristics. Quiescent cells reside in the G0 phase of the cell cycle and therefore posses ability to re-enter the cell cycle upon normal physiological stimulation [84, 85]. Thus, although quiescence refers to a state of growth arrest, this type of withdrawal from cycling does not involve a prolongation of the G1-phase. Actually, the G1-phase of cell cycle possesses a restriction point that can serve to determine cell fate. Cells can exit G1 before the checkpoint, in mammals called the “restriction-point” and become quiescent, but passage through restriction-point is considered irrevocable commitment toward replication and division [84, 85]. A unique constellation of a phenotypic signature for quiescent cells is lacking, but is slowly emerging based upon investigation of genetics, epigenetic and transcriptional profiling of the resting state. On a molecular level, a major players of quiescence is the previously mentioned tumor suppressor Rb. Indeed, the quiescent population in hematopoietic stem cells vanishes if all three Rb family proteins are genetically deleted [84–86]. Consistent with a cell cycle arrest phenotype, p21, p27 and p57 are upregulated in quiescent cells. Prevailing thinking posited that quiescent cells ought to maintain expression of key cell cycle regulators in preparation to enter the cell cycle in response to appropriate inductive stimulation. Expression and synthesis of cell cycle regulators requires energy consumption from the cells, thus implies increases metabolic activity. This metabolic demand is contradictory to the main quiescence attribute of low energy consumption. Indeed, quiescent cells regulate cellular processes in a less energy demanding fashion, miRNA-mediated regulation of transcription, in particular the microRNA 16 family [87, 88]. Since miRNA families target multiple genes in common pathways, miRNA’s have been proposed as major regulators of quiescence. Transcriptional regulation of a quiescent phenotype has been extensively studied in multiple types of stem cells [89], revealing a “blueprint” for creating senescence involving down regulation of genes promoting cell cycle progression, DNA-replication and metabolic pathways. Specifically, major cyclins (B1, A2 and E3)[84–86, 90, 91], Survivin [92] and cytochrome c are suppressed in conjunction with up-regulation of genes important for differentiation and fate decision-making (FOXO3 and EZH1)[84–86, 90, 91]. Observing that epigenetic modification influences gene expression, epigenetic profiling of quiescence has begun in earnest. Studies conducted in embryonic stem cells reveal that presence of bivalent domains in the proximity of transcriptional sites are key regulators of gene expression. Two such domains have been shown to be relevant in a quiescent phenotype, namely H3K4me3 and H3K27me3. In muscle stem cells and human fetal stem cells manipulation of a great number of genes are marked by H3K4me3, which indicates active transcription sites in a phenotype with low transcriptional activity [93].
Overall, the black box of quiescence is slowly revealing the mysteries hiding within, offering new hope for comprehensive understanding of myocyte quiescence that will provide a exciting opportunities for manipulation of cell cycle entry and division to enhance myocardial function and regeneration. Unlike senescent cells, quiescent cells provide a tremendous platform for new cell formation, since the cells retain origin and lineage commitment, lack hurdles such as DNA-damage and risks for adverse effects and essentially require a push in the right direction. Unfortunately, quiescence has never been rigorously studied in myocytes. The term “myocyte quiescence” in the literature is based upon a lack of DNA-synthesis or incorporation of BrdU. However, a distinction between myocytes in G1 versus G0 has never been possible due to lack of accurate myocyte models that provide distinction between these phases.
If you can’t explain it easily, then you don’t understand it well enough Albert Einstein
For decades, the heart was considered a postmitotic or recently a mostly postmitotic organ. A postmitotic cell refers to a cell that has completed mitosis, not surrended the capacity for cell cycling activity. Since processes such as hypertrophic growth and DNA-synthesis and repair require cell cycle proteins and machinery, could those features be interpreted as indicative of the myocardium being in an ongoing state of “premitotic” life and the heart as a “premitotic” rather than “postmitotic”? Comprehending the distinction between possibilities of premitosis versus postmitosis are crucial for defining targets to manipulate in order to promote myocyte division.
The ever-elusive goal of myocardial regeneration is closer today than ever and currently enjoying a renaissance with the discovery of new rules for the reparative and regenerative potential of the heart and stem cells. However, ambition and enthusiasm surrounding myocardial regeneration seem to be exacerbating an intellectual disconnect between “myocyte division” and “myocyte cell cycle”. Currently, the field lacks straightforward mechanistic postulates on how the number of dividing myocytes can be increased. If the aim is to force a binucleated myocyte to divide, what exactly is it that we want such a myocyte to do? If the aim is to predominantly focus on mononucleated myocytes, then are we aiming for quiescent mononucleated cells, mononucleated cells that are presumably arrested in G1 or do we want to rejuvenate senescent cells and reinforce division? If a mononucleated myocyte population is in G1, is it due to a checkpoint inhibitory block or a lack of stimulatory regulators? Are these two pathways additive or is one superior to the other? Overall, after all our pretentions to mastery of myocardial biology fall away, we must confess that we can’t easily explain regulation of the myocyte cell cycle just yet. Efficient and successful manipulation of myocyte division requires insight in all phases and processes of the myocyte cell cycle; quiescence, G1, S, G2/M, checkpoints, senescence and perhaps most importantly a temporal assessment of the duration of a myocyte cell cycle. Future studies must be aimed at generating reporter models that provides real-time distinction between the different stages of the cell cycle, duration of these phases in conjunction with ploidy and regulatory pathways.
Figure 1.
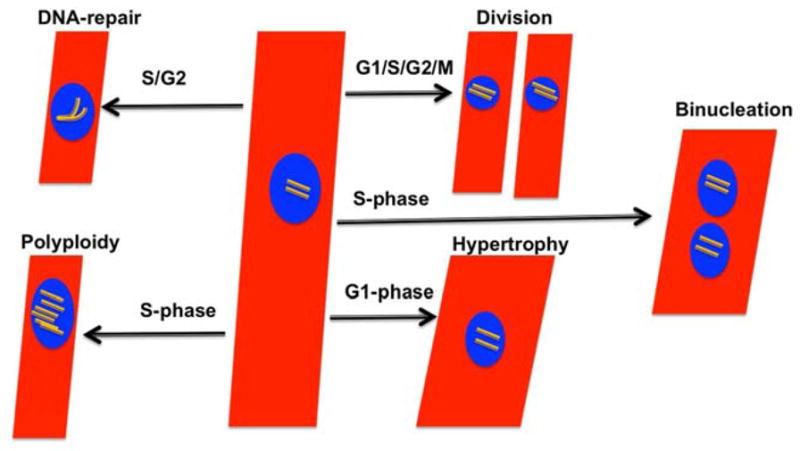
The heart mostly Pre-mitotic or Mostiy Post-mitotic? A schematic demonstration of natural and or pathological processes requiring cell cycle activity in myocytes. In addition to cell division, myocytes require cell cycle activity for binucleation, hypertrophic growth, DNA-synthesis and DNA-repair process.
Table 1.
A summary of previous work on myocyte division & senescence.
| Myocyte Cell Cycle
| ||
|---|---|---|
| Target | Intervention | Phenotype |
| Cyclin D | Mutant mice | Hypertrophic Ventricle Ventricular Septal Defect |
| Overexpression | Increased DNA-synthesis in adult hearts | |
|
| ||
| CDK2 CDK4 |
Knock-out mice | Embryonic Lethal due to cardiac defects |
|
| ||
| E2F3 | Knock-out mice | Congestive cardiac failure |
|
| ||
| C-myc | Knock-out mice | Cardiac developmental retardation |
| Overexpression | Hyperplastic ventricle, in adult mice hypertrophy | |
|
| ||
| HIF1α | Knock-out mice | Hyperplastic heart |
| Myocyte Senescence
| ||
|---|---|---|
| Target | Intervention | Phenotype |
| Mitochondrial Catalases | Overexpression | Youthful cardiac phenotype |
|
| ||
| P66shc | Deletion | Youthful cardiac phenotype |
|
| ||
| Angiotensin II | Local overexpression | Ages hearts Dilated cardiomyopathy |
|
| ||
| mTORC | Inhibition using Rapamycin | Reversal of aging |
| Overstimulation using growth factors | Cardiac senescence | |
| Inhibition by Pras40 | Ameliorated hypertrophy | |
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Crescenzi M, Soddu S, Tato F. Mitotic cycle reactivation in terminally differentiated cells by adenovirus infection. J Cell Physiol. 1995;162(1):26–35. doi: 10.1002/jcp.1041620105. [DOI] [PubMed] [Google Scholar]
- 2.Latella L, et al. Reconstitution of cyclin D1-associated kinase activity drives terminally differentiated cells into the cell cycle. Mol Cell Biol. 2001;21(16):5631–43. doi: 10.1128/MCB.21.16.5631-5643.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacLellan W, SChneider MD. Cell Cycle Reactivation in Cardiac Myocytes. Landes Bioscience. 2003:01/25/2003. Gene Expression. [Google Scholar]
- 4.Li JM, Poolman RA, Brooks G. Role of G1 phase cyclins and cyclin-dependent kinases during cardiomyocyte hypertrophic growth in rats. Am J Physiol. 1998;275(3 Pt 2):H814–22. doi: 10.1152/ajpheart.1998.275.3.H814. [DOI] [PubMed] [Google Scholar]
- 5.Nozato T, et al. G1 cyclins are involved in the mechanism of cardiac myocyte hypertrophy induced by angiotensin II. Jpn Circ J. 2000;64(8):595–601. doi: 10.1253/jcj.64.595. [DOI] [PubMed] [Google Scholar]
- 6.Wolfram JA, et al. The role of E2F1 in the development of hypertrophic cardiomyopathy. Int J Clin Exp Pathol. 4(5):521–5. [PMC free article] [PubMed] [Google Scholar]
- 7.Kirschenbaum LAMD. Regulators of Cardiac Cell Growth, DIfferentiation, and Apoptosis. Heart Failure Reviews. 1997;2(2):117–124. [Google Scholar]
- 8.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9(4):297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 9.Foster SS, et al. Cell cycle- and DNA repair pathway-specific effects of apoptosis on tumor suppression. Proc Natl Acad Sci U S A. 109(25):9953–8. doi: 10.1073/pnas.1120476109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren B, et al. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002;16(2):245–56. doi: 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartz EI, et al. Cell cycle activation in postmitotic neurons is essential for DNA repair. Cell Cycle. 2007;6(3):318–29. doi: 10.4161/cc.6.3.3752. [DOI] [PubMed] [Google Scholar]
- 12.Kozlovskis-Wade PL, Smets MJ, Myerburg RJ. The effect of nicotine on DNA repair in adult myocytes. J Mol Cell Cardiol. 1998;30(8):1483–91. doi: 10.1006/jmcc.1998.0712. [DOI] [PubMed] [Google Scholar]
- 13.Blagosklonny MV. Cell senescence: hypertrophic arrest beyond the restriction point. J Cell Physiol. 2006;209(3):592–7. doi: 10.1002/jcp.20750. [DOI] [PubMed] [Google Scholar]
- 14.Blagosklonny MV. Cell cycle arrest is not senescence. Aging (Albany NY) 3(2):94–101. doi: 10.18632/aging.100281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Velarde MC, Demaria M, Campisi J. Senescent cells and their secretory phenotype as targets for cancer therapy. Interdiscip Top Gerontol. 38:17–27. doi: 10.1159/000343572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campisi J. Cell biology: The beginning of the end. Nature. 505(7481):35–6. doi: 10.1038/nature12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li F, Wang X, Gerdes AM. Formation of binucleated cardiac myocytes in rat heart: II. Cytoskeletal organisation. J Mol Cell Cardiol. 1997;29(6):1553–65. doi: 10.1006/jmcc.1997.0403. [DOI] [PubMed] [Google Scholar]
- 19.Clubb FJ, Jr, Bishop SP. Formation of binucleated myocardial cells in the neonatal rat. An index for growth hypertrophy. Lab Invest. 1984;50(5):571–7. [PubMed] [Google Scholar]
- 20.Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87(2):521–44. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sano M, Schneider MD. Cyclins that don’t cycle--cyclin T/cyclin-dependent kinase-9 determines cardiac muscle cell size. Cell Cycle. 2003;2(2):99–104. [PubMed] [Google Scholar]
- 22.MacLellan WR, Schneider MD. Genetic dissection of cardiac growth control pathways. Annu Rev Physiol. 2000;62:289–319. doi: 10.1146/annurev.physiol.62.1.289. [DOI] [PubMed] [Google Scholar]
- 23.Wagner M, Mascareno E, Siddiqui MA. Cardiac hypertrophy: signal transduction, transcriptional adaptation, and altered growth control. Ann N Y Acad Sci. 1999;874:1–10. doi: 10.1111/j.1749-6632.1999.tb09219.x. [DOI] [PubMed] [Google Scholar]
- 24.Puri PL, et al. The molecular basis of myocardial hypertrophy. Ann Ital Med Int. 1994;9(3):160–5. [PubMed] [Google Scholar]
- 25.deAlmeida A, Sedmera D. Fibroblast Growth Factor-2 regulates proliferation of cardiac myocytes in normal and hypoplastic left ventricles in the developing chick. Cardiol Young. 2009;19(2):159–69. doi: 10.1017/S1047951109003552. [DOI] [PubMed] [Google Scholar]
- 26.Mima T, et al. Fibroblast growth factor receptor is required for in vivo cardiac myocyte proliferation at early embryonic stages of heart development. Proc Natl Acad Sci U S A. 1995;92(2):467–71. doi: 10.1073/pnas.92.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kajstura J, et al. The IGF-1-IGF-1 receptor system modulates myocyte proliferation but not myocyte cellular hypertrophy in vitro. Exp Cell Res. 1994;215(2):273–83. doi: 10.1006/excr.1994.1343. [DOI] [PubMed] [Google Scholar]
- 28.Reiss K, et al. Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proc Natl Acad Sci U S A. 1996;93(16):8630–5. doi: 10.1073/pnas.93.16.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Condorelli G, et al. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci U S A. 2002;99(19):12333–8. doi: 10.1073/pnas.172376399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gude N, et al. Akt promotes increased cardiomyocyte cycling and expansion of the cardiac progenitor cell population. Circ Res. 2006;99(4):381–8. doi: 10.1161/01.RES.0000236754.21499.1c. [DOI] [PubMed] [Google Scholar]
- 31.Shiraishi I, et al. Nuclear targeting of Akt enhances kinase activity and survival of cardiomyocytes. Circ Res. 2004;94(7):884–91. doi: 10.1161/01.RES.0000124394.01180.BE. [DOI] [PubMed] [Google Scholar]
- 32.Torella D, et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. 2004;94(4):514–24. doi: 10.1161/01.RES.0000117306.10142.50. [DOI] [PubMed] [Google Scholar]
- 33.Liu J, Kipreos ET. Evolution of cyclin-dependent kinases (CDKs) and CDK-activating kinases (CAKs): differential conservation of CAKs in yeast and metazoa. Mol Biol Evol. 2000;17(7):1061–74. doi: 10.1093/oxfordjournals.molbev.a026387. [DOI] [PubMed] [Google Scholar]
- 34.Berthet C, Kaldis P. Cdk2 and Cdk4 cooperatively control the expression of Cdc2. Cell Div. 2006;1:10. doi: 10.1186/1747-1028-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woo RA, Poon RY. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle. 2003;2(4):316–24. [PubMed] [Google Scholar]
- 36.Ciemerych MA, et al. Development of mice expressing a single D-type cyclin. Genes Dev. 2002;16(24):3277–89. doi: 10.1101/gad.1023602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diehl JA, Sherr CJ. A dominant-negative cyclin D1 mutant prevents nuclear import of cyclin-dependent kinase 4 (CDK4) and its phosphorylation by CDK-activating kinase. Mol Cell Biol. 1997;17(12):7362–74. doi: 10.1128/mcb.17.12.7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang MJ, et al. Cyclins and cyclin dependent kinases during cardiac development. Mol Cells. 1997;7(3):360–6. [PubMed] [Google Scholar]
- 39.Brooks G, Poolman RA, Li JM. Arresting developments in the cardiac myocyte cell cycle: role of cyclin-dependent kinase inhibitors. Cardiovasc Res. 1998;39(2):301–11. doi: 10.1016/s0008-6363(98)00125-4. [DOI] [PubMed] [Google Scholar]
- 40.Tane S, et al. CDK inhibitors, p21(Cip1) and p27(Kip1), participate in cell cycle exit of mammalian cardiomyocytes. Biochem Biophys Res Commun. 443(3):1105–9. doi: 10.1016/j.bbrc.2013.12.109. [DOI] [PubMed] [Google Scholar]
- 41.Vara D, et al. Inhibition of E2F abrogates the development of cardiac myocyte hypertrophy. J Biol Chem. 2003;278(24):21388–94. doi: 10.1074/jbc.M212612200. [DOI] [PubMed] [Google Scholar]
- 42.Timmers C, et al. E2f1, E2f2, and E2f3 control E2F target expression and cellular proliferation via a p53-dependent negative feedback loop. Mol Cell Biol. 2007;27(1):65–78. doi: 10.1128/MCB.02147-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsai SY, et al. Mouse development with a single E2F activator. Nature. 2008;454(7208):1137–41. doi: 10.1038/nature07066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Falcone G, Tato F, Alema S. Distinctive effects of the viral oncogenes myc, erb, fps, and src on the differentiation program of quail myogenic cells. Proc Natl Acad Sci U S A. 1985;82(2):426–30. doi: 10.1073/pnas.82.2.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Endo T, Nadal-Ginard B. Transcriptional and posttranscriptional control of c-myc during myogenesis: its mRNA remains inducible in differentiated cells and does not suppress the differentiated phenotype. Mol Cell Biol. 1986;6(5):1412–21. doi: 10.1128/mcb.6.5.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kindy MS, Sonenshein GE. Regulation of oncogene expression in cultured aortic smooth muscle cells. Post-transcriptional control of c-myc mRNA. J Biol Chem. 1986;261(27):12865–8. [PubMed] [Google Scholar]
- 47.Claycomb WC, Lanson NA., Jr Proto-oncogene expression in proliferating and differentiating cardiac and skeletal muscle. Biochem J. 1987;247(3):701–6. doi: 10.1042/bj2470701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saule S, et al. Heart tumors specifically induced in young avian embryos by the v-myc oncogene. Proc Natl Acad Sci U S A. 1987;84(22):7982–6. doi: 10.1073/pnas.84.22.7982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schneider MD, Olson EN. Control of myogenic differentiation by cellular oncogenes. Mol Neurobiol. 1988;2(1):1–39. doi: 10.1007/BF02935631. [DOI] [PubMed] [Google Scholar]
- 50.Dang CV. MYC on the path to cancer. Cell. 149(1):22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gomez-Casares MT, et al. MYC antagonizes the differentiation induced by imatinib in chronic myeloid leukemia cells through downregulation of p27(KIP1.) Oncogene. 32(17):2239–46. doi: 10.1038/onc.2012.246. [DOI] [PubMed] [Google Scholar]
- 52.Davis AC, et al. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993;7(4):671–82. doi: 10.1101/gad.7.4.671. [DOI] [PubMed] [Google Scholar]
- 53.Green NK, et al. Transfection of cardiac muscle: effects of overexpression of c-myc and c-fos proto-oncogene proteins in primary cultures of neonatal rat cardiac myocytes. Clin Sci (Lond) 1997;92(2):181–8. doi: 10.1042/cs0920181. [DOI] [PubMed] [Google Scholar]
- 54.Kotch LE, et al. Defective vascularization of HIF-1alpha-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev Biol. 1999;209(2):254–67. doi: 10.1006/dbio.1999.9253. [DOI] [PubMed] [Google Scholar]
- 55.Huang Y, et al. Cardiac myocyte-specific HIF-1alpha deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. Faseb J. 2004;18(10):1138–40. doi: 10.1096/fj.04-1510fje. [DOI] [PubMed] [Google Scholar]
- 56.Koshiji M, et al. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. Embo J. 2004;23(9):1949–56. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24(17):2796–809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 58.Ikeda MA, Jakoi L, Nevins JR. A unique role for the Rb protein in controlling E2F accumulation during cell growth and differentiation. Proc Natl Acad Sci U S A. 1996;93(8):3215–20. doi: 10.1073/pnas.93.8.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feliers D, Frank MA, Riley DJ. Activation of cyclin D1-Cdk4 and Cdk4- directed phosphorylation of RB protein in diabetic mesangial hypertrophy. Diabetes. 2002;51(11):3290–9. doi: 10.2337/diabetes.51.11.3290. [DOI] [PubMed] [Google Scholar]
- 60.Taneja P, et al. Transgenic and knockout mice models to reveal the functions of tumor suppressor genes. Clin Med Insights Oncol. 5:235–57. doi: 10.4137/CMO.S7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sdek P, et al. Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes. J Cell Biol. 194(3):407–23. doi: 10.1083/jcb.201012049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Senyo SE, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 493(7432):433–6. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kajstura J, et al. Myocyte turnover in the aging human heart. Circ Res. 107(11):1374–86. doi: 10.1161/CIRCRESAHA.110.231498. [DOI] [PubMed] [Google Scholar]
- 64.Eulalio A, et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 492(7429):376–81. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 65.Mahmoud AI, et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 497(7448):249–53. doi: 10.1038/nature12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 192(4):547–56. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc Med. 2009;19(7):213–20. doi: 10.1016/j.tcm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 69.Nosek J, et al. Mitochondrial telomeres as molecular markers for identification of the opportunistic yeast pathogen Candida parapsilosis. J Clin Microbiol. 2002;40(4):1283–9. doi: 10.1128/JCM.40.4.1283-1289.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gertz M, Steegborn C. The mitochondrial apoptosis pathway and p66Shc--a regulatory redox enzyme or an adapter protein snuggling around? Cell Cycle. 9(22):4425–6. doi: 10.4161/cc.9.22.14053. [DOI] [PubMed] [Google Scholar]
- 71.Zhou YY, et al. Constitutive beta2-adrenergic signalling enhances sarcoplasmic reticulum Ca2+ cycling to augment contraction in mouse heart. J Physiol. 1999;521(Pt 2):351–61. doi: 10.1111/j.1469-7793.1999.00351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Edwards A, Pallone TL. Mechanisms underlying angiotensin II-induced calcium oscillations. Am J Physiol Renal Physiol. 2008;295(2):F568–84. doi: 10.1152/ajprenal.00107.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heymes C, et al. Cardiac senescence is associated with enhanced expression of angiotensin II receptor subtypes. Endocrinology. 1998;139(5):2579–87. doi: 10.1210/endo.139.5.6023. [DOI] [PubMed] [Google Scholar]
- 74.Flynn JM, et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. 12(5):851–62. doi: 10.1111/acel.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wolf G. Calorie restriction increases life span: a molecular mechanism. Nutr Rev. 2006;64(2 Pt 1):89–92. doi: 10.1301/nr.2006.feb.89-92. [DOI] [PubMed] [Google Scholar]
- 76.Melnik BC, John SM, Schmitz G. Over-stimulation of insulin/IGF-1 signaling by western diet may promote diseases of civilization: lessons learnt from laron syndrome. Nutr Metab (Lond) 8:41. doi: 10.1186/1743-7075-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Volkers M, et al. PRAS40 prevents development of diabetic cardiomyopathy and improves hepatic insulin sensitivity in obesity. EMBO Mol Med. 6(1):57–65. doi: 10.1002/emmm.201303183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Volkers M, Sussman M. mTOR/PRAS40 interaction: hypertrophy or proliferation. Cell Cycle. 12(23):3579–80. doi: 10.4161/cc.26822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Volkers M, et al. Pathological hypertrophy amelioration by PRAS40-mediated inhibition of mTORC1. Proc Natl Acad Sci U S A. 110(31):12661–6. doi: 10.1073/pnas.1301455110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mohsin S, et al. Rejuvenation of human cardiac progenitor cells with Pim-1 kinase. Circ Res. 113(10):1169–79. doi: 10.1161/CIRCRESAHA.113.302302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mohsin S, et al. Human cardiac progenitor cells engineered with Pim-I kinase enhance myocardial repair. J Am Coll Cardiol. 60(14):1278–87. doi: 10.1016/j.jacc.2012.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fischer KM, et al. Enhancement of myocardial regeneration through genetic engineering of cardiac progenitor cells expressing Pim-1 kinase. Circulation. 2009;120(21):2077–87. doi: 10.1161/CIRCULATIONAHA.109.884403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Khan M, et al. Cardiac Progenitor Cells Engineered With betaARKct Have Enhanced beta-Adrenergic Tolerance. Mol Ther. 22(1):178–85. doi: 10.1038/mt.2013.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wells A, et al. The dormancy dilemma: quiescence versus balanced proliferation. Cancer Res. 73(13):3811–6. doi: 10.1158/0008-5472.CAN-13-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. 14(6):329–40. doi: 10.1038/nrm3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Quadrato G, Di Giovanni S. Gatekeeper between quiescence and differentiation: p53 in axonal outgrowth and neurogenesis. Int Rev Neurobiol. 105:71–89. doi: 10.1016/B978-0-12-398309-1.00005-6. [DOI] [PubMed] [Google Scholar]
- 87.Truesdell SS, et al. MicroRNA-mediated mRNA translation activation in quiescent cells and oocytes involves recruitment of a nuclear microRNP. Sci Rep. 2:842. doi: 10.1038/srep00842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guo CJ, et al. Effects of upregulated expression of microRNA-16 on biological properties of culture-activated hepatic stellate cells. Apoptosis. 2009;14(11):1331–40. doi: 10.1007/s10495-009-0401-3. [DOI] [PubMed] [Google Scholar]
- 89.Subramaniam S, et al. Distinct transcriptional networks in quiescent myoblasts: a role for Wnt signaling in reversible vs. irreversible arrest. PLoS One. 8(6):e65097. doi: 10.1371/journal.pone.0065097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Oh IH, Humphries RK. Concise review: Multidimensional regulation of the hematopoietic stem cell state. Stem Cells. 30(1):82–8. doi: 10.1002/stem.776. [DOI] [PubMed] [Google Scholar]
- 91.Li L, Bhatia R. Stem cell quiescence. Clin Cancer Res. 17(15):4936–41. doi: 10.1158/1078-0432.CCR-10-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fukuda S, Pelus LM. Elevation of Survivin levels by hematopoietic growth factors occurs in quiescent CD34+ hematopoietic stem and progenitor cells before cell cycle entry. Cell Cycle. 2002;1(5):322–6. [PubMed] [Google Scholar]
- 93.Srivastava S, Mishra RK, Dhawan J. Regulation of cellular chromatin state: insights from quiescence and differentiation. Organogenesis. 6(1):37–47. doi: 10.4161/org.6.1.11337. [DOI] [PMC free article] [PubMed] [Google Scholar]