

Abstract
There is a growing interest on glycol-split low-molecular weight heparins (gs-LMWHs), obtained by periodate oxidation of LMWHs, optionally followed by borohydride reduction, as potential anticancer and anti-inflammatory drugs. However, their structural characterization is still a challenging task, mainly because of the high microheterogeneity of the starting material. In addition, susceptibility to oxidation of some end-groups of LMWHs induces additional heterogeneity, making analysis of gs-LMWHs more complex. In our previous study we showed that 1,6-anhydro-D-mannosamine N-sulfate was affected by periodate, while its epimer 1,6-anhydro-D-glucosamine N-sulfate was resistant. In order to understand the apparently anomalous behavior of terminal 1,6-anhydro-D-mannosamine N-sulfate residues, in the present work we have studied by NMR spectroscopy and LC/MS the behavior of the reducing end amino sugar residues of the tetrasaccharides, isolated from the LMWH enoxaparin, in the presence of periodate. Their molecular mechanics conformational characterisation has been also performed. We have shown that the C(2)–C(3) bond of the 1,6-anhydro-D-mannosamine residue can be split by periodate despite the N-substitution. Moreover, we have found that both terminal D-mannosamine N-sulfate and D-glucosamine N-sulfate, lacking the 1,6-anhydro-bridge, can be also oxidized by periodate but with significantly lower rate. The present results suggest that the cis-e-/a-position of OH and NHSO3− groups of N-sulfated 1,6-anhydro-D-mannosamine is not the only factor that makes these end residues susceptible to the oxidation. The 1,6-anhydro-bridge that “blocks” the ring conformation appears another crucial factor for oxidation to occur. Moreover, we have shown that controlling the reaction time could permit to selectively split non-sulfated iduronic acids of enoxaparin chains without oxidizing terminal amino sugar residues, a finding that may be useful to obtain more structurally homogeneous gs-LMHWs.
Keywords: Enoxaparin; Low-molecular weight heparins; Glycol-split heparins; Periodate oxidation; 1, 6-Anhydro-D-mannosamine
1. Introduction
Heparin belongs to the glycosaminoglycan family and it is constituted by repeating disaccharide building blocks containing 1,4-linked uronic acid (L-iduronic or D-glucuronic) and D-glucosamine. Regular “fully sulfated” regions, containing 2-O-sulfated iduronic acid (I2S) and N-sulfate-glucosamine-6-O-sulfate (ANS6S) are prevalent within heparin chains, but are interspersed by undersulfated sequences bearing non-sulfated iduronic (I) or glucuronic (G) acids and N-acetylated glucosamine residues (ANAc). The specific pentasaccharide sequence N-acetyl-D-glucosamine-6-O-sulfate-α-(1→4)-D-glucuronic acid-β-(1→4)-D-glucosamine N,3-O,6-O-trisulfate-α(1→4)-L-iduronic acid 2-O-sulfate-α(1→4)-D-glucosamine N,6-O-disulfate (ANAc6S-G-ANS3S6S-I2S-ANS6S), present only in some of the chains, constitutes the antithrombin-binding region (ATBR), essential for a high anticoagulant and antithrombotic activity of heparins.1,2
Glycol-split (gs) derivatives of heparins with potential application in anti-cancer and anti-inflammation therapies3,4 are usually obtained by oxidation of vicinal hydroxyl groups of non-sulfated uronic acid residues followed by a reduction step in order to stabilize the generated aldehydes.5 The new medical uses of gs-heparins are favored by the loss of anticoagulant activity associated with glycol-splitting of the D-glucuronic acid residue within the ATBR sequence, which is essential for strong binding to, and activation of, antithrombin (AT).6,7 Other biological activities of heparins, which are not critically dependent on the intact AT-binding sequence, usually are not significantly impaired by glycol-splitting. Moreover, given the increased local mobility induced by gs residues, the flexibility of gs-heparin chains is higher than for unmodified heparins, facilitating in some cases their interactions with proteins.8,9 Since low-molecular weight heparins (LMWHs) are considered to be pharmacologically more attractive than unfractionated heparins, due to their better bioavailability, also several gs-LMWHs are currently being developed.10,11
In spite of such a growing interest on gs-LMWHs, the structural characterization of these non-anticoagulant experimental drugs is still a challenging task, firstly, because of the high microheterogeneity of the starting material (varying also for different origins and manufacture processes). When LMWHs are used as starting materials, the end-groups generated during the depolymerisation processes induce additional heterogeneity, making the analysis even more complex.12
In our previous work we found that not only the internal non-sulfated uronic acids, but also several terminal residues characteristic for LMWHs are susceptible to periodate oxidation.12 The most interesting and unexpected behavior was observed for terminal enoxaparin residues. Enoxaparin oligosaccharides bear various amino sugars at their reducing end (RE): glucosamine N-sulfate (ANS(6S), ~9% of total amino sugar content13) and unnatural residues formed during depolymerisation of heparin under alkaline conditions14, namely mannosamine N-sulfate (MNS(6S)), 1,6-anhydro-glucosamine N-sulfate (1,6aANS) and 1,6-anhydro-mannosamine N-sulfate (1,6aMNS), about 3, 2 and 2.5% of total amino sugar content13). It was shown that unlike for 1,6aANS residues, the NMR cross peaks of its epimer 1,6aMNS13,15 dissappeared in the HSQC spectrum of enoxaparin upon glycol-splitting.12 Interestingly, LC/MS analysis of both intact gs-enoxaparin and its heparinase-digest indicated the generation of unknown structures by glycol-splitting. Based on the LC/MS and NMR data, we hypothesized that the splitting of the C(2)-C(3) bond bearing a N-sulfated amino group at the C(2) and a hydroxyl group at the C(3) of 1,6aMNS could generate the structures shown in Fig. 1.12 The oxidation by periodate of amino sugars having vicinal OH- and NH2-groups is a well-known process leading to the splitting of the corresponding C–C bond.16 However, the presence of different N-substituents induces a variety of oxidative behavior.16,17 For example, N-alkyl-amino alcohols are also susceptible to periodate oxidation but the reaction rate largely varies for secondary and tertiary amines.16 However, when amino-group is N-sulfated the nucleophilic properties of nitrogen are expected to be diminished, so that periodate oxidation of N-sulfated hexosamines appears unexpected.
Figure 1.
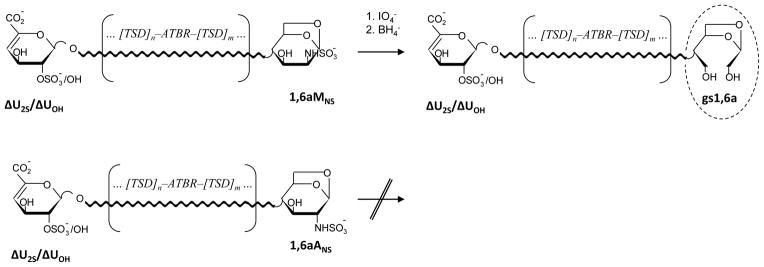
Behavior of N-sulfated 1,6-anhydro-D-mannosamine (1,6aMNS) and 1,6-anhydro-D-glucosamine (1,6aANS) residues upon periodate oxidation/borohydride reduction, as previously hypothesized12
ΔU2S and ΔUOH – 2-O-sulfated and non-sulfated 4,5-unsaturated uronic acids, gs1,6a – glycol-split-1,6-anhydro-sugar, TSD – trisulfated disaccharide (I2S–ANS6S) prevalently present within heparin chains
The present study is focused on the behavior of terminal amino sugar residues of enoxaparin tetrasaccharides upon periodate oxidation/borohydride reduction. In order to directly prove that the unknown structures found in gs-enoxaparin by LC/MS analysis in our previous work12 were generated by periodate oxidation of 1,6aMNS, we isolated from enoxaparin a fraction containing the two isomeric pentasulfated tetrasaccharides ΔU2S-ANS6S-I2S-1,6aANS and ΔU2S-ANS6S-I2S-1,6aMNS and studied their behavior under glycol-splitting conditions by NMR spectroscopy and LC/MS. To verify the role of the 1,6-anhydro-bridge in the susceptibility towards oxidation a fraction containing the hexasulfatated tetrasaccharides ΔU2S-ANS6S-I2S-ANS6S and ΔU2S-ANS6S-I2S-MNS6S have been also studied in the presence of periodate. Molecular mechanics characterisation of all target tetrasaccharides has permitted to determine their average conformation in solution, and indicated how their structural differences can influence their behavior towards periodate oxidation.
2. Results
2.1. NMR and LC/MS study of susceptibility of enoxaparin 1,6-anhydro-amino sugars to periodate oxidation
Model compounds were needed to verify whether periodate can split terminal 1,6aMNS residues of some enoxaparin oligosaccharides. Since no oligosaccharide standards containing 1,6-anhydro-sugars are available, we performed a multi-step fractionation of enoxaparin components to isolate a fraction containing two isomeric pentasulfated tetrasaccharides ΔU2S-ANS6S-I2S-1,6aANS and ΔU2S-ANS6S-I2S-1,6aMNS (Scheme 1). The tetrasaccharide fraction was chosen because its structural characterization is easier with respect to longer-chain oligosaccharides. As first step, SEC-fractionation of the enoxaparin sample was carried out to obtain a fraction enriched in tetrasaccharides, which was, then, sub-fractionated by ion-pair reversed phase high-performance liquid chromatography (IPRP-HPLC) to isolate the target tetrasaccharides (Scheme 1). The obtained fraction was fully characterized by 2D NMR and LC/MS analysis. The corresponding spectral data are reported in Fig. S1.
Scheme 1.

Isolation procedure of two isomeric pentasulfated tetrasaccharides ΔU2S-ANS6S-I2S-1,6aANS and ΔU2S-ANS6S-I2S-1,6aMNS
The periodate treatment of the isolated mixture of ΔU2S-ANS6S-I2S-1,6aANS and ΔU2S-ANS6S-I2S-1,6aMNS was performed directly in the NMR tube in order to monitor the behavior of the corresponding anomeric signals. The absence of ANS-G disaccharide unit within the isolated tetrasaccharides permitted to monitor the anomeric signal of 1,6aMNS by monodimensional 1H NMR spectroscopy. In the case of more complex fractions or unfractionated enoxaparin, the 1,6aMNS signal (at 5.57–5.59 ppm)13,15 overlaps with the anomeric signal of ANS followed by G (at 5.58 ppm)18 in the 1H NMR spectrum. Even if their differentiation is possible by 2D HSQC NMR spectroscopy, it could not permit monitoring reaction kinetics because of much longer acquisition times. In Fig. 2 the 1H NMR spectra of the pentasulfated tetrasaccharide mixture in presence of periodate after different time periods are shown. It can be noted that the chemical shifts of the 1,6-anhydro-residues (1,6aMNS 5.56 ppm and 1,6aANS 5.60 ppm) are slightly shifted with respect to the published values found for enoxaparin (1,6aMNS 5.59 ppm and 1,6aANS 5.63 ppm)13, probably due to the shorter length of these oligosaccharides. In fact, the observed values are similar to those of some disaccharides with 1,6-anhydro-terminal units (1,6aMNS 5.57 ppm and 1,6aANS 5.61 ppm).15 The anomeric 1,6aMNS signal showed a notable intensity decrease after 5 h of reaction, and after 20 h it was present only in trace amount (Fig. 2). Together with the disappearance of 1,6aMNS peak, a new signal appeared (indicated as * in the Fig. 2), likely corresponding to the anomeric signal of I2S adjacent to the modified 1,6aMNS, at 5.30 ppm (see below).
Figure 2.

Anomeric region of the 1H NMR spectra of ΔU2S-ANS6S-I2S-1,6aANS and ΔU2S-ANS6S-I2S-1,6aMNS mixture before and after addition of periodate
Symbol * indicates the anomeric signal of I2S adjacent the modified 1,6aMNS
LC/MS analysis of the reaction mixture after 40 h (performed as described in the experimental section) showed one of the isomeric pentasulfated tetrasaccharides unaffected (Fig. S2), and it was indentified as 1,6aANS-containing isomer because all its signals were preserved in the NMR spectrum (Fig. 2). On the contrary, the LC/MS peak of the second isomer, bearing the 1,6aMNS residue, disappeared, and a new peak with m/z 486.4881 appeared in the LC/MS chromatogram (Fig. S2). The odd value of the nominal mass (975 Da) suggests the presence of a single nitrogen atom. Because the internal ANS6S residue was shown to be unaffected by NMR (Fig. 2), the structure of new compound likely corresponds to that of the tetrasaccharide bearing an oxidized and split 1,6-anhydro-sugar residue (Fig. 3). Interestingly, in the MS spectrum such a residue appeared in free (theoretical m/z 486.4861(2−)) as well as hydrated and methyl acetal forms (formed by both methanol and water present in the eluent), proving the presence of two aldehyde groups in its molecule. The MS spectrum, the proposed structure, and errors between measured m/z values and calculated ones are shown in Fig. 3. Monoisotopic exact mass values calculated for each ion form are also reported in Table S1.
Figure 3.

Mass spectrum of the compound generated from ΔU2S-ANS6S-I2S-1,6aMNS by periodate oxidation
Arrows indicate the formation of adducts with sodium ions
The solution recovered from the NMR tube was treated with sodium borohydride to stabilize the formed aldehydes and, then, analyzed by NMR and LC/MS. HSQC NMR spectroscopy showed that all the cross-peaks in the anomeric region, except for 1,6aMNS and the adjacent I2S preceding it (I2S-(1,6aMNS)), maintained the same positions as in the original mixture of the two pentasulfated tetrasaccharides (Fig. 4). The anomeric signal of I2S linked to the modified terminal residue shifted from 5.37 (blue spot of I2S-(1,6aMNS) in HSQC spectrum, Fig. 4a) to 5.31 ppm (red spot of I2S-(gs1,6a) in HSQC spectrum) as shown by TOCSY spectroscopy (Fig. S3). Notably, while the anomeric 1,6aMNS cross-peak disappeared, a new signal at 5.04/105.9 ppm was observed in the region typical for anomeric signals of gs-units (4.8–5.0/105–107 ppm).12 Its correlation with the cross-peak at 3.72/64.0 ppm, typical for CH2OH groups (that was confirmed by HSQC-DEPT experiment, not shown), supports the presence of a gs-residue. The other three signals that appeared in the “ring region” after the periodate/borohydride reaction (Fig. 4b), namely, at 4.10/69.5 ppm, 3.94/82.1 ppm and 3.75/64.7 ppm (gs1,6a H3/C3) are correlated with each other in the TOCSY spectrum (indicated as gs1,6a in Fig. s 4 and S3). The latter signal was assigned by HSQC-DEPT experiment (not shown) to the second CH2OH group of the gs-unit, arising from the C(3) of the starting 1,6aMNS residue.
Figure 4.

HSQC spectra of ΔU2S-ANS6S-I2S-1,6aANS and ΔU2S-ANS6S-I2S-1,6aMNS mixture before (blue spots) and after (red spots) periodate/borohydride treatment
(a) – anomeric region, (b) – “ring” region. In panel (c) the structures of the unaffected tetrasaccharide ΔU2S-ANS6S-I2S-1,6aANS and the gs one ΔU2S-ANS6S-I2S-gs1,6a are shown. The first two residues ΔU2S and ANS6S within their sequence (cross-peaks shown in black) show very similar chemical shifts in the anomeric region of the spectrum, whereas the I2S and terminal 1,6aMNS/1,6aANS (shown in red) differ from each other. Chemical shifts of I2S residues differ from each other due to the so-called “sequence effect”
The LC/MS chromatograms of the pentasulfated tetrasaccharide mixture before and after periodate/borohydride treatment are shown in Fig. 5. Combining NMR and LC/MS data several conclusions can be made. First of all, as in the case of the solution treated only with periodate, the 1,6aANS-containing isomer remains unaffected (m/z 526.983(2−)). Our knowledge about the elution order of heparin oligosaccharides in IPRP mode is in agreement with this result. It has been noted that 1,6aMNS-containing oligosaccharides were eluted before their 1,6aANS isomers (unpublished data from our group). In fact, the peak with a shorter retention time (RT 55.0 min) disappeared from the chromatogram, while the second one (RT 58.2 min) remained unaffected (Fig. 5). The new peak (RT 32.9 min) observed in the chromatogram after borohydride treatment corresponded to the reduced form (m/z 488.506(2−)) of the compound generated by periodate oxidation (m/z 486.488(2−), Fig. 3). The increase of the nominal mass value by 4 Da after reduction is in agreement with the proposed structure with two aldehyde groups. As shown in our previous work, the exact mass and the isotope pattern corresponds to the proposed structure with the gs1,6a-residue.12
Figure 5.

LC/MS chromatogram of the ΔU2S-ANS6S-I2S-1,6aANS and ΔU2S-ANS6S-I2S-1,6aMNS mixture (a) and the same mixture after periodate/borohydride treatment (b). In panel (c) the corresponding mass spectra are shown
Peak at 70 min in panel (b) is a system signal (due to the gradient)
2.2. Behavior of terminal N-sulfated mannosamine and glucosamine residues in presence of periodate
To verify whether the 1,6-anhydro-bridge or the configuration of C(2) of the 1,6aMNS residue favours the ring splitting, we performed the same oxidation experiment with a mixture containing the isomeric hexasulfated tetrasaccharides ΔU2S-ANS6S-I2S-ANS6S and ΔU2S-ANS6S-I2S-MNS6S lacking in the 1,6-anhydro-bridge. The two isomers were isolated from the same enoxaparin tetrasaccharide fraction used for isolation of the 1,6-anhydro-tetrasaccharides (Scheme 1), as described in the experimental section, and characterized by NMR and LC/MS (Fig. S1).
Their behavior in presence of periodate was studied directly in the NMR tube by HSQC NMR spectroscopy. Unlike for 1,6-anhydro-tetrasaccharides, monodimensional 1H NMR could not be used because of the overlapping of the anomeric signal of MNS6S (α-anomer) with that of the internal ANS6S (Fig. S4). Since after 20 h no changes were observed in the 2D HSQC spectrum, an additional portion of periodate was added, and the reaction was carried out for further 20 h. However, even after 40 h, the volumes of the cross-peaks of α-anomers of both N-sulfated D-glucosamine (ANS6Sα) and D-mannosamine (MNS6Sα) as well as the signal area ratio ANS6Sα(H1/C1)/MNS6Sα(H1/C1) did not significantly change (Fig. S4), as, at first glance, the LC/MS profile of the hexasulfated tetrasaccharide mixture. Both the isomeric peaks were still present in the chromatogram after 40 h, along with a new peak with m/z 506.47(2−) observed only in trace amount (Fig. 6). The observed m/z value 506.47(2−) of the new compound corresponds to ΔU2S-ANS6S-I2S-Ram, where Ram is a remnant formed by simultaneous splitting of C(1)–C(2) and C(2)–C(3) bonds of the terminal amino sugar (Fig. 6c). After borohydride treatment both ΔU2S-ANS6S-I2S-ANS6S and ΔU2S-ANS6S-I2S-MNS6S (not oxidized by periodate) appeared in the LC/MS chromatogram in their alditol forms (not shown). The new compound ΔU2S-ANS6S-I2S-Ram was also reduced (m/z 507.47(2−)) (not shown). A mass increase of 2 Da after its borohydride reduction, together with the presence of only a mono-methyl acetal form of ΔU2S-ANS6S-I2S-Ram (m/z 522.48(2−)) (Fig. 6c), confirms the presence of one aldehyde group in its structure.
Figure 6.

LC/MS chromatograms of isomeric hexasulfated tetrasaccharides ΔU2S-ANS6S-I2S-ANS6S and ΔU2S-ANS6S-I2S-MNS6S before (a) and after (b) periodate treatment (40 h). In panel (c) we show the mass spectrum of the peak generated by periodate oxidation after 5 days but not after 40h because for the latter case the peak intensity was too low for exact mass determination.
Peak broadening may be explained by presence of numerous forms of the oxidized tetrasaccharide because aldehyde groups reversibly react with water and methanol present in the mobile phase
This compound could be generated by both isomers. However, a change in the peak intensity ratio of the two hexasulfated tetrasaccharides after addition of periodate suggested a prevalent oxidation of the isomer with a longer RT (73.8 min) (Fig. 6). Based on our knowledge about the elution order of enoxaparin oligosaccharides (unpublished data of our group), this component is the MNS6S-bearing isomer. Because of a slow reaction rate and insufficient chromatographic resolution it was difficult to establish whether also terminal ANS6S residue was affected by periodate. The possibility of its oxidation was independently verified using a disaccharide ΔU2S-ANS6S, isolated in our previous work.12 The disaccharide was found to be partially oxidized by 0.1 M sodium periodate: a new minor peak, corresponding to the ΔU2S-Ram (m/z 436,9701), appeared in the LC/MS chromatogram after 20 h of oxidation (not shown).
We also carried out the oxidation of hexasulfated tetrasaccharides for longer time (5 days). Even if the intensity of ΔU2S-ANS6S-I2S-Ram was slightly increased with respect to the results shown in Fig. 6, about 90 % of tetrasaccharides were still unaffected (not shown). All together these data indicate that both terminal residues, MNS6S and ANS6S, may be partially affected by periodate under drastic conditions (long reaction time and high periodate concentration). The oxidation of the MNS6S-containing isomer appeared to be relatively faster, probably due to the a-/e-cis-position of NHSO3− and OH groups at C(2) and C(3) predicted by molecular mechanics Fig. 7d.
Figure 7.
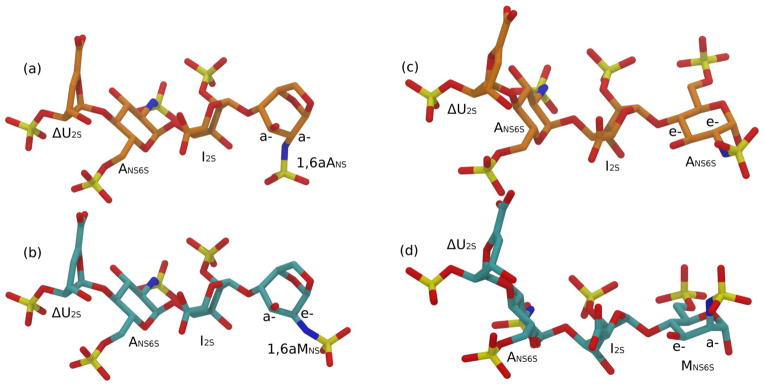
Conformation predicted for the tetrasaccharides ΔU2S-ANS6S-I2S-1,6aANS (a), ΔU2S-ANS6S- I2S-1,6aMNS (b), ΔU2S-ANS6S-I2S-ANS6S (c) and ΔU2S-ANS6S-I2S-MNS6S (d)
2.3. Molecular mechanics conformational characterisation of isomeric pentasulfated 1,6-anhydro-tetrasaccharides and their corresponding hexasulfated tetrasaccharides without the 1,6-anhydro-bridge
The conformations of the two isomeric tetrasaccharides ΔU2S-ANS6S-I2S-1,6aANS and ΔU2S-ANS6S-I2S-1,6aMNS, differing in the C(2) configuration of the terminal RE residue, were estimated assuming the same intra-residue and inter-glycosidic junction conformations previously determined for selected heparin-like octasaccharides isolated from an enoxaparin sample, and characterized in solution by NMR-NOESY and 3JHH coupling constants techniques.19 In particular the ΔU2S and ANS6S residues are described in 1H2 and 4C1 conformation, respectively. The I2S residue is drawn in 1C4 chair even if a significant (but minor) contribution of the 2S0 conformation is still present. In fact, I2S surrounded by two ANS6S residues, as frequently found in heparin-like sequences, showed the two forms: 1C4 and 2S0 populated approximately at 60 and 40%, respectively.20 Guerrini et al.19 reported in supporting information selected 3JHH coupling constants for the residues in the tetrasaccharide ΔU2S-ANS6S-I2S-1,6aMNS identical to that studied in this work. Notably, the I2S residue is characterized by the following values of the coupling constants 3JH1–H2, 3JH2–H3, 3JH3–H4: 2.5, 5.0 and 3.4 Hz, respectively. In accord to Ferro et al.20, these values correspond to a mixed conformation 1C4 and 2S0 at about 65% and 35%, respectively, confirming that the prevalent conformation of I2S within the studied sequence is 1C4. The terminal residues (1,6aANS and 1,6aMNS) are driven by 1,6-anhydro-bridge to assume 1C4 chair. In the studied tetrasaccharide models the 1,6-anhydro-ring extends above the pyranose cycle, as previously reported for enoxaparin isomeric octasaccharides, having the same I2S-1,6aMNS and I2S-1,6aANS terminal disaccharide units as the tetrasaccharides under study. The conformations of the following interglycosidic bonds: ΔU2S-ANS6S, ANS6S-I, and I2S-1,6aMNS or I2S-1,6aANS in the octasaccharides previously studied19 (matching the H1–H4 and/or H1–H3 interglycosidic NOEs), were used to estimate the analogous glycosidic bonds in the present tetrasaccharides, that in term of ϕ/ψ torsional angles are: 10/−62°, −38/−34°, and 88/3°, respectively. The conformations of both tetrasaccharides were then refined by energy minimization, as reported in the experimental section. The glycosidic torsional states obtained after the energy minimization procedure, reported in Table 1, were qualitatively supported by the ϕ/ψ molecular mechanics torsional angle maps drawn in Fig. S5. The obtained tetrasaccharide structures are shown in Fig. 7 (a, b). The same approach was applied to estimate the conformation of two regular hexasulfated tetrasaccharides ΔU2S-ANS6S-I2S-ANS6S and ΔU2S-ANS6S-I2S-MNS6S, lacking the 1,6-anhydro-bridge. In this case the conformation of both RE residues (ANS6S and MNS6S) are described in 4C1 conformation, as previously observed.21 The optimized hexasulfated tetrasaccharide structures are reported in Fig. 7c and Fig. 7d. The torsional angle states for the torsions ΔU2S-ANS6S and ANS6S-I2S are indistinguishable from the corresponding ones in the 1,6-anhydro-tetrasaccharides, while significant differences were observed at the level of the I2S-ANS6S (or I2S-MNS6S) torsions compared to the I2S-1,6aANS (or I2S-1,6aMNS) ones (Table 1). Qualitative comparison of the glycosidic torsional maps (Fig. S5) showed that hexasulfated tetrasaccharides, lacking the 1,6-anhydro-bridge, are less sterically hindered than the corresponding pentasulfated 1,6-anhydro-tetrasaccharides. On the contrary, changing the ANS6S at the RE with a MNS6S did not significantly alter the RE glycosidic conformation.
Table 1.
Glycosidic torsional states ϕ/ψ expressed in degree for the isomeric pentasulfated tetrasaccharides ΔU2S-ANS6S-I2S-1,6aMNS and ΔU2S-ANS6S-I2S-1,6aANS and hexasulfated tetrasaccharides ΔU2S-ANS6S-I2S-MNS6S and ΔU2S-ANS6S-I2S-ANS6S. Glycosidic torsional states ϕ/ψ for ΔU2S-ANS6S and ANS6S-I2S are the same for all the four tetrasaccharides
| Glycosidic torsional states ϕ/ψ | |||||
|---|---|---|---|---|---|
| ΔU2S-ANS6S | ANS6S-I2S | I2S-1,6aMNS | I2S-1,6aANS | I2S-MNS6S | I2S-ANS6S |
| 0/−31 | −39/−27 | 48/30 | 48/30 | 33/11 | 33/13 |
For all the target tetrasaccharides, in the predicted conformations, the electrostatic charge distribution, described as a set of point-like charges located at each atom position, was calculated applying the AM1-BCC scheme,22 which is proved to perform better than other empirical or semi-empirical (i.e. Gasteiger) set of charges in quantitative structure-activity relationship (QSAR) study.23 The method allows a fast calculation of point-like charges based on the semi-empirical AM1 approach, followed by the additive correction BCC (Bond Corrected Charges) that reproduces the standard HF/6–31G* Electrostatic Potential on an ensemble of about 2700 organic compound.22 The atomic charges for the four compounds under study were also estimated fitting the electrostatic potential obtained by the quanto-chemical theory HF/6–31G* (see experimental part). The determined dipole and quadrupole components calculated by two methods are in agreement with each other (Table S2). Interestingly, the obtained results show significant differences in term of the electric dipole, and quadrupole moments, for each epimer tetrasaccharide pair, differing by the C(2) configuration of the RE residue (Table S2).
Even if it is outside of the scope of this work, qualitative correlation between the electrostatic charge distribution of the four tetrasaccharides, and their RT in IPRP-HPLC separation could be underlined. Such a correlation was shown to take place for other molecules by several QSAR (Quantitative Structure Activity Relationship) approaches.24,25 Considering that the electrostatic interaction between analyte and stationary phase is one of the dominant forces in the ion-pair reversed phase chromatographic mechanism, it appears reasonable to discuss how the electrostatic charge distribution could drive the IPRP-HPLC separation. Generally, hexasulfated tetrasaccharides are eluted after pentasulfated ones (Figs. 5a and 6a) due to the greater number of negatively charged sulfate groups (total charge −8.0 instead of −7.0) and, as result, stronger interaction with the stationary phase, dynamically modified by the positively charged dibutylamine cation. For the tetrasaccharides with the same number of sulfate groups (i.e. the same total charge), the value of the electric dipole appeared to distinguish the two epimers on the ability to interact with the stationary phase, driving their separation. In this case the higher dipole moment magnitude of the molecule correlates with the stronger interaction with the stationary phase. The 1,6aANS-bearing tetrasaccharide with a dipole moment length (11.9 eÅ), greater than that of 1,6aMNS-bearing isomer (9.0 eÅ), is characterized by a longer RT (Fig. 5). The same observation can be done for the isomeric hexasulfated tetrasaccharides: the isomer ΔU2S-ANS6S-I2S-ANS6S, characterized by lower dipole moment (8.5 eÅ, Table S2), is eluted with a shorter RT (73.5 min, Fig. 6) than ΔU2S-ANS6S-I2S-MNS6S (dipole moment 9.3 eÅ; RT 73.8 min, Fig. 6).
3. Discussion
Based on the well-known mechanism of periodate oxidation that includes the formation of a cyclic intermediate,16,17 and the predicted conformations of the tetrasaccharides under study (Fig. 7), the observed oxidation of the N-sulfated 1,6-anhydro-D-mannosamine may be explained by the e-/a-cis-position of NHSO3− and OH groups at the C(2) and C(3), respectively (Fig. 7b). It is likely that the coordination of nitrogen atom by periodate ion could lead to N-desulfation, formation of a cyclic intermediate and further splitting of the C(2)–C(3) bond. In the case of 1,6aANS the a-/a-transposition of these groups (Fig. 7a) should not allow the formation of a periodate cyclic intermediate, and, as a consequence, the 1,6aANS-terminal residue should not be oxidized. We also performed the oxidation of enoxaparin during different time periods (1, 2, 3, 4, 6 and 8 h) in order to compare the behavior of 1,6aMNS residues with that of non-sulfated uronic acids. 2D HSQC NMR analysis has shown that the complete oxidation of non-sulfated iduronic acids occurred in 2 h, while glucuronic acids appeared more resistant under the applied conditions, and both glucuronic acids and 1,6aMNS residues were only partially oxidized after 8 h of reaction (Fig. S6). Such a high susceptibility to periodate of iduronic acid may be associated with its higher conformational lability.20,26 The difference in the oxidation kinetics of different units could permit to control the glycol-splitting reaction.
It is worth noting that C(2) NHSO3− and C(3) OH groups of the internal ANS6S residue of all the target tetrasaccharides as well as RE residue of ΔU2S-ANS6S-I2S-ANS6S are in e-/e-trans -position that could allow the formation of a cyclic periodate intermediate. However, the internal ones were found resistant to the oxidation, probably due to the effect of bulky and electrostatic repulsive groups of the adjacent 1,4-linked residues. The partial or trace slow oxidation of RE ANS6S and MNS6S is likely to be associated with their hemiacetalic or acyclic hydrated aldehyde forms, whose C(1) hydroxyl groups can partially contribute to the formation of cyclic periodate intermediates, involving the adjacent C(2) N-sulfated groups.
Notably, we have previously observed the presence of ΔU2S-ANS6S-I2S-Ram in trace amount in the heparinase-digests of gs-heparins5 and in relatively high quantity in the digest of gs-enoxaparin.12 In the first case its formation was hypothesized to be the result of oxidation of non-N-substituted amino sugars that could be present in heparins, while in the case of gs-enoxaparin its generation was mainly associated with oxidation of terminal N-sulfated mannosamine. The present study suggests that the formation of ΔU2S-ANS6S-I2S-Ram may be also associated with a partial oxidation of RE hexosamines of heparin and enoxaparin chains. Moreover, its higher quantity in the gs-enoxaparin digest is likely to be explained by its lower molecular weight and, consequently, higher amount of RE terminal N-sulfated amino sugars (~16% of total glucosamine content13) with respect to unfractionated heparins (<3%).27
The observed difference in the reaction rate between 1,6aMNS residue and terminal MNS6S residue suggests that the cis-position of OH and NHSO3− groups of terminal residues (1,6aMNS or MNS6S, Fig. 7) is not a sufficient requirement for the oxidation. It is likely that the 1,6-anhydro-bridge present in ΔU2S-ANS6S-I2S-1,6aMNS induces a rigid ring conformation more favourable for oxidation to occur. Also the differences in residue electrostatic charge between 1,6aMNS (charge = −1) and MNS6S (charge = −2) may also explain the preference for the negatively charged periodate to react with the former substrate.
4. Conclusions
The present work proves that enoxaparin oligosaccharides can be split not only at the level of internal non-sulfated uronic acids but also at their RE residues when they are represented by 1,6aMNS or MNS6S/ANS6S, contributing to the increased heterogeneity of gs products. Even if examples of periodate oxidation of N-substituted α-amino alcohols are known16,17 this result is quite surprising because N-sulfation should significantly decrease the nucleophilic properties of the nitrogen atom, weakening its interaction with iodine electrophile during the formation of a cyclic intermediate. Unlike RE ANS6S residues, the internal ANS6S ones are resistant to periodate oxidation, at least under the applied conditions, confirming that heparin chains do not undergo splitting at the level of internal N-sulfated D-glucosamine residues when treated with periodate.
Importantly, an incomplete and slow oxidation of the terminal ANS6S and MNS6S may occur only under extremely drastic conditions (very long reaction times and high periodate concentration). On the contrary, the oxidation of 1,6aMNS seems to be favoured by a more rigid structure with respect to MNS6S. Interestingly, the kinetics of this process is similar to that of the oxidation of glucuronic acids but it is much slower than glycol-splitting of non-sulfated iduronic acids. These findings may be useful to optimize the reaction conditions during preparation of gs-LMHWs.
The obtained data underline a possible correlation between the molecule electric dipole moment, calculated using molecular mechanics techniques, and retention time in the ion-pair reversed phase mode of LC separation. For each pair of the isomeric tetrasaccharides, the higher the electric dipole moment magnitude value, the longer appears the retention time. These data may give a possibility to predict the chromatographic behavior of enoxaparin oligosaccharides and to distinguish epimers in the ion-pair reversed phase mode of separation.
5. Experimental
5.1. Reagents
Enoxaparin sodium was a commercial product (Clexane, Sanofi Aventis, Italy). Sodium periodate (>99%), dibutylamine (DBA, >99.5%), methanol (LC-MS grade), acetic acid (glacial, 99.9%), ammonium chloride (> 99.5%) were purchased from Sigma-Aldrich, sodium borohydride (95%), from Riedel-de Haën. Deionized (conductivity less than 0.06 μS) and filtered (Millipore filter 0.22 μm) water was used for sample dilution and for mobile phases. Ethanol (96%) was from Girelli, Italy. Disaccharide ΔU2S-ANS6S was isolated from the heparinase-digest of gs-enoxaparin by SEC.12
5.2. Multi-step fractionation of enoxaparin
The first-step fractionation was performed using size exclusion chromatography (Scheme 1). 2 ml of 120 mg/ml solution were loaded onto the column packed with Biogel P6 and eluted with 0.25 M ammonium chloride at a flow-rate 1.8 ml/min. Based on the detection at 210 nm, the tetrasaccharide fraction was isolated and, then desalted on a TSK gel HW40S column (100 × 5 cm) using water – ethanol 9:1 (v/v) as a mobile phase delivered at a flow-rate 4.8 ml/min. After desalting, the sample was concentrated under reduced pressure at 35°C and freeze-dried. The obtained fraction was then sub-fractionated by IPRP-HPLC (Scheme 1) in order to isolate the isomeric tetrasaccharides of interest. The separation was carried out on a preparative HPLC/UV system “Knauer Smartline 1000”, using conditions described in Table 2. Three runs were performed. A fraction eluted in the range of RT 50 – 56 min (Scheme 1) was concentrated; then, the solution was dialyzed using a 100–500 Da cut-off membrane in order to eliminate the excess of the DBA acetate. The same purification procedure was performed for the fraction eluted in the range 60 – 65 min (Scheme 1). After desalting, the samples was freeze-dried and characterized by NMR and LC/MS (Fig. S1) before further study. The sample from the first fraction contained pentasulfated 1,6-anhydro-tetrasaccharides ΔU2S-ANS6S-I2S-1,6aANS and ΔU2S-ANS6S-I2S-1,6aMNS, while from the second one a mixture of the hexasulfated tetrasaccharides ΔU2S-ANS6S-I2S-ANS6S and ΔU2S-ANS6S-I2S-MNS6S was obtained.
Table 2.
IPRP-HPLC/UV conditions for isolation of tetrasaccharides from total tetrasaccharide fraction of enoxaparin
| Column | C18 Knauer 250 × 8 mm, 5 μm | |||||
|---|---|---|---|---|---|---|
| Eluents | Eluent A: 10 mM DBA, 10 mM CH3COOH in H2O-CH3OH = 43:57 (v/v) | |||||
| Eluent B: 10 mM DBA, 10 mM CH3COOH in H2O-CH3OH = 50:50 (v/v) | ||||||
| Eluent C: 10 mM DBA, 10 mM CH3COOH in H2O-CH3OH = 80:20 (v/v) | ||||||
|
| ||||||
| Gradient | 0 – 45 min | isocratic step 100 % A | ||||
| 45 – 45.2 min | linear gradient from 100%A to 100%B | |||||
| 45.2 – 60 min | isocratic step 100 %B | |||||
| 60 – 60.2 min | linear gradient from 100%B to 100%C | |||||
| 60.2 – 65 min | isocratic step 100 %C | |||||
| 65 – 65.2 min | linear gradient from 100%C to 100%A | |||||
| 65.2 – 90 min | isocratic step 100 % A (column equilibrating) | |||||
|
| ||||||
| Flow | 2,5 ml/min | |||||
|
| ||||||
| UV detection | 232, 210 nm | |||||
|
| ||||||
| Sample amount | 1 mg | |||||
5.3. Periodate oxidation/borohydride reduction of the tetrasaccharide mixtures
All the content (~ 1 mg) of the first fraction containing 1,6-anhydro-tetrasaccharides was dissolved in 230 μl of D2O, and 5 mg of sodium periodate were added directly into the NMR tube. Final concentration of periodate ion corresponds to 0.1 M. The reaction was monitored as described below (section 5.4). After 40 h, 2 μl of the solution containing tetrasaccharide mixture and periodate was diluted with 50 μl of water and directly analyzed by LC/MS (section 5.5). 12 mg of sodium borohydride were added to the rest of the solution to reduce the formed aldehyde groups. The reaction was carried out for 20 h in the dark, at 4°C. The sample was then desalted using a 100–500 Da cut-off membrane and freeze-dried for further structural characterization by NMR (see 5.4) and LC/MS (see 5.5).
The oxidation of the hexasulfated tetrasaccharide-containing mixture was also performed directly in the NMR tube. About 1 mg of the material was dissolved in 230 μl of D2O and 5 mg of sodium periodate (0,1 M final concentration) were added. Because no oxidation was observed after 20 h, an additional portion of periodate (5 mg of sodium periodate) was added (final concentration of periodate ion is 0.2 M). The final 1H and HSQC spectra as well as LC/MS chromatograms were acquired after 40 h. LC/MS analysis was repeated after 5 days. The further reduction was carried out as described above for 1,6-anhydro-tetrasaccharides.
The periodate oxidation of ΔU2S-ANS6S was performed by adding different volumes (230 and 460 μl) of 0.1 M sodium periodate to 1 mg of the disaccharide. 230 μl of 0.1 M correspond to 5 mg of periodate, used for the oxidation of tetrasaccharides. After 20 h the LC/MS analysis was performed as described in the section 5.5.
The oxidation of the total enoxaparin sample was performed in batch by adding 325 μl of 0.1 NaIO4 to the solid material (~5.2 mg) and left at 4°C in the dark for different time periods (1, 2, 3, 4, 6 and 8 h). The oxidation was stopped by adding 25 μl of ethylene glycol, and after further 3 h the solution was treated by sodium borohydride.12 Each sample was desalted using a 100–500 Da cut-off membrane and freeze-dried for further HSQC NMR analysis.
5.4. NMR study
1H monodimensional NMR spectra of tetrasaccharide mixtures were recorded at 30 °C on a Bruker Avance 600 MHz spectrometer equipped with 5 mm TCI cryoprobe (Karlsruhe, Germany), the 1H spectra were performed with 128 scans with 12s of repetition time. For kinetic observation 1H spectra were performed by 2D like pulse sequence with 36 increments by 33 minutes interval for each experiment.
2D HSQC spectra of tetrasaccharide mixtures before and after periodate oxidation/borohydride reduction were made on Avance 500 and 600 MHz using standard Bruker TOPSPIN 3.0 software. Spectra were recorded using a spectral width of 8 and 80 ppm in the proton and carbon dimensions, respectively. The carrier frequencies for proton and carbon were 4.7 and 80 ppm. Spectra were acquired into a time domain of 1024 complex points, using 32 scans for each of 320 increments. The repetition time and the acquisition time were set at 2 and 0.13 s with a total measurement of 6 h, 10 min. Fourier transformation were applied on matrix size of 1024×320 data points after a squared cosine function and zero filled to 4096×2048.
2D-TOCSY spectra was acquired using 128 scans per series of 1024×384 data points and a mixing time of 90 ms. A zero filling in F1 (4096×2048) and a shifted (π/3) squared cosine function was applied prior to Fourier transformation. NMR 3 mm tubes were used for each experiment.
5.5. LC/MS analysis
LC/MS operating conditions used for the analysis of the isolated tetrasaccharides before and after oxidation/reduction (Fig. 5,6 and S2) are reported in Table 3. Analysis of the undesalted tetrasaccharide solution after the periodate oxidation, carried out directly in the NMR tube, was performed using a modified gradient in order to selectively elute salt clusters (mainly periodate salt) and avoid the suppression of the signals. Initially, 0 % of eluent B was held for the first 15 min to eliminate the excess of the salt, then, a linear gradient from 0 to 40 %B was applied to elute the oligosaccharides. Conditions optimized for the heparinase-digested gs-heparins5 were used for the analysis of the disaccharide before and after its oxidation.
Table 3.
LC/MS operating conditions
| Column | C18 (Eurospher II 100–5 C18 (250 × 4,6 mm, 5 μm, Knauer) | |
|---|---|---|
| Eluents | Eluent A 10 mM DBA, 10 mM CH3COOH in H2O-CH3OH = 9:1 (v/v) | |
| Eluent B 10 mM DBA, 10 mM CH3COOH in CH3OH | ||
|
| ||
| Gradient | 0 – 18 min | isocratic step 33 % B |
| 18 – 19 min | linear gradient from 33 to 35%B | |
| 19 – 65 min | isocratic step 33 %B | |
| 65 – 66 min | linear gradient from 35 to 45%B | |
| 66 – 80 min | isocratic step 45 %B | |
| 80 – 82 min | linear gradient 80%B | |
| 82 – 84 min | isocratic step 80%B | |
| 84 – 85 min | linear gradient from 80 to 33 %B | |
| 85 – 105 min | isocratic step 33%B | |
|
| ||
| Flow | 1 ml/min, split 1:2 before ESI-source | |
|
| ||
| UV detection | 232 nm | |
The MS spectrometric conditions were as follows: ESI in negative ion mode, drying gas temperature +180°C, drying gas flow-rate 7.0 l/min, nebulizer pressure 0.9 bar, capillary voltage +3.2 kV. The mass spectra of the oligosaccharides were acquired in a scan mode (m/z scan range 200 – 2000).
5.6. Molecular modelling conformational characterisation
The conformational characterisation of the tetrasaccharides was done starting from previously built models of octasaccharides isolated from enoxaparin.19 The tetrasaccharide conformations were then refined by potential energy minimization using the “Maestro/Macromodel 9.8” software, the Force-Field applied is the Amber* with including Homans parameter for carbohydrates, where the atomtype assigned for each described atom belongs to the Macromodel atomtype set. Nonbonded interactions were treated using the standard cut-off technique, setting it to: 12.0, 7.0 and 4.0 Å for electrostatic, van der Waals and hydrogen bond interactions respectively. The water solvent environment around the molecule is implicitly described by a continuum with a fixed dielectric constant value 80.0. The energy minimization procedure (batchmin) used 5000 minimization steps, or an energy gradient threshold of 10−3 KJ·mol−1 Å−1. The energy minimization algorithm applied was the default method (PRGC) implemented in “Macromodel-9.8” software. The energy color maps (Fig. S5) were calculated by scanning ϕ/ψ torsional angles by steps of 15°, and applying an energy minimization (10K step or a gradient threshold condition of 10−3 Kjmol−1Å−1) after each step. The non bonded interactions cut-off is set to 4.0 Å for electrostatic, van der Waals and hydrogen bond interactions. The explored angle intervals for both ϕ/ψ is [−180, 180°], except for the junction: ANS6S-I2S and I2S-1,6aANS, were more restricted interval are considered: [−155, 155°] for both ϕ/ψ in the former, and [−180, 180°] / [−100, 180°] for the latter, as underlined by the white areas in upper and lower right maps. For each tetrasaccharide conformation point-like charges are located at each atom positions and calculated as AM1-BCC level,22 using the Ambertool 1.2 package. In order to confirm the AM1-BCC results, we calculated the atomic charges fitting the electrostatic potential surface at the quanto-chemical level of theory HF/631G*, employing the quanto-chemical package GAMESS (General Atomic and Molecular Electronic Structure System).28 In particular, the HF/631G* wave function on each molecular mechanic predicted conformation is calculated using the standard setting of the single point energy calculation in GAMESS software (Self Consistent Field convergence criteria defined by the density matrix threshold: CONV = 1.0E-5). The electrostatic potential surface and the point-like charges fitted on it are then calculated using the “electrostatic potential analysis procedure” included in GAMESS (ELPOT Keyword), constraining it to reproduce the molecular charge, dipole and quadrupole moments, in accord to Jakalian et al.22
Supplementary Material
1,6-Andydro-bearing tetrasaccharides were isolated from enoxaparin sample
Their susceptibility to periodate oxidation were evaluated by NMR and LC/MS
1,6-Anhydro-bridge is crucial for oxidation of 1,6-anhydro-N-sulfo-mannosamine
Reducing end N-sulfate hexosamine are affected by NaIO4 under drastic conditions
It is possible to selectively split non-sulfated iduronic acids of enoxaparin chains
Acknowledgments
The authors thank Dr. Giuseppe Cassinelli and Prof. Benito Casu for writing assistance and critical reading of the manuscript. The work was supported by the National Institute of Health (grant number R01-CA138535) and The Italian Association for Cancer Research (AIRC, grant number IG10569).
Abbreviations
- gs-LMWHs
glycol-split low-molecular weight heparins
- I
L-iduronic acid
- G
D-glucuronic acid
- I2S
L-iduronic acid 2-O-sulfate
- ANS6S
D-glucosamine-N,6-O-disulfate
- ANAc
N-acetylated D-glucosamine residues
- ANS3S6S
D-glucosamine N,3-O,6-O-trisulfate
- MNS6S
D-mannosamine-N,6-O-disulfate
- 1,6aANS
1,6-anhydro-D-glucosamine N-sulfate
- 1, 6aMNS
1,6-anhydro-D-mannosamine N-sulfate
- ΔU2S
4,5-unsaturated uronic acid 2-O-sulfate
- Ram
remnant of the oxidized terminal amino sugar
- ATBR
antithrombin-binding region
- RE
reducing end
- NRE
non-reducing end
- gs1, 6a
split 1,6-anhydro-amino sugar
- NMR
nuclear magnetic resonance
- 2D HSQC
bidimensional heteronuclear single quantum coherence spectroscopy
- TOCSY
total correlation spectroscopy
- SEC
size-exclusion chromatography
- IPRP-HPLC
ion-pair reversed phase high performance liquid chromatography
- ESI-Q-TOF-MS
electrospray ionization – quadrupole – time-of-flight mass spectrometer
- RT
retention time
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Linhardt RJ. J Med Chem. 2003;46:2551–2564. doi: 10.1021/jm030176m. [DOI] [PubMed] [Google Scholar]
- 2.Casu B, Lindahl U. Adv Carbohydr Chem Biochem. 2001;57:159–206. doi: 10.1016/s0065-2318(01)57017-1. [DOI] [PubMed] [Google Scholar]
- 3.Casu B, Naggi A, Torri G. Matrix Biology. 2010;29:442–452. doi: 10.1016/j.matbio.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ritchie JP, Ramani VC, Ren Y, Naggi A, Torri G, Casu B, Penco S, Pisano C, Carminati P, Tortoreto M, Zunino F, Vlodavsky I, Sanderson RD, Yang Y. Clin Cancer Res. 2011;17:1382–1393. doi: 10.1158/1078-0432.CCR-10-2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alekseeva A, Casu BM, Torri G, Pierro S, Naggi A. Anal Biochem. 2013;434:112–122. doi: 10.1016/j.ab.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mousa SA, Linhardt R, Francis JL, Amirkhosravi A. Thromb Hemost. 2006;96:816–821. doi: 10.1160/th06-05-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Islam T, Butler M, Sikkander SA, Toida T, Linhardt RJ. Carbohydr Res. 2002;337:2239–2243. doi: 10.1016/s0008-6215(02)00229-x. [DOI] [PubMed] [Google Scholar]
- 8.Casu B, Guerrini M, Naggi A, Perez M, Torri G, Ribatti D, Carminati P, Giannini G, Penco S, Pisano C, Belleri M, Resanti M, Presta M. Biochemistry. 2002;41:10519–10528. doi: 10.1021/bi020118n. [DOI] [PubMed] [Google Scholar]
- 9.Casu B, Naggi A. Pure Appl Chem. 2003;75:157–166. [Google Scholar]
- 10.Zhou H, Roy S, Cochan E, Zouaoui R, Chu CL, Duffner J, Zhao G, Smith S, Galcheva-Gargova Z, Karlgren J, Dussault N, Kwan RYQ, Moy E, Barnes M, Long A, Hohan C, Qi YW, Shriver Z, Ganguly T, Schultes B, Venkataraman G, Kishimoto TK. PLosONE. 2011;6:e21106. doi: 10.1371/journal.pone.0021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leitgeb AM, Blomqvist K, Cho-Ngwa F, Samje M, Nde P, Titanji V, Wahlgren M. Trop Med Hyg. 2011;84:390–396. doi: 10.4269/ajtmh.2011.10-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alekseeva A, Casu B, Cassinelli G, Guerrini M, Torri G, Naggi A. Anal Bioanal Chem. 2014;406:249–265. doi: 10.1007/s00216-013-7446-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guerrini M, Guglieri S, Naggi A, Sasisekharan R, Torri G. Sem Thromb Hemost. 2007;33:478–487. doi: 10.1055/s-2007-982078. [DOI] [PubMed] [Google Scholar]
- 14.Mourier P, Viskov C. 2005/0119477 A1. US Pat. 2005
- 15.Mascellani G, Guerrini M, Torri G, Liverani L, Spelta F, Bianchini P. Carbohydr Res. 2007;342:835–842. doi: 10.1016/j.carres.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 16.Dryhurst G. Analytical and structural applications. 1. Pergamon press; London: 1970. Periodate oxidation of diol and other functional groups. [Google Scholar]
- 17.Perlin AS. Adv Carbohydr Chem Biochem. 2008;60:183–250. doi: 10.1016/S0065-2318(06)60005-X. [DOI] [PubMed] [Google Scholar]
- 18.Guerrini M, Naggi A, Guglieri S, Santarsiero R, Torri G. Anal Biochem. 2005;337:35–47. doi: 10.1016/j.ab.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Guerrini M, Elli S, Gaudesi D, Torri G, Casu B, Mourier P, Herman F, Boudier C, Lorenz M, Viskov C. J Med Chem. 2010;53:8030–8040. doi: 10.1021/jm100771s. [DOI] [PubMed] [Google Scholar]
- 20.Ferro DR, Provasoli A, Ragazzi M, Casu B, Torri G, Bossennec V, Perly B, Sinay P, Petitou M, Choay J. Carbohydr Res. 1990;195:157–167. doi: 10.1016/0008-6215(90)84164-p. [DOI] [PubMed] [Google Scholar]
- 21.Yamada S, Watanabe M, Sugahara K. Carbohydr Res. 1998;309:261–268. doi: 10.1016/s0008-6215(98)00144-x. [DOI] [PubMed] [Google Scholar]
- 22.Jakalian A, Jack DB, Bayly CI. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 23.Tsai KC, Chen YC, Hsiao NW, Wang CL, Lin CL, Lee YC, Li M, Wang B. Eur J Med Chem. 2010;45:1544–1551. doi: 10.1016/j.ejmech.2009.12.063. [DOI] [PubMed] [Google Scholar]
- 24.Karelson M, Lobanov SV, Katritzky AR. Chem Rev. 1996;96:1027–1043. doi: 10.1021/cr950202r. [DOI] [PubMed] [Google Scholar]
- 25.Song M, Breneman CM, Bi J, Sukumar N, Bennett KP, Cramer S, Tugcu N. J Chem Inf Comput Sci. 2002;42:1347–1357. doi: 10.1021/ci025580t. [DOI] [PubMed] [Google Scholar]
- 26.Ferro DR, Provasoli A, Ragazzi M, Torri G, Casu B, Gatti G, Jacquinet J-C, Sinay P, Petitou M, Choay J. J Am Chem Soc. 1986;108:6773–6778. [Google Scholar]
- 27.Desai UR, Linhardt RJ. J Pharm Sci. 1995;84:212–215. doi: 10.1002/jps.2600840218. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA. General Atomic and Molecular Electronic Structure System. J Comput Chem. 1993;14:1347–1363. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.