

Abstract
Calpain mediated cleavage of CDK5 natural precursor p35 causes a stable complex formation of CDK5/p25, which leads to hyperphosphorylation of tau. Thus inhibition of this complex is a viable target for numerous acute and chronic neurodegenerative diseases involving tau protein, including Alzheimer’s disease. Since CDK5 has the highest sequence homology with its mitotic counterpart CDK2, our primary goal was to design selective CDK5/p25 inhibitors targeting neurodegeneration. A novel structure-based virtual screening protocol comprised of e-pharmacophore models and virtual screening work-flow was used to identify nine compounds from a commercial database containing 2.84 million compounds. An ATP non-competitive and selective thieno[3,2-c]quinolin-4(5H)-one inhibitor (10) with ligand efficiency (LE) of 0.3 was identified as the lead molecule. Further SAR optimization led to the discovery of several low micromolar inhibitors with good selectivity. The research represents a new class of potent ATP non-competitive CDK5/p25 inhibitors with good CDK2/E selectivity.
Keywords: Alzheimer’s disease, CDK5/p25 inhibitor, ATP non-competitive, Virtual screening, E-pharmacophore
1. Introduction
Alzheimer’s disease (AD) is one of the most dreaded forms of progressive neurodegenerative diseases of the modern era. According to a recent report, AD is identified as the sixth leading causes of death in United States and is estimated to affect about 14 million individuals by the year 2050.1 The only drugs available to address cognitive impairment are actylcholinesterase (ACh) inhibitors, such as aricept, exelon and razadyne. These drugs do not cure AD, but improve the cognitive functions by increasing the ACh levels in the neurons. Currently several other drugs with different pharmacological pathways are undergoing clinical trials in AD patients, such as γ-secretase inhibitors, β-secretase inhibitors and Aβ immune-modulators. Further research is still needed to find a cure for this unmet medical challenge.
The two main histopathological hallmarks have been identified in neurodegeneration leading to AD are the formation of amyloid senile plaques (SP) and neurofibrillary tangles (NFT).2 SPs are extra-cellular and mainly composed of β-amyloid (Aβ) peptides, whereas intra-cellular NFTs are considered to be the downstream products of the hyper-phosphorylated tau, a microtubule associated protein.2,3 One of the many mechanistic pathways leading to hyperphosphorylation of tau suggests the involvement of an atypical cyclin-dependent kinase (CDK), CDK5.4–6 CDK5 is a proline-directed serine/threonine kinase (STK), primarily expressed in the CNS and unlike its other mitotic counterparts, regulates neuronal development rather than cell division.7,8 Another major difference is that it does not require phosphorylation of the ‘T-loop’ to achieve an active conformation like other CDKs.7 CDK5 is involved in post mitotic neuronal survival, migration and neurogenesis.9 The post mitotic neuronal expression requires its activation by non-cyclin activators p35 and p39. In the biochemical process, the membrane bound natural precursor p35 is cleaved by calpain to a longer lived cytosolic protein p25, leading to the formation of a hyperactive p25-CDK5 complex.10 Such cleavage is reported as one of the key reasons for NFT formation leading to AD.4–6
Therefore inhibition of the p25-CDK5 complex is a viable target for AD, by blocking the hyperphosphorylation of tau and subsequent NFT formation. CDK5 deregulation is also indicated in other neurodegenerative diseases such as Huntington’s chorea, stroke, Parkinson’s disease, Lou Gehrig’s disease and neurological disorders like major depression and abuse.4,11–13 In non-neuronal systems it is indicated in the control of glucose metabolism in pancreatic beta cells14 and in tumorigenesis of the pancreas.15 All these therapeutic indications made it a very attractive target to investigate in depth.
We chose to design CDK5/p25 inhibitors as a target against neurodegeneration leading to AD. One of our major goals was to design CDK5/p25 inhibitors selective over CDK2. Since we were targeting neurodegeneration we wanted to avoid any undesired cell cycle mediated apoptotic effects of CDK2 inhibition. The task was very challenging, because the two kinases have very high levels of sequence homology (60%) and commonality in the number of residues (27 out of 29) in the ATP binding domains. The only two differing amino acid residues in the catalytic domain for CDK5 are Cys83 and Asp84, whereas for CDK2 these are Leu83 and His84.
Despite these similarities, selectivity was achieved previously by different research groups, utilizing the ligand-based structure-activity relationship (SAR) strategies, where the lead compounds were identified by high throughput screening and optimized by docking into a homology model.16–18
The prospect of structure-based inhibitor design received a shot in the arm with the availability of X-ray crystal structures of CDK5/p25.7,19,20 The apo-protein structure (PDB id: 1H4L), which was in its active conformation showed no phosphorylation at T-loop (specifically at Ser159). The four other structures (PDB id: 1UNG, 1UNH, 1UNL and 3O0G)19,20 have embedded ligands and all are crystallized from a mutant CDK5/p25 form in which Asp144 of the wild form was mutated with Asn144. Structures of the known ligands with available PDB ids are described in Figure 1. Even with the availability of the crystal structures, to-date only a few groups have utilized structure-based design using simple docking protocols to identify leads.21–23
Figure 1.

Examples of known CDK5/p25 inhibitors with Protein Data Bank id (PDB id)
Structure based virtual screening is a successful computational tool often used as complimentary to high throughput biological screening to identify hits.24,25 In view of the potential therapeutic importance and available crystal structures, CDK5/p25 is an attractive target for structure based inhibitor design. However, no prospective virtual screening studies have been reported to-date. A recent three dimensional quantitative structure-activity relationship (3D-QSAR) study26 has been reported aimed towards understanding the SAR requirements of previously disclosed CDK5/p25 inhibitors.27 Another recent virtual screening effort28 described the validity of the docking model of previously known inhibitors and it fell short of disclosing any new structural core. In our current research, an in-silico approach was used to identify novel selective CDK5/p25 inhibitors using structure-based virtual screening. The computational approach was divided into four distinct segments: protein selection, primary screening, structure-based virtual screening and compound selection as described in the schematic representation (Figure 2). To identify de-novo templates we decided to use structure based e-pharmacophore models29,30 coupled with a docking based virtual screening workflow to screen a commercially available database containing 2.84 million compounds with 4.28 million conformations. In this approach we further imposed a selectivity ratio constraint and subsequently used clustering and similarity analysis to identify 9 compounds. Based on the kinase data compound 10 was identified as the lead with a 0.3 LE and four-fold selectivity over CDK2/cyclin E. The lead showed non-competitive inhibition with ATP, whereas a truncated analog showed competitive inhibition. Further SAR development identified several additional analogs with selective and ATP non-competitive inhibition. The results provided the potential for exploring this new structural class of selective non-ATP competitive CDK5/p25 inhibitors in detail.
Figure 2.

In-silico scheme for virtual screening study
2. Results and Discussion
2.1. Protein preparation and selection
Several previous computational studies have explored the importance of water molecules in the active sites and showed that they were pivotal for ligand binding in virtual-screening frameworks.31,32 In this study, we explored the significance of water molecules in CDK5/p25 crystal structures (PDB id: 3O0G, 1UNL, 1UNG and 1UNH).19,20 We compared the docking scores of the co-crystallized ligands docked in the proteins having conserved water molecules within 5Å of the ligand binding distances and proteins with removed water in the binding domain. The ligands showed better docking scores (GlideScores) with the water-bound forms (Table 1). We used the same analogy for the CDK2 protein structures and kept the highly conserved waters in the 5Å of their ligand binding domain.
Table 1.
XP-docking scores of ligands in proteins with and without water
| CDK/p25 protein structures | Status of water in the active sites | Glide scores |
|---|---|---|
|
| ||
| 3O0G | Water bound | − 10.570 |
| No water | − 9.925 | |
|
| ||
| 1UNL | Water bound | − 9.968 |
| No water | − 9.554 | |
|
| ||
| 1UNG | Water bound | − 9.208 |
| No water | − 8.419 | |
|
| ||
| 1UNH | Water bound | − 8.263 |
| No water | − 7.441 | |
Subsequently, an ensemble-docking of the CDK5/p25 crystal bound ligands was performed to find a suitable protein structure to be used for the virtual screening study. We found that in all cases; only the co-crystallized ligands were docked well with the originating proteins (see supporting information, Table T1). This made us decide that no single protein structures could be used to dock all the ligands and consequently we chose all four CDK5/p25 crystal structures for the virtual screening framework. On the other hand, the selection of CDK2 protein structures were bit difficult, because of the presence of large number of protein structures in the PDB. We primarily looked for available CDK2 structures with a resolution of ≤ 2.5Å between the years of 1996–2011 and ended up with 148 hits. It was then narrowed down through similarity searching33 and hierarchical clustering34 to six structures (PDB id: 1AQ1, 2W17, 1OIT, 2A4L, 2VTP and 1R78).35–40 While analyzing the data from ensemble-docking of these six co-crystallized ligands, we found that all six ligands docked well only with 2VTP (see supporting information, Table T2). Consequently 2VTP was chosen as the CDK2 protein for virtual screening workflow.
2.2. Primary screening and validation
For quick screening of a large database, we extracted the pharmacophoric features required for CDK5/p25 inhibition from the known x-ray structures (PDB id: 1UNL, 1UNH, 1UNG, 3O0G)19,20 and derived e-pharmacophore models29,30 using Maestro.41 The structure-based pharmacophore model generated from 1UNL contained seven features: two hydrogen bond acceptors (A2, A4, red spheres), two hydrogen bond donors (D5, D6, blue spheres), one hydrophobic feature (H8, green sphere) and two ring aromatics (R11, R12, red rings) (Figure 3). Similarly, the pharmacophoric models were also generated from 1UNH as A3D4H8R10; from 1UNG as A1A2D5H6R8R9; and from 3O0G as A1A2D3D4R7R8 (Figure 3).
Figure 3.

E-pharmacophoric features of 1UNG, 1UNH, 1UNL and 3O0G
These e-pharmacophore models were further refined by generating some additional models incorporating the true hydrogen bonding (H-bonding) interactions of the ligands as described in the PDB structures (1UNL, 1UNH, 1UNG, 3O0G). In total twelve hypotheses: five from 1UNL, two from 1UNH, three from 1UNG and two from 3O0G (see supporting information, Table T3) were created. The next goal was the validation of these twelve hypotheses to select a concise list for virtual screening study.
Hypotheses validation was achieved in two stages, first by building and screening a Phase database42 containing known CDK5/p25 inhibitors,18,20,43–59 followed by Glide docking60 of the screened hits in CDK2 crystal structure (PDB id: 2VTP). We chose more precise XP- docking method for our purposes.61
For each hypothesis the compounds with relatively poor docking fit (GlideScores ≥ −5.0) in CDK2 structure and good experimental activity (IC50 ≥ 0.6 μM) in CDK5/p25 were selected. Finally we ranked order the hypotheses on the basis of number of hits (Figure 4). One hypothesis from each of 1UNH, 1UNG and 3O0G and three from 1UNL were selected (Table 2) to screen the larger commercial database.62
Figure 4.
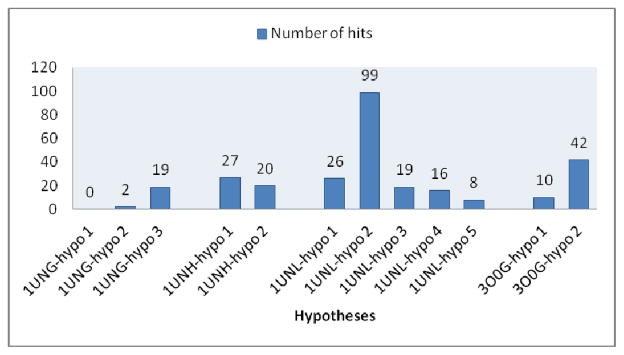
Hypotheses rank order
Table 2.
Selection of Hypotheses
| Selected Hypotheses | Pharmacophore Features |
|---|---|
|
| |
| 1UNG-hypo 3 | A2D5R8R9 |
| 1UNH-hypo 1 | A3D4H8R10 |
| 1UNL-hypo 1 | A2D5D6R11R12 |
| 1UNL-hypo 2 | A2D6H8R11R12 |
| 1UNL-hypo 3 | A2A4H8D6R11R12 |
| 3O0G-hypo 2 | A1A2D3D4R7R8 |
2.3. Structure-based virtual screening
In our virtual screening study we used a database62 containing 2.84 million commercially available compounds with 4.28 million conformations. The conformations in the database were generated using ligprep,63 Phase42 and ADME (absorption, distribution, metabolism, and excretion) property screens.64 We used it as is without any further refinements.
As an initial step, we decided to screen the database using Phase based quick pharmacophore match65 with our selected six hypotheses as an alternative to the computationally exhaustive docking based virtual screening workflow.
These six sets of hits from the Phase pharmacophore match were docked into CDK2 crystal structure (PDB id: 2VTP) using XP Glide.60 In several previous studies GlideScores were used as a predictive tool to rank score compounds in virtual screening.61,66 We judiciously incorporated it in our scheme. We selected a cut-off CDK2 GlideScores ≥ −6.0 as a benchmark for compounds to be less active in CDK2. To understand this principle, a number > −6.0, say −5.0 will have relatively poor docking in the CDK2 crystal. Hence, predictively it will translate to poor activity in CDK2. With this threshold in place we selected a total of 21,284 potential low-CDK2-active compounds.
Compounds identified with the low CDK2 GlideScores were then docked into CDK5/p25 crystal structures (PDB id: 1UNL, 1UNH, 1UNG, 3O0G) using XP Glide. The set of compounds identified from the initial Phase screening of 1UNG-hypotheses, docked in 1UNG. Similar correlation went for the compounds identified from the other three protein-generated hypotheses as well. We used a cut-off CDK5/p25 GlideScores ≤ −7.5 to identify compounds as potential CDK5/p25 active compounds. In principle, a number < −7.5, say – 8.0 means better docking scores with predictively better CDK5 activity. Consequently, we identified 1741 potential CDK5/p25 selective compounds with poor CDK2 GlideScores (≥ −6.0) and good CDK5/p25 GlideScores (≤ −7.5). We further refined the selection of compounds by imposing a selectivity constraint (≥ 1.5) as defined below:
In total we identified 884 computationally defined active CDK5/p25 compounds with selectivity against CDK2.
2.4. Selection of compounds
The selected compounds were statistically analyzed by using 2-dimesional (2-D) Radial fingerprinting,67 in Canvas.68 The hierarchical clustering of the fingerprints identified 49 diverse sets of clusters using Tanimoto similarity and Ward’s cluster linkage method.34 We eventually identified 62 compounds from these 49 diverse sets, and further optimized through a second round of 2-D fingerprinting and clustering to a group of 17 compounds. Final selection of 9 compounds was done by knowledge based screening. These final selections along with the intermediate carboxylic acid (14) are depicted in Figure 5. The sources and the calculated properties such as, docking scores, physicochemical and ADME properties, like logP (octanol/water), log [brain]/[blood] (logBB); a predictor of a compound’s ability to cross blood-brain barrier and number of metabolic sites (#metab) calculated by QuikProp64 are listed in Table 3. The values of the intermediate carboxylic acid (14) were also calculated and included in Table 3. The log BB for a good oral CNS drug is usually between −3.0 and 1.0. All the selected compounds showed good predicted blood-brain barrier (BBB) permeability.
Figure 5.

Compounds selected from virtual-screening study and intermediate 14
Table 3.
Sources and calculated properties of virtual screening hits
| Compound # | CDK5/ p25 Glide Score | CDK2 Glide Score | Sources | LogP (o/w) | LogBB | # Metab |
|---|---|---|---|---|---|---|
| 5 | −7.827 | −3.673 | Synthesized in-house | 2.066 | −0.823 | 2 |
| 6 | −8.201 | −4.069 | Chem Bridge | 4.071 | −1.193 | 3 |
| 7 | −9.221 | −4.775 | Ryan Sci. | 3.685 | −1.664 | 5 |
| 8 | −7.582 | −3.973 | Ryan Sci. | 1.233 | −1.767 | 2 |
| 9 | −8.007 | −4.332 | Chem Div | 2.557 | −2.107 | 6 |
| 10 | −9.469 | −5.539 | Synthesized in-house | 2.548 | −1.020 | 4 |
| 11 | −8.612 | −5.240 | Chem Bridge | 4.134 | −1.023 | 5 |
| 12 | −8.908 | −5.570 | Ryan Sci. | 1.522 | −1.329 | 5 |
| 13 | −7.864 | −5.065 | Chem Div | 3.802 | −1.115 | 3 |
| 14 | −9.730 | −8.117 | Synthesized in-house | 1.418 | −1.027 | 2 |
2.5. Biological evaluation
The nine compounds identified from the virtual screening were tested in a γ-33P ATP filter binding assay70,71 using full length hCDK5/p25 and hCDK2/E. The intermediate carboxylic acid (14) was also tested and shown to be active at 50 μM concentrations. The in-vitro potencies of the selected compounds are reported in Table 4.
Table 4.
In-vitro CDK5/p25 and CDK2/E assay
| Compound # | CDK5/p25 % inhibitiona @ 50 μM | CDK2/E % inhibitiona @ 50 μM | CDK5/p25 IC50 (μM)b | CDK2/E IC50 (μM)b |
|---|---|---|---|---|
| 5 | 20.0 | 24.2 | ||
| 6 | 43.1 | 32.3 | ||
| 7 | 23.6 | 29.9 | ||
| 8 | 12.7 | 14.8 | ||
| 9 | 25.2 | 38.7 | ||
| 10 | 88.2 | 77.0 | 7.02 | 25.8 |
| 11 | 17.2 | 8.0 | ||
| 12 | 48.3 | 58.2 | 107 | 81.7 |
| 13 | 14.7 | 5.3 | ||
| 14 | 64.8 | 23.5 | 52.1 | 14.5% inhc |
| 1 | 0.188 | 0.028 | ||
| 4 | 0.655 | 0.417 |
At least two independent experiments were performed to generate the average numbers @ 10 μM ATP and 50 μM compound concentrations
Assays were performed @ ATP (Km) concentrations of 30 μM for CDK5/p25 and 65 μM for CDK2/E and at least two independent experiments were performed to generate the average values
% inhibitions were generated @ 50 μM compound and ATP Km concentrations
It was observed that among the tested compounds 5, 7, 8 were equipotent in both the kinases, whereas compounds 9, 12 and 13 showed preferential activity towards CDK2/E. The success of the computational model was confirmed by the observed CDK5/p25 selectivity of compounds 6, 10, 11 and 14.
We used 1 and 4 as the standards for the assay validation and found that both compounds showed sub-micromolar activities for CDK5/p25 as reported earlier,19,21 but showed selectivity towards CDK2/E.
Compound 10 showed a reasonably good potency for CDK5/p25 (IC50=7 μM, Ki = 3.38 μM) with a calculated ligand efficiency72 (LE) of 0.3 and a moderate 4 fold selectivity over CDK2/E (IC50=25 μM). On the other hand, the carboxylic acid (14) showed moderate activity in CDK5/p25 (IC50=52 μM) and a marginal selectivity over CDK2/E (14% inhibition @ 50 μM concentration).
The modest selectivity of 10 can be explained by comparing its docking poses in the two enzymes. In CDK5/p25 it showed H-bonding interactions with Asp86 and Asn144 (Figure 6A), whereas in CDK2, the only interaction was with a water molecule (represented by black dotted line in Figure 6B). Two other residues (Thr14 and Asp145) were within 3.5 Å distances (represented by red dotted line in Figure 6B), but they were only capable of producing very week H-bonding interactions with 10. The selectivity of the carboxylic acid (14) can be explained by its H-bonding interactions with Cys83 and Lys33 in CDK5/p25 (Figure 6C) compared to the only interactions with Leu83 in case for CDK2 (Figure 6D). We used Pymol69 to represent the docking poses in Figure 6.
Figure 6.

H-bonding interactions of 10 and 14 with CDK5/p25 (A, C) and CDK2 (B, D). (generated with Pymol.69)
Virtual screening workflow and subsequent kinase assays identified a novel thieno[3,2-c]quinolin-4(5H)-one derivative (10) with LE=0.3 and an intermediate (14) with moderate activity in the same series. This led us to choose 10 as the lead structure to proceed with its mechanistic evaluation and further SAR development. In the ATP competitive binding assay,71 10 showed non-competitive inhibition as represented by the unchanged IC50 values with increased ATP concentrations (Figure 7A). The nature of the double-reciprocal (Lineweaver-Burke) plot in Figure 7B also reiterates the non-competitive binding. On the contrary when we tested the carboxylic acid (14), we observed competitive inhibition shown by both the ATP concentration curve and the Lineweaver-Burke plot (Figure 8A and B). The docking poses of the molecules as depicted in figure 6 can explain some of these observations. In the protonated form the 2-pyridine amide (10) showed a traditional backbone interaction with Asp86. But the additional deep pocket type-II interaction with Asn144 (Figure 6A) can explain its ATP non-competitive inhibition. On the other hand the carboxylic acid (14) failed to reach deep into the pocket and consequently showed competitive inhibition (Figure 6C). To further test this hypothesis we synthesized several 2- (ortho-) substituted analogs which resulted in this deep pocket interaction and we were pleased to observe the similar non-competitive inhibitions for these SAR compounds.
Figure 7.

IC50 curves (A) and Lineweaver-Burk Plot (B) of 10 in CDK5/p25 at different ATP concentrations.
Figure 8.

IC50 curves (A) and Lineweaver-Burk Plot (B) of 14 in CDK5/p25 at different ATP concentrations.
2.6. Chemistry and SAR
Our initial focus was to prepare the 2,4-diaminothiazole standard (4) and the virtual screening hits 5 and 10. The synthesis of 2,4-diaminothiazole derivative (4) was accomplished previously in three steps.22 We developed an efficient one-pot synthesis of 4 (Scheme 1) from cyanamide, substituted phenyl isothiocyanate (15) and a β-halo acetophenone (16).73
Scheme 1.
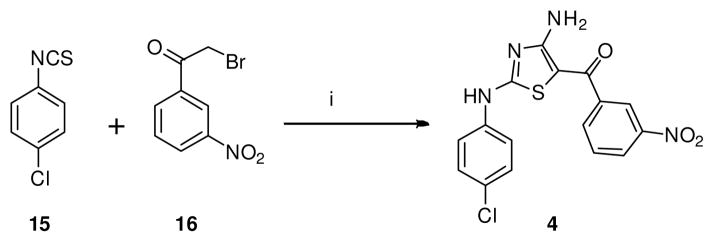
One-pot synthesis of 2,4 diaminothiazole derivative (4). Reagents and conditions: (i) Cyanamide, KOtBu, ACN, t-BuOH, 30 °C, 2 h, 59%.
The synthesis of 2-((4-((4-chlorophenyl)amino)-6-methoxy-1,3,5-triazin-2-yl)amino)ethanol (5) was effectively achieved by nucleophilic substitutions (SNAr) of 2,4-dichloro-6-methoxy-1,3,5-triazine (17) successively with 4-chloroaniline and ethanolamine in one-pot (Scheme 2). The syntheses of 4-oxo-4,5-dihydrothieno[3,2-c]quinoline-2-carboxylic acid (14) was achieved in six steps by using a sequence of Friedel–Crafts, Vilsmeier–Haack, SNAr and cyclization reactions as previously described by our group (Scheme 3).74 We synthesized 10 by an activated ester coupling of 14 with 5-methylpyridin-2-amine.
Scheme 2.
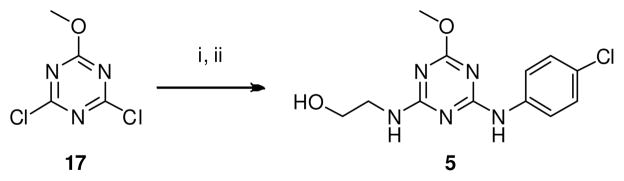
One-pot synthesis of triazine derivative (5). Reagents and conditions: (i) 4-Chloroaniline, iPr2NEt, DCM, 30 °C, 15 min; (ii) Ethanolamine, 30 °C, 5 h, 70%
Scheme 3.
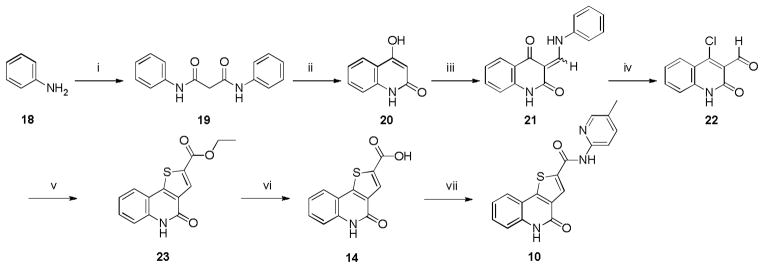
Synthesis of 10 and 14. Reagents and conditions: (i) Diethylmalonate, cat. DMF, 140 °C, 8 h, 95%; (ii) PPA, 150 °C, 3 h, 70%; (iii) Triethyl orthoformate, ethylene glycol, mw, 145 °C, 20 min, 82%; (iv) POCl3, DMF, 8 h, 90%; (v) Ethyl 2-mercaptoacetate, K2CO3, anhyd EtOH, 80 °C, 3 h, 88%; (vi) LiOH, MeOH, H2O, 12 h, 96%; (vii) 5-Methylpyridin-2-amine, HBTU, iPr2NEt, DMF, 30 °C, 12 h, 43%.
The SAR compounds 24 and 25 were synthesized from the carboxylic acid (14) using convergent analog synthesis (Scheme 4).74 The methyl ester (24) was synthesized by reacting 14 with TMS-diazomethane. The syntheses of the amide derivatives (25) were achieved by reacting the acid chloride, generated in-situ with the corresponding amines.74 We observed that a major byproduct (25w) was isolated in the reaction of the acid chloride with 2-aminopyridine (Scheme 4). In certain cases where the substituted amides contain Boc-protection (25h and 25j), the deprotection was carried out with HCl in dioxane to yield the HCl salts (25i and 25k). The free carboxylic acid derivatives (25s and 25u) were prepared by the saponification of the methyl esters (25r and 25t) with aqueous NaOH. Spectra of the known compounds were compared with the reported values.74 All the new compounds were characterized by 1H and 13C NMR, HRMS and the purities were determined by HPLC (see supporting information).
Scheme 4.

Convergent syntheses of 25a–h, j, l–r, t, v–x. Reagents and conditions: (i) TMS diazomethane, CH2Cl2, MeOH, 81%; (ii) Oxalyl chloride, CH2Cl2, 3h, 0–30°C; (iii) Amines, Hunig’s base, CH2Cl2, 8h, 23–86%; (iv) 2-aminopyridine, Hunig’s base, CH2Cl2, 8h, 23–30%.
Compounds were tested in γ-33P ATP filter binding assay using full length hCDK5/p25 and hCDK2/E.70,71 The primary goal of the SAR was to improve the activity of CDK5/p25 and selectivity against CDK2/E. We also synthesized several analogs exploiting the probable deep pocket H-bonding interactions to understand the ATP non-competitive binding as in 10.
In the SAR effort we focused our emphasis mainly on the Cring substitutions as shown in Table 5. We utilized convergent analog synthesis to systematically explore different substitutions like esters, aliphatic amides, solubilizing groups, aromatic and hetero-aromatic amides.
Table 5.
In-vitro CDK5/p25 and CDK2/E assay of SAR compounds
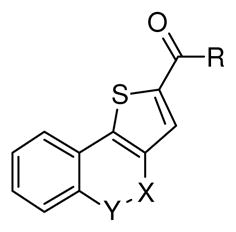
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R | X | Y | CDK5/p25 inhibition @50μMa | CDK5/p25 IC50 (μM)a,b | CDK2/E inhibition @50μMa | CDK2/E IC50 (μM)a,b |
| 24 | -OCH3 | C=O | NH | 48.2% | 58.1 | 7.9% | |
| 25a | -NH2 | C=O | NH | 29.0% | 21.8% | ||
| 25b | -NH(iPr) | C=O | NH | No inhibition | 12% | ||
| 25c | -NH(cyPr) | C=O | NH | No inhibition | 10% | ||
| 25d | -NH(cyHex) | C=O | NH | No inhibition | No inhibition | ||
| 25e | -NH(1-adamentyl) | C=O | NH | No inhibition | No inhibition | ||
| 25f |
|
C=O | NH | 24.9% | 15.9% | ||
| 25g |
|
C=O | NH | 16.6% | 8.8% | ||
| 25h |
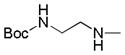
|
C=O | NH | 23.3% | 12.3% | ||
| 25i |
|
C=O | NH | 15.3% | 4.9% | ||
| 25j |
|
C=O | NH | 13.1% | 6.5% | ||
| 25k |
|
C=O | NH | 12.8% | 3.7% | ||
| 25l |
|
C=O | NH | 83.8% | 29.2 | 72.2% | 35.3 |
| 25m |
|
C=O | NH | 87.5% | 5.2 | 58.7% | 27.4 |
| 25n |
|
C=O | NH | 61.7% | 15.6 | 42.7% | 53.8 |
| 25o |
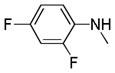
|
C=O | NH | 87.7% | 3.6 | 82.41% | 35.4 |
| 25p |
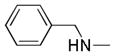
|
C=O | NH | No inhibition | No inhibition | ||
| 25q |
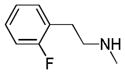
|
C=O | NH | No inhibition | No inhibition | ||
| 25r |
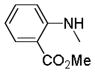
|
C=O | NH | 89.9% | 3.0 | 74.8% | 25.5 |
| 25s |
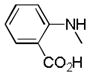
|
C=O | NH | 70.6% | 4.3 | No inhibition | |
| 25t |
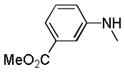
|
C=O | NH | 93.6% | 1.6 | 97% | 6.3 |
| 25u |
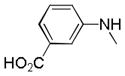
|
C=O | NH | 27.4% | No inhibition | ||
| 25v |
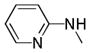
|
C=O | NH | 89.9% | 4.3 | 86% | 9.9 |
| 25w |
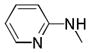
|
=C-Cl | N | 75.4% | 3.8 | 21.8% | |
| 25x |
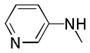
|
C=O | NH | 80.7% | 10.6 | 36.7% | |
Assays were performed @ ATP (Km) concentrations (30 μM for CDK5/p25 and 65 μM for CDK2/E) and at least two independent experiments were performed to generate the average values
Gap signifies IC50 could not be determined because of very low % inhibition at 50 μM
The ester (24) and the simple aliphatic amide (25a) showed no significant activity either in the CDK5/p25 or in CDK2/E assays. The similar activity trend was observed in case for the aliphatic amides (25b–e) and the amides with water soluble groups (25f–k). The activity started to improve drastically as we started exploring aromatic amides (25l–u) indicating, requirement of the aromatic interactions for improved activity. We observed that the over-extension of the aromatics with linkers (25p and 25q) caused disruption of aromatic interactions and resulted in inactive compounds. Comparing 25m and 25o, we observed an increased CDK2/E selectivity for 25o from 5 fold in case for 25m to >10 fold. This can be justified by the H-bonding of the ortho- F group in 25o with Asp86 which resulted in the deep pocket interaction of the thienoquinolone moiety as observed previously with 10. We prepared analogs 25r and 25s by exploiting this effect of H-boding interaction of the ortho- groups and we observed incresed selectivity for both compounds. 25s showed the best selectivity profile in the series with at least a >13-fold (no inhibition at 50 μM) selectivity and a low micromolar activity in CDK5/p25 (IC50 = 4.3 μM). Both 2-pyridyl compounds (25v and 25w) showed good activities in CDK5/p25 (for 25v IC50 = 4.3 μM; for 25w IC50 = 3.8 μM), and the by-product thieno[3,2-c]quinoline (25w) showed much improved selectivty. These improvent of selectivities did not go beyond the ortho- substitutions. As we explored the 3- (meta-) substituted compounds (25n, 25t, 25u and 25x), none of the compounds provide improved selectivity. From this SAR studies we identified three compounds (25o, 25s and 25w) with low micromolar potencies and improved selectivities to study further in ATP competitive binding assay.71 All three of the compounds showed ATP non-competitive inhibition (see supporting information, Figure F1–3). The physicochemical and ADME properties, like logP (octanol/water), log [brain]/[blood] (logBB) and number of metabolic sites (#metab) calculated by QuikProp64 are listed in Table 6. These compounds showed very good virtual profile to be successful as CNS drug candidates with low metabolic liabilities.
Table 6.
Calculated properties of 25o, 25s and 25w
| Compound # | Molecular weight | LogP (o/w) | LogBB | # Metab |
|---|---|---|---|---|
| 25o | 356.346 | 3.202 | −0.588 | 2 |
| 25s | 364.375 | 2.204 | −1.655 | 3 |
| 25w | 339.798 | 3.79 | −0.182 | 3 |
These three most potent and selective compounds were screened for selectivity in several cell cycle and tau related serine/threonine and tyrosine kinases (Table 7). In general all three CDK5/p25 inhibitors showed selectivity against the cell cycle kinases, but 25o and 25s showed some promiscuity over few tau related kinases like GSK3α and GSK3β. The best selectivity profile was observed for 25w.
Table 7.
Selectivity profile (% inhibition at 20 μM)a of 25o, 25s and 25w
| Kinases | 25o | 25s | 25w | ATP Km (μM) |
|---|---|---|---|---|
| CDK1/B | 56 | No inhibition | 35 | 5 |
| CDK2/A | 58 | 50 | 45 | 10 |
| CDK2/E | 27 | 5 | 6 | 65 |
| CDK4/D1 | 8 | No inhibition | No inhibition | 90 |
| CDK6/D1 | 26 | No inhibition | No inhibition | 30 |
| CK1a1 | 22 | 41 | 19 | 10 |
| DYRK1/DYRK1A | 12 | 23 | 4 | 30 |
| GSK3α | 65 | 79 | 22 | 30 |
| GSK3β | 53 | 81 | 41 | 5 |
| KDR/VEGFR2 | 95 | 88 | 44 | 30 |
| PKA | 3 | 89 | No inhibition | 20 |
| PKCα | 8 | No inhibition | 1 | 10 |
| PKCε | 32 | 7 | 4 | 35 |
| WEE1 | 11 | No inhibition | 2 | 50 |
Assays were performed @ ATP (Km) concentrations and at least two independent experiments were performed to generate the average values
3. Conclusions
In summary, we used a virtual screening strategy to identify CDK5/p25 inhibitors with selectivity over CDK2/E. In this effort we utilized a unique combination of e-pharmacophore based Phase screening and Glide docking with subsequent insertion of a selectivity constraint to identify a thieno[3,2-c]quinolin-4(5H)-one lead (10) with 0.3 LE. The ATP non-competitive inhibition of 10 led us to develop further SAR exploration using convergent analog synthesis. Consequently we identified three low micromolar ATP non-competitive CDK5/p25 inhibitors 25o, 25s and 25w with much greater CDK2/E selectivity. All three compounds showed good selectivity over cell cycle kinases, while 25w showed the best selectivity profile across the selected panel of cell cycle and tau related kinases. Biochemically, there are clear indications about the involvement of CDK5/p25 complex in the hyperphosphorylation of tau with the downstream formation of NFT leading to Alzheimer’s disease. The known kinase inhibitors selected to-date for evaluation in AD, such as R-(−) roscovitine, flavopiridol, indirubin-3′-oxime and kenpaullone showed lack of selectivity and are termed as pan-kinase inhibitors. Here we are reporting a novel series of compounds with ATP non-competitive CDK5/p25 inhibitions and selectivity over cell cycle kinases. Additionally, the good predicted BBB permeabilities, physicochemical properties and low metabolic liabilities generate real potential for the compounds to be evaluated in future research for developing a candidate for AD.
4. Experimental
4.1. Computational
4.1.1. General
All calculations were performed on a Linux workstation equipped with four parallel Intel Xenon X5460 processors (3.16 GHz) with 8GB total RAM.
4.1.2. Ligand Preparation
LigPrep63 was used to produce low energy 3D structures of compounds. The ionization/tautomeric states were generated using Epik. The chiralities of the compounds were retained from the original state. All the conformations were minimized using OPLS-2005 force field and at the most 32 conformations per ligands were generated.
4.1.3. Protein Preparation
Each protein crystallographic structure was loaded from the RCSB Protein Data Bank (PDB) and prepared by using Protein Preparation Wizard.75 For the dimer, the ligand bound fragment was selected for processing. It was then pre-processed by assigning the bond orders, added hydrogen, filled in the missing loops and the side chains using Prime. Waters were deleted beyond 5Å from the ligand and ionization/tautomeric states were generated at pH 7.0±4.0 using Epik. Subsequently the protein was refined by optimizing the hydrogen bonds (H-bonds) and the sample water orientations. Finally the Impref-minimization was carried out using OPLS 2005 force field.
4.1.4. Hypothesis generation
The initial pharmacophoric hypotheses were generated from the PDB structures using E-Pharmacophore.30 All the subsequent hypotheses were generated using Phase.65,76
4.1.5. Phase database generation
A database was generated from known CDK5/p25 inhibitors,18,20,43–59 using Phase.42 The ligand conformations were generated by Phase ligand processing using the following protocol: i) different ionization/tautomeric states were generated at pH 7.0±2.0 using Epik and the high-energy states were removed; ii) stereochemical information was obtained from the 3D geometry of the ligands; iii) for unspecified stereocenters, 4 low-energy stereoisomers were retained; iv) for 5/6 membered rings, up to 1 conformation per structure was retained; v) duplicate structures were skipped; vi) a maximum number of 100 conformers were generated per molecule and retained up to 10 conformations per rotatable bond.
4.1.6. Phase Database Screening
Both the Phase databases (the database built from known CDK5/p25 inhibitors and the commercial database) were screened to find pharmacophore matches65,76 using existing conformers. For the known compound database, at the most 1 hit per molecule and 1000 hits in total were set to be returned. On the other hand, for the commercial database, the return limit was set at 1 hit per molecule and 10000 hits in total. In both the cases, the hits with align scores >1.2, vector scores <-1.0 and volume scores <0.0 were rejected.
4.1.7. Ligand docking
All the docking calculations were performed with XP-Glide60,61 and run in the Virtual Screening Workflow framework. The ligands were pre-processed using LigPrep. Docking grids were generated by Glide using the co-crystallized ligand at the center of the grid box. The compounds were docked flexibly by using penalization for non-planar amide bond conformations and after docking kept 100% of the best compounds with all good scoring states.
4.1.8. Fingerprinting, Similarity and Clustering Analysis
The statistical analysis of the virtual screening hits were performed using Canvas68 in a sequential manner as follows: i) creation of fingerprints using Radial fingerprinting;67 ii) similarity analysis of the fingerprints were done using Tanimoto similarity;33 iii) hierarchical clustering by Ward’s cluster linkage method.34
4.2. Chemistry
4.2.1. General
1H and 13C NMR spectra were obtained on Bruker model AMX 500 and Avance 400 NMR spectrometers with standard pulse sequences, operating at 500 and 400 MHz for 1H and 125 and 100 MHz for 13C. The residual DMSO-d6 solvent signals (DMSO-d6: δH = 2.50 ppm and δC = 39.51 ppm) were used as internal reference. The chemical shifts (δ) were expressed in ppm. Multiplicities were described as singlet (s), doublet (d), triplet (t), multiplet (m) and broad resonance (br). The coupling constants (J) were expressed in Hz. High-resolution mass spectra (HRMS) were recorded on either a Bruker Daltonics micro-TOF mass spectrometer or a Micromass Q-TOF Agilent G1969A mass spectrometer with electro spray ionization (ESI) interface. IR spectra were recorded using an Agilent model Cary 630 FT-IR.
R (−) roscovitine (1) was purchased from Enzo Life Sciences, (www.enzolifesciences.com). 1-(4-(((4-chlorophenyl)thio)methyl)-6-hydroxypyrimidin-2-yl)-3-(4-methoxyphenyl)guanidine (6) and 4-(4,6-dimethyl-2-(pyridin-2-yl)-1H-indol-3-yl)butanoic acid (11) were purchased from ChemBridge, (www.chembridge.com). N-(5-((2-oxo-2-(p-tolylamino)ethyl)thio)-1,3,4-thiadiazol-2-yl)-3-phenylpropanamide (7), N-(3-(1H-pyrazol-1-yl)propyl)-2-oxo-1,2,3,4-tetrahydroquinoline-6-sulfonamide (8) and 2-((4-((4-methoxyphenyl)amino)pteridin-2-yl)amino)ethanol (12) were purchased from Ryan Scientific, Inc., (https://ryansci.com). 2-((3-(4-methoxybenzyl)-4-oxo-3,4-dihydropteridin-2-yl)thio)-N-(3-methyl-1H-pyrazol-5-yl)acetamide (9) and 2-((4-((3,4-dimethylphenyl)amino)-1-phenyl-1H-pyrazolo[3,4- d]pyrimidin-6-yl)amino)ethanol (13) were purchased from ChemDiv, (www.chemdiv.com). All the purchased standards were characterized by 1H NMR and HRMS. The purity was checked by HPLC and have purity of >95%. Column chromatography was carried out on silica gel (70–230 mesh, Merck). TLC was performed on silica gel 60 F254 plates. All the anhydrous solvents were purchased in sure-seal® bottles from Aldrich. All the reagents were used without further purification unless otherwise noted. A Biotage® Initiator microwave was used for all microwave (MW) reactions. All the HPLC analysis were performed on Waters W2690/5, attached with a 996 PDA detector, using X-tera C-18, 3×100 column and using a gradient system consisting of water (0.1% formic acid) and acetonitrile (0.1% formic acid).
4.2.2. (4-amino-2-((4-chlorophenyl)amino)thiazol-5-yl)(3-nitrophenyl)methanone (4)
To a stirred suspension of cyanamide (0.0505g, 1.2 mmol) in 40 mL of CH3CN and KOtBu (0.135 g, 1.2 mmol) was added a solution of 4- chlorophenyl isothiocyanate (0.17 g, 1 mmol) in 5 mL of butanol. The mixture was stirred at ambient temperature for 30 min and a suspension of 2-bromo-3′-nitroacetophenone (16) (0.6 g, 2.9 mmol) in 10 mL of CH3CN was added and stirred for 2h. The precipitated solid was triturated with water, extracted with EtOAC and layers were separated. The organics were extracted with saturated brine, dried over anhyd. Na2SO4, filtered, and purified on silica gel using CH2Cl2:EtOAc (8:2) to yield the title compound as a yellow solid (0.22 g, 59%). 1H NMR (400 MHz, DMSO) δ 11.02 (s, 1H), 8.45 (s, 2H, ex), 8.40 (s, 1H, ex), 8.35 (d, J = 8.3 Hz, 2H), 8.14 (d, J = 7.5 Hz, 1H), 7.80 (t, J = 8.0 Hz, 1H), 7.68 (d, J = 8.3 Hz, 2H), 7.42 (d, J = 8.4 Hz, 2H). 13C NMR (100 MHz, DMSO) δ 179.42, 166.89, 166.27, 147.65, 142.74, 138.35, 133.20, 130.35, 128.96, 127.03, 125.07, 121.41, 120.48, 92.22. HRMS (ESI-TOF) m/z calcd for C16H10ClN4O3S [M−H]− : 373.0162. Found: 373.0164. HPLC: retention time 19.27 min; purity >99%.
4.2.3. 2-((4-((4-chlorophenyl)amino)-6-methoxy-1,3,5-triazin-2-yl)amino)ethanol (5)
To a stirred solution of 2,4-dichloro-6-methoxy-1,3,5-triazine (17) (0.4 g, 2.22 mmol) and Hunig’s base (0.43 g, 3.33 mmol) in 50 mL of CH2Cl2 at 0 °C was added 4-chloroaniline (0.34 g, 2.66 mmol). The mixture was stirred at ambient temperature for 30 min and a solution of ethanol amine (212 μL, 2.66 mmol) and Hunig’s base (0.43 g, 3.33 mmol) in 40 mL of CH2Cl2 was added to the stirred reaction. The mixture was stirred for 8h and monitored by TLC. It was then concentrated in vacuo, diluted with EtOAc and extracted with saturated NaHCO3. The organics were extracted with saturated brine, dried over anhyd. Na2SO4, filtered, and purified over silica gel using CH2Cl2:EtOAc (8:2) to yield the title compound as a white solid (0.461 g, 70%). 1H NMR (400 MHz, DMSO) δ 9.79 – 9.40 (m, 1H, ex), 7.77 (d, J = 8.7 Hz, 2H), 7.43 (brs, 1H), 7.31 (d, J = 8.7 Hz, 2H), 4.68 (d, J = 5.1 Hz, 1H, ex), 3.81 (s, 3H), 3.61 – 3.44 (m, 2H), 3.42 – 3.33 (m, 2H). 13C NMR (100 MHz, DMSO) δ 170.36, 166.84, 165.10, 138.92, 128.23, 125.48, 121.15, 59.64, 53.58, 42.88. HRMS (ESI-TOF) m/z calcd for C12H13ClN5O2 [M−H]− : 294.0758. Found: 294.0783. HPLC: retention time 13.57 min; purity >99%.
4.2.4. 5-Dihydrothieno[3,2-c]quinoline-2-carboxylic acid (14)
Synthesis of the carboxylic acid was achieved by our previously described procedure.74 1H NMR (400 MHz, DMSO-d6) δ 13.64 (s, 1H, ex), 11.86 (s, 1H, ex), 8.02 (s, 1H), 7.87 (d, J = 7.9 Hz, 1H), 7.53 (t, J = 7.6 Hz, 1H), 7.41 (d, J = 8.4 Hz, 1H), 7.24 (t, J = 7.6 Hz, 1H). 13C NMR (100 MHz, DMSO- d6) δ 162.52, 157.78, 149.48, 137.14, 133.88, 130.94, 130.77, 130.27, 123.92, 122.65, 116.38, 115.60. FTIR νmax (cm−1): 3090, 2983, 2067, 1685, 1644, 1587, 1541, 1404, 1159, 839, 743. HRMS (ESI-TOF) m/z calcd for C12H8NO3S [M+H]+: 246.0225. Found: 246.0244. HPLC: retention time 12.58 min; purity >99%.
4.2.5. N-(5-methylpyridin-2-yl)-4-oxo-4,5-dihydrothieno[3,2-c]quinoline-2-carboxamide (10)
To a stirred suspension of 5-dihydrothieno[3,2-c]quinoline-2-carboxylic acid (14) (0.18 g, 0.73 mmol) in 10 mL of DMF was added HBTU (0.279 g, 0.73 mmol) and Hunig’s base (0.472 g, 3.65 mmol). The mixture was stirred at ambient temperature for 30 min and a solution of 5-methylpyridin-2-amine (0.118g, 1.1 mmol) in 2 mL of DMF was added to the reaction mixture. The mixture was stirred for 8h and monitored by TLC. It was then concentrated in vacuo to a slurry, filtered through a Strata™ X-C pre-packed column and purified over silica gel using CH2Cl2:MeOH with 1% NH3 (95:5) to yield the title compound as a white solid (0.083 g, 34%). 1H NMR (400 MHz, DMSO) δ 11.83 (s, 1H, ex), 11.20 (s, 1H, ex), 8.75 (s, 1H), 8.25 (s, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.92 (d, J = 7.9 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.55 (t, J = 7.7 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.26 (t, J = 7.6 Hz, 1H), 2.29 (s, 3H). 13C NMR (125 MHz, DMSO) δ 159.79, 157.93, 149.41, 148.80, 147.78, 138.92, 138.49, 136.97, 131.53, 130.57, 129.05, 127.65, 124.02, 122.60, 116.32, 115.66, 114.46, 17.30. HRMS (ESI-TOF) m/z calcd for C18H14N3O2S [M+H]+: 336.0807. Found: 336.0818. HPLC: retention time 14.29 min; purity >99%.
4.2.6. Methyl 4-oxo-4,5-dihydrothieno[3,2-c]quinoline-2-carboxylate (24)
The methyl ester was synthesized by our previously described procedure.74 1H NMR (400 MHz, DMSO-d6) δ 11.91 (s, 1H), 8.08 (s, 1H), 7.91 (d, J = 7.9 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 7.26 (t, J = 7.0 Hz, 1H), 3.90 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 161.51, 157.72, 149.83, 137.28, 131.71, 131.10, 130.91, 124.09, 122.79, 116.46, 115.47, 52.85. HRMS (ESI-TOF) m/z calcd for C13H8NO3S [M−H]− : 258.0225. Found: 258.0234. HPLC: retention time 14.60 min; purity >99%.
4.2.7. General procedure for synthesis of carboxamides (25)
To a stirred suspension of 5-dihydrothieno[3,2-c]quinoline-2-carboxylic acid (14) (0.025g, 0.1 mmol) in 5 mL of CH2Cl2 was added oxalyl chloride (0.02 g, 0.15 mmol) and 2 drops of DMF. The reaction mixture was stirred for 2h and then concentrated in vacuo. The dried suspension of the acid chloride in 10 mL of CH2Cl2 was added to a stirred solution of the respective amines (0.03 mmol) and Hunig’s base (in 5 mL of CH2Cl2) and the reaction mixture was stirred for 12h. It was then concentrated to dryness and purified by column chromatography on silica gel using CH2Cl2:MeOH with 1% NH3 (95:5) to give the target compounds. 25r, 25t, 25v, 25w, 25x are described for the first time, whereas 25a–h, 25j and 25l–q were synthesized following our previously described methodology.74
4.2.7.1. Methyl 2-(4-oxo-4,5-dihydrothieno[3,2-c]quinoline-2-carboxamido)benzoate (25r)
Yield: 88% of a white solid. 1H NMR (400 MHz, DMSO) δ 11.85 (s, 1H, ex), 11.60 (s, 1H, ex), 8.35 (d, J = 8.4 Hz, 1H), 8.26 (s, 1H), 8.00 (d, J = 7.9 Hz, 1H), 7.93 (d, J = 7.9 Hz, 1H), 7.68 (t, J = 8.0 Hz, 1H), 7.55 (d, J = 7.7 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.28 (t, J = 7.7 Hz, 2H), 3.93 (s, 3H). 13C NMR (100 MHz, DMSO) δ 167.69, 158.93, 157.74, 148.81, 138.98, 136.92, 133.95, 131.16, 130.58, 125.83, 123.87, 122.60, 121.41, 118.25, 116.30, 115.52, 52.50. HRMS (ESI-TOF) m/z calcd for C20H13N2O4S [M−H]− : 377.0596. Found: 377.0595. HPLC: retention time 18.32 min; purity >99%.
4.2.7.2. Methyl 3-(4-oxo-4,5-dihydrothieno[3,2- c]quinoline-2-carboxamido)benzoate (25t)
Yield: 55% of a white solid. 1H NMR (400 MHz, DMSO) δ 11.84 (s, 1H, ex), 10.75 (s, 1H, ex), 8.66 (s, 1H), 8.44 (s, 1H), 8.11 (d, J = 8.3 Hz, 1H), 7.93 (d, J = 7.9 Hz, 1H), 7.71 (d, J = 7.7 Hz, 1H), 7.61 – 7.49 (m, 2H), 7.45 (d, J = 8.3 Hz, 1H), 7.27 (t, J = 7.6 Hz, 1H), 3.88 (s, 3H). 13C NMR (100 MHz, DMSO) δ 166.01, 159.57, 158.01, 148.77, 138.98, 136.97, 131.39, 130.63, 130.12, 129.28, 127.12, 124.45, 124.05, 122.70, 120.57, 116.40, 115.70, 52.23. HRMS (ESI-TOF) m/z calcd for C20H13N2O4S [M−H]− : 377.0596. Found: 377.0596. HPLC: retention time 17.34 min; purity >99%.
4.2.7.3. 4-Oxo-N-(pyridin-2-yl)-4,5-dihydrothieno[3,2-c]quinoline-2-carboxamide (25v)
Yield: 30% of a white solid. 1H NMR (500 MHz, DMSO) δ 11.84 (s, 1H, ex), 11.29 (s, 1H, ex), 8.79 (s, 1H), 8.44 (d, J = 3.3 Hz, 1H), 8.15 (d, J = 8.3 Hz, 1H), 7.95 (d, J = 7.9 Hz, 1H), 7.87 (t, J = 7.8 Hz, 1H), 7.57 (t, J = 7.8 Hz, 1H), 7.45 (d, J = 8.2 Hz, 1H), 7.28 (t, J = 7.5 Hz, 1H), 7.24 – 7.17 (m, 1H). 13C NMR (125 MHz, DMSO) δ 160.02, 157.94, 151.64, 148.94, 148.04, 138.76, 138.19, 137.01, 131.54, 130.64, 127.91, 124.06, 122.64, 120.06, 116.35, 115.65, 114.92. HRMS (ESI-TOF) m/z calcd for C17H10N3O2S [M−H]− : 320.0494. Found: 320.0498. HPLC: retention time 11.52 min; purity >99%.
4.2.7.4. 4-chloro-N-(pyridin-2-yl)thieno[3,2-c]quinoline-2-carboxamide (25w)
Yield: 23% of a white solid. 1H NMR (400 MHz, DMSO) δ 11.55 (s, 1H, ex), 9.04 (s, 1H), 8.45 (d, J = 4.5 Hz, 1H), 8.35 (d, J = 8.0 Hz, 1H), 8.19 (d, J = 8.3 Hz, 1H), 8.10 (d, J = 8.3 Hz, 1H), 7.88 (t, J = 7.6 Hz, 2H), 7.78 (t, J = 7.5 Hz, 1H), 7.22 (dd, J = 7.3, 4.8 Hz, 1H). 13C NMR (100 MHz, DMSO) δ 159.79, 151.59, 148.23, 148.10, 145.44, 143.24, 141.03, 138.32, 131.78, 130.77, 128.94, 128.32, 126.16, 124.10, 122.53, 120.21, 114.78. HRMS (ESI-TOF) m/z calcd for C17H9ClN3OS [M−H]− : 338.0155. Found: 338.0158. HPLC: retention time 18.97 min; purity >97%.
4.2.7.5. 4-oxo-N-(pyridin-3-yl)-4,5-dihydrothieno[3,2-c]quinoline-2-carboxamide (25x)
Yield: 41% of a white solid. 1H NMR (400 MHz, DMSO) δ 11.83 (s, 1H, ex), 10.74 (s, 1H, ex), 8.95 (s, 1H), 8.64 (s, 1H), 8.34 (d, J = 4.6 Hz, 1H), 8.19 (d, J = 8.3 Hz, 1H), 7.94 (d, J = 7.9 Hz, 1H), 7.56 (t, J = 7.7 Hz, 1H), 7.49 – 7.38 (m, 2H), 7.28 (t, J = 7.5 Hz, 1H). 13C NMR (125 MHz, DMSO) δ 159.78, 158.02, 148.85, 144.88, 141.75, 138.66, 137.01, 135.28, 131.37, 130.74, 127.31, 127.21, 124.10, 123.68, 122.76, 116.45, 115.69. HRMS (ESI-TOF) m/z calcd for C17H10N3O2S [M−H]− : 320.0494. Found: 320.0502. HPLC: retention time 10.97 min; purity >99%.
4.2.8. Representative procedures for the synthesis of 25i and 25k
4.2.8.1. N-(2-aminoethyl)-4-oxo-4,5-dihydrothieno[3,2-c]quinoline-2-carboxamide (25i)
To a stirred suspension of tert-butyl (2-(4-oxo-4,5-dihydrothieno[3,2-c]quinoline-2-carboxamido)ethyl)carbamate (25h) (0.03g, 0.075 mmol) in 5 mL of CH2Cl2 was added 4M HCl in dioxane (500 μL) and the mixture was stirred for 8h at ambient temperature. The precipitated solid was filtered, washed with Et2O and dried under vacuum to yield the title compound as a white solid (0.024 g, 96%). 1H NMR (400 MHz, DMSO) δ 9.08 (s, 1H, ex), 8.35 (s, 1H), 7.90 (d, J = 8.0 Hz, 2H, 1 ex), 7.54 (t, J = 7.9 Hz, 1H), 7.44 (d, J = 8.4 Hz, 1H), 7.26 (t, J = 7.8 Hz, 1H), 3.65 – 3.41 (m, 2H), 3.00 (t, J = 6.6 Hz, 2H). 13C NMR (125 MHz, DMSO) δ 161.18, 157.93, 147.95, 138.85, 136.84, 131.21, 130.40, 126.18, 123.83, 122.61, 116.34, 115.71, 38.45, 37.13. HRMS (ESI-TOF) m/z calcd for C14H12N3O2S [M−H]− : 286.0650. Found: 286.0644. HPLC: retention time 8.46 min; purity >99%.
4.2.8.2. N-(3-aminopropyl)-4-oxo-4,5-dihydrothieno[3,2-c]quinoline-2-carboxamide (25k)
Yield: 92% of a white solid. 1H NMR (400 MHz, DMSO) δ 11.82(s, 1H, ex), 9.03 (s, 1H, ex), 8.33 (s, 1H), 8.06 – 7.70 (d, J = 7.9 Hz, 2H, 1 ex), 7.53 (t, J = 7.7 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.25 (t, J = 7.5 Hz, 1H), 3.43 – 3.33 (m, 2H), 2.86 (t, J = 6.7 Hz, 2H), 1.90 – 1.76 (m, 2H). 13C NMR (125 MHz, DMSO) δ 160.83, 157.97, 147.91, 139.23, 136.82, 131.27, 130.39, 125.69, 123.84, 122.64, 116.36, 115.76, 36.45, 36.06, 27.20. HRMS (ESI-TOF) m/z calcd for C15H14N3O2S [M−H]− : 300.0807. Found: 300.0812. HPLC: retention time 8.71 min; purity >99%.
4.2.9. Representative procedures for the synthesis of 25s and 25u
4.2.9.1. 2-(4-oxo-4,5-dihydrothieno[3,2-c]quinoline-2-carboxamido)benzoic acid (25s)
To a stirred suspension of methyl 2-(4-oxo-4,5-dihydrothieno[3,2-c]quinoline-2-carboxamido)benzoate (25r) (0.025g, 0.066 mmol) in 5 mL of MeOH was added 1 M NaOH (1 mL) and the mixture was stirred for 30 min at ambient temperature. It was then quenched with 10% HCl. The precipitated solid was filtered, washed successively with H2O and Et2O and dried under vacuum to yield the title compound as a white solid (0.022 g, 92%). 1H NMR (400 MHz, DMSO) δ 13.87 (s, 1H, ex), 12.48 (s, 1H, ex), 11.91 (s, 1H, ex), 8.51 (d, J = 8.3 Hz, 1H), 8.19 (s, 1H), 8.06 (d, J = 7.8 Hz, 1H), 7.95 (d, J = 7.9 Hz, 1H), 7.66 (t, J = 8.0 Hz, 1H), 7.57 (t, J = 7.9 Hz, 1H), 7.45 (d, J = 8.3 Hz, 1H), 7.32 – 7.19 (m, 2H). 13C NMR (125 MHz, DMSO) δ 169.47, 158.52, 157.44, 148.48, 139.75, 138.71, 136.64, 133.74, 130.86, 130.36, 125.25, 123.62, 123.03, 122.34, 119.88, 117.10, 116.04, 115.24. HRMS (ESI-TOF) m/z calcd for C19H11N2O4S [M−H]− : 363.0440. Found: 363.0443. HPLC: retention time 17.36 min; purity >99%.
4.2.9.2. 3-(4-oxo-4,5-dihydrothieno[3,2-c]quinoline-2-carboxamido)benzoic acid (25u)
Yield: 71% of a white solid. 1H NMR (400 MHz, DMSO) δ 13.04 (s, 1H, ex), 11.86 (s, 1H, ex), 10.74 (s, 1H, ex), 8.67 (s, 1H), 8.42 (s, 1H), 8.08 (d, J = 8.1 Hz, 1H), 7.95 (d, J = 7.9 Hz, 1H), 7.70 (d, J = 7.7 Hz, 1H), 7.61 – 7.41 (m, 3H), 7.28 (t, J = 7.6 Hz, 1H). 13C NMR (125 MHz, DMSO) δ 167.05, 159.52, 158.00, 148.71, 139.08, 138.80, 136.96, 131.36, 130.61, 129.03, 127.05, 124.67, 124.08, 122.69, 120.86, 116.39, 115.70. HRMS (ESI-TOF) m/z calcd for C19H13N2O4S [M+H]+: 365.0596. Found: 365.0591. HPLC: retention time 15.41 min; purity >99%.
4.3. Biological Assays
4.3.1. CDK5/p25 and CDK2/E assays
Compounds were tested in a γ-33P ATP filter binding assay using full length hCDK5/p25 and hCDK2/E at Reaction biology Corp. (www.reactionbiology.com)70,71 according to their standard protocol. Initially all the compounds were screened at 50 μM concentration with an ATP concentration of 10 μM. The compounds with >50% inhibition were rescreened at ATP Km concentrations (30 μM for CDK5/p25 and 65 μM for CDK2/E). At least two independent experiments were performed to generate the average values for the % inhibitions or IC50s (generated from the dose-response curves).
4.3.2. ATP competitive binding assay
ATP competitive binding assays were performed at Reaction Biology Corp. (www.reactionbiology.com)71 according to their standard procedure.
4.3.3. Kinase selectivity assays
The selectivity profile was evaluated against a short and diverse panel of 14 different serine/threonine and tyrosine kinases at ATP-Km concentrations according to the standard protocol of Reaction Biology Corp. (www.reactionbiology.com).71 The % inhibition assays were performed @ 20 μM compound and ATP-Km concentrations. At least two independent experiments were performed to generate the average values.
Supplementary Material
Acknowledgments
This study was supported in part by Grant Number P20GM104931 from the National Institute of General Medical Sciences (NIGMS), a component of the National Institutes of Health (NIH) and its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIGMS or NIH. This investigation was conducted in a facility constructed with support from research facilities improvement program C06RR14503 from the NIH National Center for Research Resources. The authors would like to thank Dr. Bharati Avula for generating the HRMS data. We would also like to thank Professor Ronald F. Borne for careful review of this manuscript. AC would like to acknowledge Dr. David Watson, Dr. Francisco Leon and Dr. Kuldeep K. Roy for their helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Stephen J. Cutler, Email: cutler@olemiss.edu.
John S. Williamson, Email: mcjsw@olemiss.edu.
References and Notes
- 1.Association, Alzheimer’s . Alzheimer’s Dementia. 2013;9:208. doi: 10.1016/j.jalz.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Querfurth HW, LaFerla FM. N Engl J Med. 2010;362:329. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 3.Dimakopoulos AC. Curr Alzheimer Res. 2005;2:19. doi: 10.2174/1567205052772795. [DOI] [PubMed] [Google Scholar]
- 4.Cruz JC, Tsai LH. Trends Mol Med. 2004;10:452. doi: 10.1016/j.molmed.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Lee S, Hall GF, Shea TB. J Alzheimer’s Dis. 2011;26:355. doi: 10.3233/JAD-2011-102016. [DOI] [PubMed] [Google Scholar]
- 6.Maccioni RB, Otth C, Concha II, Munoz JP. Eur J Biochem. 2001;268:1518. doi: 10.1046/j.1432-1033.2001.02024.x. [DOI] [PubMed] [Google Scholar]
- 7.Tarricone C, Dhavan R, Peng J, Areces LB, Tsai LH, Musacchio A. Mol Cell. 2001;8:657. doi: 10.1016/s1097-2765(01)00343-4. [DOI] [PubMed] [Google Scholar]
- 8.Tsai L-H, Delalle I, Caviness VS, Jr, Chae T, Harlow E. Nature. 1994;371:419. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- 9.Jessberger S, Gage FH, Eisch AJ, Lagace DC. Trends Neurosci. 2009;32:575. doi: 10.1016/j.tins.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhavan R, Tsai LH. Nat Rev Mol Cell Biol. 2001;2:749. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- 11.Cheung ZH, Ip NY. Trends Cell Biol. 2012;22:169. doi: 10.1016/j.tcb.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Patzke H, Tsai LH. Trends Neurosci. 2002;25:8. doi: 10.1016/s0166-2236(00)02000-2. [DOI] [PubMed] [Google Scholar]
- 13.Shelton SB, Johnson GVW. J Neurochem. 2004;88:1313. doi: 10.1111/j.1471-4159.2003.02328.x. [DOI] [PubMed] [Google Scholar]
- 14.Wei FY, Nagashima K, Ohshima T, Saheki Y, Lu YF, Matsushita M, Yamada Y, Mikoshiba K, Seino Y, Matsui H, Tomizawa K. Nat Med. 2005;11:1104. doi: 10.1038/nm1299. [DOI] [PubMed] [Google Scholar]
- 15.Feldmann G, Mishra A, Hong SM, Bisht S, Strock CJ, Ball DW, Goggins M, Maitra A, Nelkin BD. Cancer Res. 2010;11:4460. doi: 10.1158/0008-5472.CAN-09-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Booth RJ, Chatterjee A, Malone TC. 2001055148. WO. 2001
- 17.Helal CJ, Kang Z, Lucas JC, Gant T, Ahlijanian MK, Schachter JB, Richter KEG, Cook JM, Menniti FS, Kelly K, Mente S, Pandit J, Hosea N. Bioorg Med Chem Lett. 2009;19:5703. doi: 10.1016/j.bmcl.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 18.Rzasa RM, Kaller MR, Liu G, Magal E, Nguyen T, Osslund TD, Powers D, Santora VJ, Viswanadhan VN, Wang HL, Xiiong X, Zhong W, Norman MH. Bioorg Med Chem. 2007;15:6574. doi: 10.1016/j.bmc.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Ahn JS, Radhakrishnan ML, Mapelli M, Choi S, Tidor B, Cuny GD, Musacchio A, Yeh LA, Kosik KS. Chem Biol. 2005;12:811. doi: 10.1016/j.chembiol.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 20.Mapelli M, Massimiliano L, Crovace C, Seeliger MA, Tsai LH, Meijer L, Musacchio A. J Med Chem. 2005;48:671. doi: 10.1021/jm049323m. [DOI] [PubMed] [Google Scholar]
- 21.Jain P, Flaherty PT, Yi S, Chopra I, Bleasdell G, Lipay J, Ferandin Y, Meijer L, Madura JD. Bioorg Med Chem. 2011;19:359. doi: 10.1016/j.bmc.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 22.Laha JK, Zhang X, Qiao L, Liu M, Chatterjee S, Robinson S, Kosik KS, Cuny GD. Bioorg Med Chem Lett. 2011;21:2098. doi: 10.1016/j.bmcl.2011.01.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nair N, Kudo W, Smith MA, Abrol R, Goddard WA, III, Reddy VP. Bioorg Med Chem Lett. 2011;21:3957. doi: 10.1016/j.bmcl.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 24.Andricopulo AD, Salum LB, Abraham DJ. Curr Top Med Chem. 2009;9:771. doi: 10.2174/156802609789207127. [DOI] [PubMed] [Google Scholar]
- 25.Lyne PD. Drug Discovery Today. 2002;7:1047. doi: 10.1016/s1359-6446(02)02483-2. [DOI] [PubMed] [Google Scholar]
- 26.Haq ZU, Uddin R, Wai LK, Wadood A, Lajis NH. J Mol Model. 2011;17:1149. doi: 10.1007/s00894-010-0817-2. [DOI] [PubMed] [Google Scholar]
- 27.Shiradkar MR, Padhalingappa MB, Bhetalabhotala S, Akula KV, Tupe DA, Pinninti RR, Thummanagoti S. Bioorg Med Chem. 2007;15:6397. doi: 10.1016/j.bmc.2007.06.053. [DOI] [PubMed] [Google Scholar]
- 28.Pitchuanchom S, Boonyarat C, Forli S, Olson AJ, Yenjai C. Comput Biol Med. 2012;42:106. doi: 10.1016/j.compbiomed.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 29.Loving K, Salam NK, Sherman W. J Comput-Aided Mol Des. 2009;23:541. doi: 10.1007/s10822-009-9268-1. [DOI] [PubMed] [Google Scholar]
- 30.Salam NK, Nuti R, Sherman W. J Chem Inf Model. 2009;49:2356. doi: 10.1021/ci900212v. [DOI] [PubMed] [Google Scholar]
- 31.de Beer SBA, Vermeulen NPE, Oostenbrink C. Curr Top Med Chem. 2010;10:55. doi: 10.2174/156802610790232288. [DOI] [PubMed] [Google Scholar]
- 32.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. J Comput-Aided Mol Des. 2013;27:221. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 33.Willett P, Barnard JM, Downs GMD. J Chem Inf Model. 1998;38:983. [Google Scholar]
- 34.Sastry M, Lowrie JF, Dixon SL, Sherman W. J Chem Inf Model. 2010;50:771. doi: 10.1021/ci100062n. [DOI] [PubMed] [Google Scholar]
- 35.Anderson M, Beattie JF, Breault GA, Breed J, Byth KF, Culshaw JD, Ellston RPA, Green S, Minshull CA, Norman RA, Pauptit RA, Stanway J, Thomas AP, Jewsbury PJ. Bioorg Med Chem Lett. 2003;13:3021. doi: 10.1016/s0960-894x(03)00638-3. [DOI] [PubMed] [Google Scholar]
- 36.de Azevedo WF, Jr, Leclerc S, Meijer L, Havlicek L, Strnad M, Kim S-H. Eur J Biochem. 1997;243:518. doi: 10.1111/j.1432-1033.1997.0518a.x. [DOI] [PubMed] [Google Scholar]
- 37.Jones CD, Andrews DM, Barker AJ, Blades K, Daunt P, East S, Geh C, Graham MA, Johnson KM, Loddicka SA, McFarland HM, McGregor A, Moss L, Rudge DA, Simpson PB, Swain ML, Tam KY, Tucker JA, Walker M. Bioorg Med Chem Lett. 2008;18:6369. doi: 10.1016/j.bmcl.2008.10.102. [DOI] [PubMed] [Google Scholar]
- 38.Lawrie AM, Noble MEM, Tunnah P, Brown NR, Johnson LN, Endicott JA. Nat Struct Mol Biol. 1997;4:796. doi: 10.1038/nsb1097-796. [DOI] [PubMed] [Google Scholar]
- 39.Luk KC, Simcox ME, Schutt A, Rowan K, Thompson T, Chen Y, Kammlott U, DePinto W, Dunten P, Dermatakis A. Bioorg Med Chem Lett. 2004;14:913. doi: 10.1016/j.bmcl.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 40.Wyatt PG, Woodhead AJ, Berdini V, Boulstridge JA, Carr MG, Cross DM, Davis DJ, Devine LA, Early TR, Feltell RE, Lewis EJ, Mcmenamin RL, Navarro EF, O’Brien MA, O’Reilly M, Reule M, Saxty G, Seavers LCA, Smith D, Squires MS, Trewartha G, Walker MT, Woolford AJ. J Med Chem. 2008;51:4986. doi: 10.1021/jm800382h. [DOI] [PubMed] [Google Scholar]
- 41.Maestro, version 9.2. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- 42.Phase, version 3.3. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- 43.Gompel M, Leost M, De Kier Joffe EB, Puricelli L, Franco LH, Palermo J, Meijer L. Bioorg Med Chem Lett. 2004;14:1703. doi: 10.1016/j.bmcl.2004.01.050. [DOI] [PubMed] [Google Scholar]
- 44.Helal CJ, Sanner MA, Cooper CB, Gant T, Adam M, Lucas JC, Kang Z, Kupchinsky S, Ahlijanian MK, Tate B, Menniti FS, Kelly K, Peterson JR. Bioorg Med Chem Lett. 2004;14:5521. doi: 10.1016/j.bmcl.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 45.Larsen SD, Stachew CF, Clare PM, Cubbage Jw, Leach KL. Bioorg Med Chem Lett. 2003;13:3491. doi: 10.1016/s0960-894x(03)00726-1. [DOI] [PubMed] [Google Scholar]
- 46.Ortega MA, Montoya ME, Zarranz B, Jaso A, Aldana I, Leclerc S, Meijer L, Monge A. Bioorg Med Chem Lett. 2002;10:2177. doi: 10.1016/s0968-0896(02)00069-x. [DOI] [PubMed] [Google Scholar]
- 47.Polychronopoulos P, Magiatis P, Skaltsounis AL, Myrianthopoulos V, Mikros E, Tarricone A, Musacchio A, Roe SM, Pearl L, Leost M, Greengard P, Meijer L. J Med Chem. 2004;47:935. doi: 10.1021/jm031016d. [DOI] [PubMed] [Google Scholar]
- 48.Meijer L, Borgne A, Mulner O, Chong JPJ, Blow J, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP. Eur J Biochem. 1997;243:527. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 49.Kaller MR, Zhong W, Henley C, Magal E, Nguyen T, Powers D, Rzasa RM, Wang W, Xiong X, Norman MH. Bioorg Med Chem Lett. 2009;19:6591. doi: 10.1016/j.bmcl.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 50.Metty Y, Gompel M, Thomas V, Garnier M, Leost M, Ceballos-Picot I, Noble M, Endicott J, Vierfond J-m, Meijer L. J Med Chem. 2003;46:222. doi: 10.1021/jm020319p. [DOI] [PubMed] [Google Scholar]
- 51.Shiradkar M, Akula KV, Dasari V, Baru V, Chiningiri B, Gandhi S, Kaur R. Bioorg Med Chem. 2007;15:2601. doi: 10.1016/j.bmc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 52.Zhong W, Liu H, Kaller MR, Henley C, Magal E, Nguyen T, Osslund TD, Powers D, Rzasa RM, Wang HL, Wang W, Xiiong X, Zhang J, Norman MH. Bioorg Med Chem Lett. 2007;17:5384. doi: 10.1016/j.bmcl.2007.07.045. [DOI] [PubMed] [Google Scholar]
- 53.Becknell NC, Hudkins RL. 20070299061. US. 2007
- 54.Ip NY-Y, Ip FC-F, Fu WY, Fu G. 2011/069334. WO. 2011
- 55.Luo Y, Shu F, Wang S. 2010/051781. WO. 2010
- 56.Machacek MR, Ahearn S, Romeo PE, Siu T, Chichetti S, De Almeida G, Rivkin A. 2011/037780. WO. 2011
- 57.Routier S, César Léonce Guillaumet G, Boulahjar R, Meijer L, Chiurato M. 2010/103240. WO. 2010
- 58.Shiradkar M, Thomas J, Kanase V, Dighe R. Eur J Med Chem. 2011;46:2066. doi: 10.1016/j.ejmech.2011.02.059. [DOI] [PubMed] [Google Scholar]
- 59.Siu T, Dinsmore C, Kumarasinghe SE. 2011/049722. WO. 2011
- 60.Glide, version 5.7. Schrödinger, LLC; New York, NY: [Google Scholar]
- 61.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J Med Chem. 2006;49:6177. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 62.CAC. 2010 Database of commercially available compounds. Schrodinger, LLC; New York, NY: 2011. [Google Scholar]
- 63.LigPrep, version 2.5. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- 64.QikProp, version 3.4. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- 65.Dixon SL, Smondyrev AM, Rao SN. Chem Biol Drug Des. 2006;67:370. doi: 10.1111/j.1747-0285.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- 66.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shaw DE, Shelley M, Perry JK, Francis P, Shenkin PS. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 67.Duan J, Dixon SL, Lowrie JF, Sherman W. J Mol Graphics Modell. 2010;29:157. doi: 10.1016/j.jmgm.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 68.Canvas, version 1.4. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- 69.The PyMOL Molecular Graphics System, Version 1.5.0.4. Schrödinger, LLC; [Google Scholar]
- 70.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Nat Biotechnol. 2011;29:1039. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reaction Biology Corp; Malvern, PA 19355: ( www.reactionbiology.com) [Google Scholar]
- 72.Hopkins AL, Groom CR, Alex A. Drug Discovery Today. 2004;9:430. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 73.Ibrahim PN, Cho H, England B, Gillette S, Artis DR, Zuckerman R, Zhang C. 20060041006. US. 2006
- 74.Chatterjee A, Cutler S, Khan I, Williamson J. Mol Diversity. 2014;18:51. doi: 10.1007/s11030-013-9476-4. [DOI] [PubMed] [Google Scholar]
- 75.Schrödinger Suite 2011 Protein Preparation Wizard; Epik version 2.2. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]; Impact version 5.7. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]; Prime version 3.0. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- 76.Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. J Comput-Aided Mol Des. 2006;20:647. doi: 10.1007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.