

Abstract
Membrane proteins, especially G-protein coupled receptors (GPCRs), are interesting and important theragnostic targets since many of them serve in intracellular signaling critical for all aspects of health and disease. The potential utility of designed bivalent ligands as targeting agents for cancer diagnosis and/or therapy can be evaluated by determining their binding to the corresponding receptors. As proof of concept, GPCR cell surface proteins are shown to be targeted specifically using multivalent ligands. We designed, synthesized, and tested a series of bivalent ligands targeting the over-expressed human melanocortin 4 receptor (hMC4R) in human embryonic kidney (HEK) 293 cells. Based on our data suggesting an optimal linker length of 25±10 Å inferred from the bivalent melanocyte stimulating hormone (MSH) agonist, the truncated heptapeptide, referred to as MSH(7): Ac-Ser-Nle-Glu-His-D-Phe-Arg-Trp-NH2 was used to construct a set of bivalent ligands incorporating a hMC4R antagonist, SHU9119: Ac-Nle-c[Asp-His-2′-D-Nal-Arg-Trp-Lys]-NH2 and another set of bivalent ligands containing the SHU9119 antagonist pharmacophore on both side of the optimized linkers. These two binding motifs within the bivalent constructs were conjoined by semi-rigid (Pro-Gly)3 units with or without the flexible poly(ethylene glycol) (PEGO) moieties. Lanthanide-based competitive binding assays showed bivalent ligands binds to the hMC4R with up to 240-fold higher affinity than the corresponding linked monovalent ligands.
Keywords: Bivalent ligands, linkers, melanorcortin receptors, Eu-binding assay, solid phase synthesis
1. Introduction
Cancer is in its most aggressive state when it has metastasized to the entire body. Hence early detection is critical to the successful treatment of many human cancers. Therapies to treat cancer must selectively target these invading cells within healthy normal tissues. It is accepted that metastatic cancers include multiple genetic abnormalities that are currently targets for many bio-pharmaceutical companies. Most current drug therapies are not molecularly specific and are associated with side-effects and toxicities. There is a possibility of individually developing single molecules containing multiple pharmacophores for specific overexpressed proteins on a cancer cell that may have a differentially reduced expression on the normal cell.1 These resulting multivalent molecules could display enhanced affinity for the targeted cells.2-4 Targeting cell surface receptors can help inhibit cell surface receptor-ligand interactions or act as positive / negative effectors of downstream signal transduction. Multimeric ligands that contain a target specific agonist or antagonist pharmacophore can take toxic pay-loads directly into the tumors, destroying the cancer cells, while leaving the normal cells unharmed. An imaging agent on the multimeric ligand will be guided as a single molecule that will potently bind and image the infected area (overexpressed receptors) and can be used in non-invasive techniques for cancer detection e.g. early detection of adenomatous polyps in colon cancer.
The common feature of the bivalent ligand binding is that following the initial binding of one pharmacophore within a bivalent construct, succeeding binding is more favorable thanks to decreased loss of entropy.5 As the result, the bivalent ligands can enhance binding affinity, agonist / antagonist potency and GPCR subtype selectivity. The central dogma of GPCR pharmacology has been the concept that unlike agonists, antagonist ligands display equivalent affinities for a given receptor, regardless of the cellular environment in which the affinity is assayed.6
As a proof of the concept, we chose to mimic the cancer cells through overexpression of MC4 receptor, which is one of five types of melanocortin receptors termed MC1–5R that exhibit about 40–60% homology.7 Among the melanocortin receptors, the MC4R is of particular interest and a potential target in the research study as a major regulator of eating behavior and body weight, and suggestions have been made towards its role in the stimulation of male erectile activity.8,9 Synthetically low molecular weight agonists and antagonists selective for the different subtypes are highly warranted as remedies for treatment of various dysfunctional states, such as obesity, anorexia, impotence, and autoimmune disorders.10 Studies have suggested that bivalent ligands have receptor binding properties that differ substantially from those of the monovalent ligand, and the spacer used to link the two pharmacophores within the construct exerts a profound influence on the potency. The bivalent ACTH antagonists show potency enhancements up to approximately 25 times that of the monovalent constructs also demonstrating the role of spacer [bis(maleimide)cross-linking] and a peptide pharmacophore component within a bivalent construct.11 Handl et al. demonstrated the potential of a series of MSH-7 agonist homobivalent ligands compared to its monovalent construct that can be utilized as targeting agents for cancer imaging.3 The homobivalent ligands binds to hMC4R with increased affinity and apparent co-operativity compared to their monovalent analogues.3 The increased binding affinity and positive cooperativity were most likely not due to statistical binding, but rather to a receptor clustering mechanism, wherein multiple receptors are bound by the same multivalent ligand.12 In this study, we used a combination of agonist and antagonist pharmacophores in the design of bivalent ligands and the results could help determine organizational features of the melanocortin receptor-GPCR. We chose to construct ligands containing one copy of MSH(7), a truncated version of [Nle4-D-Phe7]-α-melanocyte stimulating hormone (NDP-α-MSH) and a very potent cyclic MC4R antagonist SHU9119.13 These two MC4R pharmacophores were separated by a series of linkers, which are different in flexibility and length. Poly(ethylene glycol) (PEGO) and (Pro-Gly)3 units were used either by themselves or by incorporations, as shown in Table 1.
Table 1. Analytical data of monovalent and bivalent ligands for hMC4R.
| liganda | molecular formula | no of atoms present in the linker | Estimated linker length (Å) | MSb | HPLCc (tR, min) | ||
|---|---|---|---|---|---|---|---|
| calcd | observed | ||||||
| 1 |
|
C96 H131 N27 O21 | 36 | 10-20 | 1999.2 | 2000.1a | 11.9 |
| 2 |
|
C117 H161 N33 O27 | 54 | 15-30 | 2460.2 | 2461.2a | 11.9 |
| 3 |
|
C140 H196 N39 O33 | 72 | 20-40 | 2922.5 | 2924.1a | 11.9 |
| 4 |
|
C103 H153 N25 O27 | 58 | 13-46 | 2173.5 | 2174.4a | 12.3 |
| 5 |
|
C68 H97 N17 O15 | 20 | 4-18 | 1392.5 | 1393.6b | 12.5 |
| 6 |
|
C82 H123 N19 O21 | 40 | 8-36 | 1709.9 | 1710.6b | 12.5 |
| 7 |
|
C148 H197 N41 O29 | 36 | 10-20 | 3014.4 | 3016.4a | N.D |
| 8 |
|
C169 H227 N47 O35 | 54 | 15-30 | 3476.9 | 3478.5a | 13.2 |
| 9 |
|
C194 H267 N53 O41 | 72 | 20-40 | 3997.5 | 3940.9a | 13.1 |
| 10 |
|
C155 H219 N39 O35 | 58 | 13-46 | 3186.6 | 3188.1a | 13.6 |
| 11 |
|
C120 H163 N31 O23 | 20 | 4-18 | 2407.7 | 2407.3a | 14.0 |
| 12 |
|
C134 H189 N33 O29 | 40 | 8-36 | 2726.1 | 2727.0a | 13.9 |
| 13 |
|
C144 H197 N40 O31 | 36 | 10-20 | 2955.3 | 2955.0a | 12.0 |
| 14 |
|
C165 H227 N46 O37 | 54 | 15-30 | 3417.0 | 3417.0a | 12.0 |
| 15 |
|
C186 H257 N52 O43 | 72 | 20-40 | 3880.2 | 3880.7a | 12.0 |
| 16 |
|
C151 H219 N38 O37 | 58 | 13-46 | 3129.4 | 3129.2a | 12.2 |
| 17 |
|
C116 H163 N30 O25 | 20 | 4-18 | 2348.6 | 2348.6a | 12.4 |
| 18 |
|
C130 H189 N32 O31 | 40 | 8-36 | 2665.4 | 2666.8a | 12.5 |
 - SHU9119;
- SHU9119;
 - MSH(7);
- MSH(7);
 - PEGO moiety;
- PEGO moiety;
 - (Pro-Gly)3
- (Pro-Gly)3
N-terminus: acetylated; C-terminus: amidated.
(M + H)+, ESI method (Finnigan, Thermoelectron, LCQ classic).
Performed on a Waters Alliance 2695 HPLC using a reverse-phase column (Jupiter 5U C18 300A; 2.2 × 2.5 cm) in gradient system (10-40% of acetonitrile containing 0.1% TFA within 30 min, 1 mL/min).
It has been proposed that the first pharmacophore binding event serves to attach the multivalent ligand to the surface, here we have evaluated the use of a tight binding pharmacophore SHU9119 in combination with a comparatively lower binding pharmacophore, MSH(7).14,15 We proposed that there would be effectively an additive enhancement of binding compared to homobivalent MSH(7) analogues, which we have shown in a previous publication, as the pharmacophore SHU9119 should bind to the receptor tightly and then linkers should provide greater opportunity for the bivalent ligand to explore more volume and thus have a higher probability to bind multiple receptors at once, hence making them capable of cross-linking adjacent receptors.3
2. Results and Discussion
2.1. Synthesis
As shown in Figure 1, bivalent ligands 7-12 and 13-18 consisting of two SHU9119 moieties and MSH(7) and SHU9119, respectively, with PEGO and/or (Pro-Gly)3 linkers were synthesized by standard solid phase synthesis using Fmoc-chemistry successfully. Monovalent ligands 1-6 were also prepared as control ligands by the same procedure.
Figure 1.

Preparation of monovalent and bivalent ligands. Reagents and conditions: (a) 1:1 or 1:4 piperidine in DMF; (b) Standard solid phase synthesis using Fmoc-chemistry; (c) PEGO attachment (Ref. 3); (d) Ac2O/pyridine (50/50); (f) TFA/EDT/thioanisole/water (91/3/3/3).
The cyclized heptapeptide SHU9119 was constructed on Rink amide Tentagel S resin and PEGO linkers were attached to the resin. The PEGO attached resin was proportionally split for syntheses of control monovalent ligands 4-6, bivalent ligands 10-12, and 16-18. For the synthesis of ligands 11 and 12, the split resin was coupled with Fmoc-Lys(Alloc)-OH and the solid phase peptide synthesis continued to complete the second SHU9119 sequence. Subsequently, part of the split resin was coupled with Fmoc-amino acids stepwise to attach the MSH(7) moiety for ligands 17-18. Attachment of SHU9119 or MSH(7) moiety in bivalent or monovalent ligands were performed routinely without difficulty by the procedures as mentioned above. Another portion of the resin was used to connect with (Pro-Gly)3 and PEGO linkers and SHU9119 or MSH(7) was attached to the resulting resin to afford longer length bivalent ligands 10 and 16. The resin was previously split into three syringe reactor portions for the attachment of Pro-Gly linkers, giving monovalent ligand resins 1-3. The attachment of the second moiety SHU9119 or MSH(7) was carried out, affording 7-9 or 13-15, respectively. The Fmoc-groups on all of the resin precursors were deprotected, and peptides were acylated, and cleaved by a mixture of TFA, EDT, thioanisole, and water (91/3/3/3) that afforded the desired ligands 1-18 as shown in Table 1. Ligands 1- 18 were purified by preparative RP-HPLC and were characterized by ESI-MS and/or MALDI-TOF MS to confirm their structures.
2.2. Binding of monovalent and bivalent ligands
The binding affinities were evaluated in a lanthanide based competitive assay (Dissociation Enhanced Lanthanide FluoroImmuno Assay: DELFIA) using optimized 10 nM standard agonist Eu-DTPA-NDP-α-MSH chelate in HEK293 cells overexpressing hMC4R. As shown in Table 2, EC50 values were calculated after computing the hill slope using the GraphPad Prism software and compared with ligands MSH(7) with EC50 ∼50 nM and SHU9119 with EC50 ∼60 pM; these binding affinities were consistent with the ones obtained from previously published data using radiolabeled binding assay.16
Table 2. Binding affinities of monovalent and bivalent ligands for hMC4R.
| ligand | Structure | aLogEC50 | bEC50 (nM) | Hill coefficient |
|---|---|---|---|---|
| 1 | Ac-(Pro-Gly)6-SHU9119-NH2 | -8.40 ± 0.06 | 4.0 | 1.1 |
| 2 | Ac-(Pro-Gly)9-SHU9119-NH2 | -8.09 ± 0.05 | 8.1 | 0.8 |
| 3 | Ac-(Pro-Gly)12-SHU9119-NH2 | -7.88 ± 0.17 | 13 | 1.1 |
| 4 | Ac-PEGO-(Pro-Gly)3-PEGO-SHU9119-NH2 | -8.43 ± 0.06 | 3.7 | 0.9 |
| 5 | Ac- PEGO-SHU9119-NH2 | -7.60 ± 0.51 | 24 | 0.6 |
| 6 | Ac- PEGO-PEGO-SHU9119-NH2 | -6.39 ± 0.46 | 400 | 0.8 |
| 7 | Ac-SHU9119-(Pro-Gly)6-SHU9119-NH2 | N.D | N.D | - |
| 8 | Ac-SHU9119-(Pro-Gly)9-SHU9119-NH2 | -8.81 ± 0.02 | 1.6 | 2.2 |
| 9 | Ac-SHU9119-(Pro-Gly)12-SHU9119-NH2 | -8.71 ± 0.02 | 2.0 | 3.0 |
| 10 | Ac-SHU9119-PEGO-(Pro-Gly)3-PEGO-SHU9119-NH2 | -8.67 ± 0.02 | 2.2 | 1.9 |
| 11 | Ac-SHU9119-PEGO-SHU9119-NH2 | -8.74 ± 0.06 | 1.8 | 2.2 |
| 12 | Ac-SHU9119-PEGO-PEGO-SHU9119-NH2 | -8.73 ± 0.02 | 1.9 | 1.9 |
| 13 | Ac-MSH(7)-(Pro-Gly)6-SHU9119-NH2 | -8.39 ± 0.12 | 4.1 | 0.7 |
| 14 | Ac-MSH(7)-(Pro-Gly)9-SHU9119-NH2 | -8.71 ± 0.08 | 2.0 | 0.8 |
| 15 | Ac-MSH(7)-(Pro-Gly)12-SHU9119-NH2 | -8.80 ± 0.04 | 1.6 | 2.0 |
| 16 | Ac-MSH(7)-PEGO-(Pro-Gly)3-PEGO-SHU9119-NH2 | -8.86 ± 0.03 | 1.3 | 2.4 |
| 17 | Ac-MSH(7)-PEGO-SHU9119-NH2 | -8.73 ± 0.02 | 1.4 | 2.7 |
| 18 | Ac-MSH(7)-PEGO-PEGO-SHU9119-NH2 | -8.77 ± 0.02 | 1.7 | 2.7 |
| 19 | Ac-(Pro-Gly)6- MSH(7)-NH2 | 82 ± 12c | ||
| 20 | Ac-(Pro-Gly)9- MSH(7)-NH2 | 236 ± 24c | ||
| 21 | Ac-(Pro-Gly)12- MSH(7)-NH2 | 188 ± 25c | ||
| 22 | Ac-MSH(7)-(Pro-Gly)6- MSH(7)-NH2 | 11 ± 2c |
The logEC50 ± SEM are logarithmic values determined from the nonlinear regression analysis of data collected from 4 independent concentration range.
Determined from Dissociation Enhanced Lanthanide FluoroImmuno Assay (DELFIA) using optimized 10 nM standard agonist Eu-DTPA-NDP-α-MSH chelate.
see reference 3
The conjugation of the linkers to the monovalent antagonist-SHU9119 reduced its high affinity (EC50 = 59 pM) up to 400 nM by 6800-fold in ligand 6. As the different lengths of linkers were attached to the antagonist pharmacophore, there was a trend observed in binding to the receptor. Apparently, the analogues containing the semi rigid (Pro-Gly)3 linker retained higher binding affinity than those with the flexible linker PEGO, and longer length of PEGO linker resulted in the loss of binding affinity (EC50 = 400 nM) in ligand 6. However, the low affinity was reversed to high affinity (EC50 = 3.7 nM, 108-fold) by the insertion of a semi rigid linker (Pro-Gly)3 in ligand 4. Ligand 1 with a shorter length of linker (10-20 Å) showed higher binding affinity (EC50 = 4.0 nM) than ligands 2 (EC50 = 8.1 nM) and 3 (EC50 = 13 nM) with a longer length of linker. This is coincident with our previous result showing linker effects on the binding affinities of MSH(7) monovalent ligands.3 It is clear that the attachment of a semi rigid (Pro-Gly)3 linker, which may assist in reducing entropy of the ligand, results in the high binding affinity of the monovalent ligands.
Contrary to the effect of a linker on monovalent ligands 1-6, there was no such clear effect observed on the bivalent ligands 8-18. Interestingly, bivalent ligands 12 and 18, which contain a flexible longer linker PEGO-PEGO like monovalent ligand 6 (EC50 = 400 nM) retained the same high binding affinities (EC50 = 1.9 nM and 1.7 nM, respectively) as the other bivalent ligands. The increases of binding affinities in 12 and 18 were more than 200-fold relative to the monovalent ligand. It may not be simple to explain how the flexible linkers did not cause disturbances of the binding affinities in the bivalent ligands, but it is possible that the attachment of the second pharmacophore help in reducing the effects of flexibility of the linker but increasing cooperative effects between two pharmacophores.
Use of the optimized linker of 15 – 30 Å, obtained from our previous experiments, fixed the translational and rotational entropies associated with the linker types for the hMC4R.3 The incorporation of a potent antagonist SHU9119 with the optimized length of a linker (Pro-Gly)9 to monovalent ligand 2 (EC50 = 8.1 nM) improved the binding affinity slightly in bivalent ligands 8 (EC50 = 1.6 nM, 5-fold) and 14 (EC50 = 2.0 nM, 4-fold) (Figure 3).
Figure 3. Comparison of the binding of monovalent ligand 2 and bivalent ligand 8 to hMC4R.

In general, the homo-bivalent ligands (8-11) and hetero-bivalent ligands (13-17) bound with slightly increased (2-13 fold) affinity for the receptor compared to the monovalent linker-SHU9119 analogues (1-5). Interestingly, all of the homo- and hetero-bivalent ligands showed the same high range of binding affinities regardless of their linkers and pharmacophores. This result may be explained simply by the first tight binding of SHU9119 that can assist crosslinking of the second pharmacophore in the vicinity. The increase of binding affinities in hetero-bivalent ligands (13-15) was shown to be more pronounced (20-118 fold) comparing to the monovalent linker MSH(7) analogues (19-21). Consistent with the results obtained in previous works, it is clear that the more pronounced enhancements can be achieved with lower affinity pharmacophores in bivalent ligands.17
The enhanced binding affinity of the bivalent ligands is attributed to apparent cooperativity. Evaluation of the Hill coefficients resulting from the monovalent and bivalent ligand bindings confirms that the bivalent ligands bind with a cooperative effect. The obtained Hill coefficient for monovalent analogues on an average was less than 1 (0.88) while for the bivalent analogues, the average was greater than 2, even with exceptionally low coefficients for mixed bivalent ligands 13 and 14. These low Hill coefficients (0.7 and 0.8 for 13 and 14, respectively) may indicate that there is no cooperativity between the two pharmacohpores due to the short and rigid linkers. Comparing to ligands 13 and 14, ligand 15 possess higher Hill coefficient (2.0), which could assist in the increase of binding affinity (EC50 = 1.6 nM, 8-fold). It was observed that bivalent ligands with higher Hill coefficient showed much more increased binding affinity than those with lower Hill coefficient. The enhanced Hill coefficient that arises from the transition from monovalent binding to bivalent binding suggested that the additional binding motif increased the probability that the bivalent ligand will find and bind more tightly to its corresponding receptor or due to increased local concentration of subsequent ligands at receptor sites.18 We have concluded the improved likelihood that multivalent ligands simultaneously cross-link receptors through combination of a low affinity pharmacophore and a high affinity pharmacophore.
3. Experimental Procedures
3.1. Materials
Nα-Fmoc-amino acids were purchased from SynPep (Dublin, CA) or from Novabiochem (San Diego, CA). Rink amide Tentagel S resin was acquired from Rapp Polymere (Tubingen, Germany). HCTU, HOBt, and HOCt were purchased from IRIS Biotech (Marktredwitz, Germany). Solvents and reagents were reagent grade and were used without further purification unless otherwise noted. The solid-phase synthesis was performed manually in fritted syringes purchased from Torviq (Niles, MI). Purification of ligands was achieved on a Waters 600 HPLC using a reverse-phase column (Vydac C-18, 15-20 μm, 22 × 250 mm). The purity of the ligands was checked by analytical PR-HPLC using a Waters Alliance 2695 separation model with a Waters 2487 dual wavelength detector (220 and 280 nm) on a reverse-phase column (Jupiter 5U C18 300A; 2.2 × 2.5 cm). The mass of the ligands was confirmed by ESI method (Finnigan, Thermoelectron, LCQ classic).
3.2. Solid-Phase Synthesis
The Tentagel Rink amide resin (0.22 mmol/g) was washed with DMF 3 times and the Nα-Fmoc group was removed with 50% or 25% piperidine in DMF for 2 min or 20 min, respectively. The resin was washed successively with DMF, DCM, DMF, and then a solution of 1.0 M HOBt in DMF, and DMF. Nα-Fmoc amino acid was coupled using preactivated 0.3 M HOBt esters in THF (3 eq. of Nα-Fmoc-amino acid, 3 eq. of HOBt and 3 eq. of DIEA) for 2 h and confirmed by Kaiser test (negative). If the coupling was not completed during 2 h (positive), the resin was washed with DMF and coupled again by the HCTU/ 2,4,6-lutidine procedure (3 eq. of Nα-Fmoc-amino acid, 3 eq. of HCTU, and 6 eq. of 2,4,6-lutidine in DMF) for 3 h or by preformed symmetric anhydride (3 eq. of Nα-Fmoc-amino acid and 1.5 eq. of DIC in DMF/DCM (1/1)) until the Kaiser test was negative. If the second coupling was not completed, the resin was washed with DMF, and the free amino group was capped with 50% Ac2O in pyridine for 10 min.
After sequential couplings of Lys(N-Alloc), Trp(Ni-Boc), Arg(Ng-Pbf), 2′-D-Nal, His(Nim-Trt), and Asp(O-Allyl), the Alloc and Allyl groups were orthogonally deprotected from Lys and Asp side chains, respectively, using Pd(0) chemistry without cleaving the terminal Fmoc group on Asp(O-Allyl). 3 eq. of palladium tetrakis-triphenyl phosphine [Pd(PPh3)4] and CHCl3/AcOH/N-methylmorpholine (37:2:1) (15 mL/g resin) was mixed under Ar.19 The catalyst was dissolved by bubbling a stream of Ar through the solution and immediately pulled up by the syringe containing the dried tentagel resin. The reaction mixture was put on the shaker and the reaction carried out for 2 h. The palladium-based reaction mixture was then drained and the resin was washed thoroughly with 3 eq. DIEA in DMF, 3 eq. sodium di-thiocarbamate in DMF, and 3 eq. HOBt in DMF. The resin was then subjected to DMF/ DCM washes to prepare the resin for lactam cyclization under microwave condition. The lactam cyclization was carried out using 3 eq. DIEA/ 3 eq. HOBt/ 3 eq. HBTU in DMF (conventional microwave; 4 sec; 4 times) until the syringe got hot; the syringe was vortexed to dissipate the heat in-between individual microwave heating. The solution changed color from pale-yellow to light orange indicating the use of the reactants and the completion of reaction. After routine DMF/DCM washing, the Fmoc-group was deprotected for next coupling with Nα-Fmoc-Nle-OH.
Glycine and Proline were attached alternatively as many times as needed to synthesize the synthetic ligands 1-3 (Figure 1). Assembly of the second peptide, SHU9119 or MSH(7), in conjunction with the Pro-Gly linker, was carried out by the solid phase peptide synthesis procedure described above to afford ligands 7-9 or ligands 13-15. Similarly, for the synthesis of ligands 5 and 6, the flexible PEGO linker was attached by first adding diglycolic anhydride and then activating the free carboxylate as an imidazole for the attachment of 4,7,10-trioxa-1,13-tridecanediamine.20 In synthetic ligands 4, 10, and 16, the PEGO was followed by repetitive Pro-Gly linker units in a sequential manner. Another PEGO attachment followed by the second SHU9119 or MSH(7) and the N-terminal acetylation, yielded resin bound protected precursors of ligands 11-12 or ligands 17-18. The resin was cleaved with a cleavage cocktail (10 mL/g resin) consisting of TFA, EDT, thioanisole, and water (91:3:3:3) for 3 h at room temperature. The solution was filtered off and the resin was washed with TFA (2 × 3 min). Combined solution was concentrated under N2 and triturated by cold Et2O. The precipitate was washed with cold Et2O three times, dried, and dissolved in water for lyophilization. The lyophilized product was dissolved in water and purified by preparative RP-HPLC and characterized.
3.3. Homogeneity tests of the monovalent, bivalent synthetic peptide analogues
The purity of each peptide was evaluated by analytical RP-HPLC with a 30 min linear gradient of 10% to 40% of acetonitrile containing 0.1% TFA (Column: Jupiter 5U C18 300A; 2.2 × 2.5 cm; flow rate 1 mL/min). The same condition was employed to calculate the average concentration of the peptide ligands by the tryptophan assay. A standard solution (10 μL of 0.5 mM D-Trp in DMSO) was co-injected with 5 μL and 10 μL sample in two separate runs and the absorbance (area under peak) of the sample against D-Trp was measured at 280 nm. The peptide concentration was then calculated by the formula described in our earlier publication.21
3.4. Cell Culture
HEK293 cells overexpressing the hMC4R were used for binding assay. The hMC4R vector was originally received from Dr. Ira Gantz, University of Michigan.22 The coding region of the hMC4R gene was expressed in pcDNA3.1 (Invitrogen, V790-20). HEK293/hMC4R cells were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% FBS.
3.5. Lanthanide Based Binding Assays
Lanthanide based competitive binding assays were conducted according to the method which has been previously described.23 In brief, HEK293/hMC4R cells were plated in black and white 96-well isoplates (Wallac, 1450-584) at a density 12,000 cells/well and were allowed to grow for 3 days. On the day of the experiment, media was aspirated from all wells. 50 μL of non-labeled ligand and 50 μL of Eu-labeled ligand (final concentration of 10 nM for Eu-NDP-α-MSH) were added to each well. Ligands were diluted in binding media (DMEM, 1 mM 1, 10-Phenanthroline, 200 mg/L Bacitracin, 0.5 mg/L Leupeptin, 0.3% BSA) and samples were tested in quadruplicate, unless otherwise noted. Cells were incubated in the presence of ligand for 1 h at 37 °C. Following the incubation, cells were washed 3 times with 250 μL wash buffer (50 mM Tris-HCl, 0.2% BSA, 30 mM NaCl). Enhancement solution (Perkin Elmer; 1244-105) was added (100 μL/well) and the plate was incubated for at least 30 min at 37 °C prior to reading. The plates were read on a Wallac VICTOR3 instrument using the standard Eu TRF measurement (340 nm excitation, 400 μsec delay, and emission collection for 400 μsec at 615 nm).
3.6. Data Analysis
Data from independent binding experiments were analyzed with GraphPad Prism Software using the sigmoidal dose-response (variable slope) classical equation for non-linear regression analysis. The dose response curves obtained were used to compute the Hill coefficients for the individual synthetic ligands and their corresponding EC50 values using the above mentioned lanthanide-based (Eu-NDP-α-MSH) competition binding assay.
Figure 2.
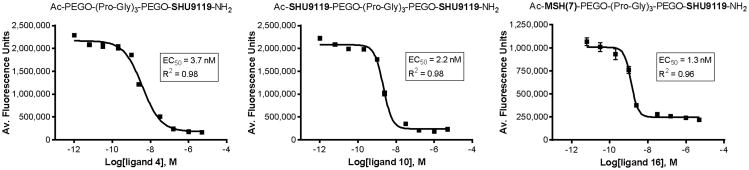
Representative binding curves resulting from a typical competitive binding assay for monovalent ligand 4 (left), bivalent ligands 10 (middle) and 16 (right). Increasing concentration of ligands were competed using 10 nM Eu-α-NDP-MSH and hMC4R. Each data point represents the average of quadruplicate sample wells, with error bars indicating the standard error of means.
Acknowledgments
This work was supported in part by grants from the U.S. Public Health Service, National Institute of Health R01 (CA097360) and R01 (CA108040). We thank Dr. Liping Xu and Kathy Brown for the hMC4R cell line.
Abbreviations
- Ac
acetyl
- ACTH
adrenocorticotropic hormone
- Alloc
allyloxycarbonyl
- Boc
t-butoxycarbonyl
- BSA
bovine serum albumin
- DIC
N,N′,-diisopropylcarbodiimide
- DIEA
diisopropylethylamine
- DMEM
Dulbecco's modified eagle medium
- DMF
N,N-dimethylformamide
- DMSO
dimethylsulfoxide
- EDT
ethanedithiol
- Fmoc
9-fluorenylcarboxy
- HCTU
2-(6-chloro-1H-benzotrialzole-1-yl)-1,1,3,3,-tetramethyluronium hexafluorophosphate
- HBTU
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate
- HEK
human embryonic kidney
- HOBt
N-hydroxybenzotriazole
- HOCt
- hMC4R
human melanocortin 4 receptor
- MSH
melanocyte stimulating hormone
- Nal
naphthylalanine
- Pbf
2,2,4,6,7-pentamethlydihydro-benzofuran-5-sulfonyl
- PEGO
poly(ethylene glycol)
- RP-HPLC
reverse phase high performance liquid chromatography
- TFA
trifluoroacetic acid
- Trt
trityl
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gillies RJ, Hruby VJ. Expert Opin Ther Targets. 2003;7:137. doi: 10.1517/14728222.7.2.137. [DOI] [PubMed] [Google Scholar]
- 2.Handl HL, Vagner J, Han H, Mash E, Hruby VJ, Gillies RJ. Expert Opin Ther Targets. 2004;8:565. doi: 10.1517/14728222.8.6.565. [DOI] [PubMed] [Google Scholar]
- 3.Handl HL, Sankaranarayanan R, Josan JS, Vagner J, Mash EA, Gilles RJ, Hruby VJ. Bioconjugate Chem. 2007;18:1101. doi: 10.1021/bc0603642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Josan JS, Handl HL, Sankaranarayanan R, Xu L, Lynch RM, Vagner J, Mash EA, Hruby VJ, Gillies RJ. Bioconjugate Chem. 2011;22:1270. doi: 10.1021/bc1004284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiessling LL, Gestwicki JE, Strong LE. Angew Chem Int Ed. 2006;45:2348. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson CP, Challis-John RA. Biochem Pharmacol. 2007;73:737. doi: 10.1016/j.bcp.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Mandrika I, Petrovska R, Wikberg J. Biochem Biophys Res Commun. 2005;326:349. doi: 10.1016/j.bbrc.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 8.Patchett AA, Martin WJ, Van der Ploeg LH. Proc Natl Acad Sci U S A. 2002;99:11381. doi: 10.1073/pnas.172378699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wessells H, Fuciarelli K, Hansen J, Hadley ME, Hruby VJ, Dorr R, Levine N. J Urol. 1998;160:389. [PubMed] [Google Scholar]
- 10.Wikberg JES. Expert Opin Ther Pat. 2001;11:61. [Google Scholar]
- 11.Lin C, Sarath G, Frank JA, Krueger RJ. Biochem Pharmacol. 1991;41:789. doi: 10.1016/0006-2952(91)90082-g. [DOI] [PubMed] [Google Scholar]
- 12.Kiessling LL, Gestwicki JE, Strong LE. Curr Opin Chem Biol. 2000;4:696. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 13.Hruby VJ, Lu D, Sharma SD, Castrucci AL, Kesterson RA, Al-Obeidi FA, Hadley ME, Cone RD. J Med Chem. 1995;38:3454. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 14.Gestwicki JE, Cairo CW, Mann DA, Owen RM, Kiessling LL. Anal Biochem. 2002;305:149. doi: 10.1006/abio.2002.5652. [DOI] [PubMed] [Google Scholar]
- 15.Hiavacek WS, Posner RG, Perelson AS. Biophysical Journal. 1999;76:3031. doi: 10.1016/S0006-3495(99)77456-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King SH, Mayorov AV, Balse-Srinivasan P, Hruby VJ, Vanderah TW. Curr Top Med Chem. 2007;7:1098. [PMC free article] [PubMed] [Google Scholar]
- 17.Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL. J Am Chem Soc. 2002;124:14922. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- 18.Thieriet N, Alsina J, Giralt E, Cuibe S, Albericio F. Tetrahedron Lett. 1997;38:7275. [Google Scholar]
- 19.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Bioorg Med Chem Lett. 2004;14:211. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 20.Vagner J, Handl HL, Monguchi Y, Uana U, Begay LJ, Mash EA, Hruby VJ, Gillies RJ. Bioconjugate Chem. 2006;17:1545. doi: 10.1021/bc060154p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Josan JS, Vagner J, Handl HL, Sankaranarayanan R, Gilles RJ, Hruby VJ. Int J Pep Res Ther. 2008;14:293. doi: 10.1007/s10989-008-9150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, Delvalle J, Yamada J. J Biol Chem. 1993;268:15174. [PubMed] [Google Scholar]
- 23.Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Anal Biochem. 2004;330:242. doi: 10.1016/j.ab.2004.04.012. [DOI] [PubMed] [Google Scholar]