

Abstract
Valosin-containing protein (VCP or p97), a member of AAA family (ATPases associated with diverse cellular activities), plays a key role in many important cellular activities. A genetic deficiency of VCP can cause inclusion body myopathy associated with Paget’s disease of bone and frontotemporal dementia (IBMPFD). Previous studies showed that the VCP N domain is essential for the regulation of nuclear entry of VCP. Here we report that IBMPFD mutations, which are mainly located in the N domain, suppress the nuclear entry of VCP. Moreover, the peptide sequence G780AGPSQ in the C-terminal region regulates the retention of VCP in the nucleus. A mutant lacking this sequence can increase the nuclear distribution of IBMPFD VCP, suggesting that this sequence is a potential molecular target for correcting the deficient nucleocytoplasmic shuttling of IBMPFD VCP proteins.
Keywords: valosin containing protein, nucleocytoplasmic shuttling, nuclear export signal, inclusion body myopathy associated with Paget’s disease of bone and frontotemporal dementia (IBMPFD)
1. Introduction
Valosin-containing protein (VCP, or p97), a member of the ATPases associated with diverse cellular activities (AAA) family, is one of the most abundant proteins in cells [1]. It plays essential roles in many cellular activities such as membrane fusion, protein degradation and DNA repair [1–3]. The traffic of VCP between the nucleus and cytoplasm is a regulated process. Previous studies suggest that both the N domain and the C-terminal region mediate the nucleocytoplasmic shuttle of VCP [4, 5]. Structurally, VCP is comprised of an N domain, two conserved ATPase domains D1 and D2, and a flexible C-terminal region [1]. The D1 domain is required for VCP to form a hexameric structure [6, 7]. The D2 domain provides the majority of the ATPase activity [8], and the N domain binds various cofactors required for its diverse functions [1, 9]. In addition, the N domain is required for the nuclear entry of VCP. Mutants bearing a deletion of the N domain are predominately present in the cytoplasm, while wild type (WT) VCP is ubiquitously distributed in the cells [4].
VCP mutations can cause inclusion body myopathy associated with Paget’s disease of bone and frontotemporal dementia (IBMPFD), an inherited human multi-organ disorder [10, 11]. All reported IBMPFD-causing mutations are single amino acid substitutions and mostly occur in the N-domain, suggesting a potential effect on the nucleocytoplasmic shuttling of VCP.
Previous studies suggest that the N domain mediated nuclear entry is regulated by the C-terminal region of VCP based on the study of its homolog, Cdc48 in saccharomyces cerevisiae. A stretch of acidic residues (E828EDDDLY834S) in the C-terminal region can interact with the nuclear localization signal sequence, RR28KKK, in the N domain to mask its signaling [5]. Tyr834 phosphorylation leading to a conformational change abolishes the attachment with RR28KKK to resume its nuclear localization signaling [5]. However, VCP does not possess a typical nuclear targeting signal sequence [5]. Even though the acidic peptide is conserved in both VCP and Cdc48, the crystal structure of VCP does not support a model for the C-terminal region to interact with the N domain directly [12]. Thus, the mechanism of the nucleocytoplasmic shuttling of VCP requires further investigation.
In this study, we analyzed the structural requirement for VCP to shuttle between the nucleus and cytoplasm using a VCP-GFP chimeric protein, and found that IBMPFD mutations (R95G, R155H or R155P, R155C, R191Q and A232E) decrease the nuclear distribution of VCP and the sequence G780AGPSQ in the C-terminal region regulates the retention of VCP in the nucleus and nucleoli. In addition, the mutants lacking this C-terminal sequence upregulates the nuclear retention of IBMPFD mutants, suggesting that this C-terminal region may serve as a molecular target for regulating the nucleocytoplasmic shuttling of VCP.
2. Methods
2.1. DNA constructs
Mammalian VCP-GFP expression constructs, WT, VCP-Δ201-806, VCP-Δ1-206, VCP-Δ780-806, VCP-Δ786-806, VCP-Δ797-806, A1, and A2, were generated as previously reported [13]. In brief, mutations were generated from pDNR-Dual-WT-VCP using QuickChange™ Site-Directed Mutagenesis Kit (Stratagene). Then pLpEGFP-constructs were produced through a recombination reaction to transfer the genes in the pDNR-Dual-constructs to the pLpEGFP vector following the manufacturer’s protocol (Life Technologies Inc. Carlsbd, CA). IBMFPD mutants of VCP were generated using the following oligonucleotides and their corresponding complimentary strands (data not shown) in the PCR cloning for individual mutants of pLpEGFP-VCP:
R95G, 5′-AGAGTTGTTCGGAATAACCTCCGAGTTGGCCTAGGAGATGTCATCAGCATC-3′;
R155C, 5′-CCGTAAAGGAGATATTTTTCTTGTCTGTGGTGGGATGCGTGCTGTGGAG-3′;
R155H, 5′-CCGTAAAGGAG ATATTTTTCTTGTCCATGGTGGGATGCGTGCTGTGGAG-3′;
R155P, 5′-CCGTAAAGGAGATATTTTTCTTGTCCCGGGTGGGATGCGTGCTGTGGAG-3′;
R191Q, 5′-CACTGTGAGGGGGAGCCAATCAAACAAGAGGATGAGGAGGAATCCTTGAAT-3′;
A232E, 5′-ACTGAGACATCCTGCGCTCTTCAAGGAGATTGGTGTAAAGCCTCCTCGGG-3′.
All introduced site-specific mutations and the deletion mutants were confirmed by DNA sequencing.
2.2. Cell culture and transfection
Human embryonic kidney (HEK)293 cells and human osteosarcoma (HOS) cells were grown in DMEM medium with 10% fetal calf serum and 2 mM glutamine. Human primary neurons (ScienCell Research Laboratories, Carlsbed, CA) were grown in neuronal medium with 10% fetal calf serum, 1% of neuronal growth supplement and 2 mM glutamine. All of these cells were cultured in an incubator with humidified 5% CO2 atmosphere. The cells were transfected with 2 μg or 0.2 μg DNA constructs per well in 6-well or 96-well plates respectively using lipofectamine 2000 (Life Technologies Inc.) following the manufacturer’s instructions and repeated for three times.
2.3. Immunoprecipitation and SDS-PAGE
HEK293 cells expressing VCP-GFP or GFP cultured in 6-well plates were washed with ice-cold PBS, and then detached with 0.05% trypsin/0.02% EDTA. After centrifugation at 500 × g for 5 min, cell pellets were lyzed in RIPA buffer supplemented with the protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Following centrifugation at 10,000 × g for 30 min at 4°C, 2 μg anti-GFP antibody (Sigma-Aldrich) and 20 μl 50% Protein-G-Sepharose beads in lysis buffer were added to 500 μl supernatants.
The cell lysates were incubated for 2 hr at 4°C, then were transferred onto a 1 ml polypropylene column (Qiagen, Valencia, CA). After washing with 10 mL TBS, proteins were eluted using 0.1 M pH 3.0 citrate acid/glycine buffer and immediately neutralized with 1 M pH 9.0 Tris-HCl. The protein concentration was determined using a BCA protein assay kit (Thermo Scientific, Waltham, MA). Protein samples were separated using 4–12% SDS-PAGE, transferred to nitrocellulose membranes, then analyzed by immunoblotting with anti-VCP and HRP-labeled secondary antibodies [13].
2.4. Hexamerization assay using native gel electrophoresis
Protein complexes were resolved using Novex® NativePAGE™ Bis-Tris gel system (Life Technologies Inc., Carlsbad CA). Cell lysates were mixed with native loading buffer, loaded to 3–16% native gel and run electrophoresis for 1.5 hr. The gel image of VCP-GFP was scanned using FUJIFILM Fluoro-Image Analyzer FLA-5000 with excitation at 473 nm, CH1 400 V and resolution at 200 μm.
2.5. ATPase assay
The ATPase activity of immunoprecipitated protein complexes was analyzed by adding 10 μl protein complexes to ATPase assay buffer containing 50 mM Tris-HCl (pH 8.0), 20 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol (DTT), 3 mM ATP for 15 min at 37°C. The inorganic phosphate released by ATP hydrolysis was measured as described [8].
2.6. Cell cycle analysis
Cells were fixed in 70% cold ethanol for 1 hr, washed 3 times with PBS containing 0.1% BSA, followed by treatment with RNase (1U/mL) for 1 hr, then were stained with 0.1% propidium iodide for 1 hr, and analyzed with flow cytometry.
2.7. Analysis of the fluorescence intensity ratio between the nucleus and cytoplasm (FIRNC)
Cells were cultured in 35 mm coverglass bottom dishes at 37°C for 24 hr. After transfection for 3d, cells were imaged using a confocal LSM510 Laser Scanning Microscope (Carl Zeiss, Germany) or Olympus IX71 inverted fluorescence microscopy equipped with DM1 CCD camera. The fluorescence intensity of VCP-GFP in the nucleus and cytoplasm was measured using Image J 1.43u (http://rsb.info.nih.gov/ij) or CellSens Dimension digital imaging analysis software 1.9 (Olympus Inc.) and statistically analyzed using SPSS ver.16.0.2 software.
2.8 Fluorescence recovery after photobleaching (FRAP) analysis
Cells were imaged with or without photobleaching using a confocal LSM510 Laser Scanning Microscope (Carl Zeiss, Germany) equipped with an Apochromat 40×/1.2 W objective and a 488 nm argon laser line for illumination. Cell images were scanned with 488 nm excitation at 2% intensity before or after bleaching. Photobleaching was conducted with laser at 100% intensity and 100 iterations using 488 nm. The entire regions of the nucleus or the cytoplasm were photobleached and sequentially imaged at 1 min interval for 15 min. The fluorescence recovery in the nucleus and cytoplasm were calculated using the Medical Image Processing, Analysis and Visualization software (MIPAV) (Center for Information Technology, National Institutes of Health).
2.9 Cell nuclear and cytoplasm fractionation
Nuclear and cytoplasmic fractions were prepared from the transfected HEK293 cells using NE-PER Reagent (Thermo Fisher Scientific Inc, Rockford, IL) following the manufacturer’s instruction. The nuclear and cytoplasmic fractions were mixed with loading buffer and resolved using 4–12% SDS-PAGE (Life Technologies Inc.). The VCP-GFP variants were detected using anti-GFP antibody. Lamin A/C and GAPDH were probed as the nuclear and cytoplasmic markers.
3. Results
3.1. VCP-GFP is functional in HEK293 cells
To study the nucleocytoplasmic shuttling of VCP, we employed VCP-GFP as a reporter. Even though it has been used to indicate the cellular localization of the VCP proteins previously [4, 13, 14], it is not clear that this fusion protein retains the biological function of VCP. Human embryonic kidney (HEK)293 cell line was used as a cell model for its high transfection efficiency and high level of protein expression. Here we showed that the expression of VCP-GFP in HEK293 cells did not affect cell morphology or the cell cycle (Fig. 1A, 1B). Since the functions of VCP require the formation of a hexameric structure [1, 15], we examined whether VCP-GFP oligomerizes with endogenous VCP. HEK293 cells expressing VCP-GFP were lyzed and the lysates were immunoprecipitated with anti-GFP antibody, then immunoprecipitated protein complexes were separated by SDS-PAGE, followed by western blotting with anti-VCP antibody. As shown in Fig. 1C (left panel), two VCP protein bands corresponding to endogenous (97 kDa) and chimeric VCP (124 kDa) were detected in cells transfected with VCP-GFP. Imaging analysis indicates that the intensity of the VCP-GFP band is about 54.3% of VCP, suggesting that VCP-GFP forms oligomers with endogenous VCP with a ratio of 1 to 2. Furthermore, native gel electrophoresis showed that a fluorescent protein band at approximately 650 to 750 kDa which is equal to the molecular mass of the heterodimeric form of VCP and VCP-GFP (Fig. 1C right panel). No monomer band of VCP-GFP (127 kDa) was observed, suggesting that all of the VCP-GFP proteins are oligomerized into larger protein complexes. In addition, the ATPase activity of the protein complexes immunoprecipitated with anti-GFP or anti-VCP antibody was analyzed. The immunoprecipitated VCP-GFP exhibits ATPase activity similar to WT VCP (Fig. 1D). Thus, VCP-GFP shows the functional characteristics of VCP in HEK293 cells.
Fig. 1.

VCP-GFP retains biological properties of VCP. (A) HEK293 cells stably expressing VCP-GFP and GFP showed similar morphology in the bright field and green fluorescence images. (B) Cells expressing VCP-GFP and GFP exhibit similar cell cycle. Cells stained with propidium iodide were analyzed using flow cytometry. Cell cycle is presented as the means and standard errors (n=8, P > 0.05). (C) VCP-GFP forms a hexameric structure. VCP-GFP co-immunoprecipited with endogenous VCP (left panel). VCP-GFP protein complexes were immunoprecipitated using anti-GFP antibody followed by Western blotting with anti-VCP antibody. VCP-GFP and VCP were shown as a 124 kDa band (similar to the calculated molecular mass of VCP-GFP) and a 97 kDa band (the molecular mass of monomeric VCP). GAPDH was probed as a loading control. The native gel fluorescence imaging of VCP-GFP showed that VCP-GFP is in a hexameric structure (right panel). The lysates of HEK293 cells expressing VCP-GFP or GFP were resolved using 3–16% native gel. The gel image was taken using a Fluoro Image Analyzer. The 650 to 750 kDa and 27 kDa bands were shown as VCP-GFP and GFP, respectively. (D) VCP-GFP retains the ATPase activity. Protein complexes were immunoprecipitated using anti-GFP or anti-VCP antibody from the lysates of HEK293 cells without transfection (Ctrl) or transfected with GFP or VCP-GFP. The ATPase activities of the immunoprecipited protein complexes were analyzed using a colorimetric ATPase assay. The ATPase activity was shown as the percentage of the ATPase activity of immunoprecipitated protein complexes in the control sample. Data are presented as mean and standard error of four experiments.
3.2. The N domain deletant and IBMPFD mutants of VCP manifest lower nuclear distribution than WT VCP
The N domain plays an important role for VCP to transport into the nucleus, and its deletion suppresses the nuclear entry of VCP (Fig. 2A) [4]. Quantitative image analysis of the N-terminal 1-206 deletion (VCP-Δ1-206) was carried out by measuring the fluorescence intensity ratio between the nucleus and cytoplasm (FIRNC), and the results showed that the nuclear distribution of VCP-Δ1-206 was 70% lower than the WT VCP-GFP (Fig. 2B). Since IBMPFD mutations predominately locate in the N domain or at the interface between N domain and D1 domain [10, 11], we hypothesize that these mutants also affect the nucleocytoplasmic shuttling of VCP. We generated 6 representative IBMPFD mutants (R95G, R155C, R155H, R155P, R191Q and A232E) using site-specific mutagenesis, and then expressed them in HEK293, HOS cells and primary neurons followed by cell imaging. Indeed, these mutants showed lower fluorescence intensity in the nucleus when compared to WT VCP in all of these three cell lines (Fig. 2C). Since bone and nervous tissues manifest major pathological change in IBMPFD patients [10], the distributions of VCP WT and IBMPFD mutants in osteoblasts and neurons are similar as in HEK293 cells suggests that the deficiency in the nuclear distribution of IBMPFD mutants may be relevant to the disease and HEK293 could serve as a cell model for the VCP nucleocytoplasmic shuttling study. Image analysis showed that the FIRNCs of all of these IBMPFD mutants were significantly lower than the WT VCP-GFP in HEK293 cells (p<0.001, n=50). Among these transfectants, R155C and R155H exhibit approximately 60% of the FIRNC of WT VCP, which was also less than R191Q (p<0.05, n=50) (Fig. 2D). This result suggests that these IBMPFD mutations suppress nucleocytoplasmic shuttling of VCP, and R155C and R155H have the most significant effect on nuclear entry.
Fig. 2.

IBMPFD mutations decrease the nuclear distribution of VCP. HEK293 cells were transiently transfected with VCP-Δ1-206, VCP-Δ201-806 and WT VCP and imaged with confocal microscopy (A) and then images were quantitatively analyzed using image J software. The statistical analysis was showed that VCP-Δ1-206 is significantly lower than WT (P<0.01, n=28). (C) IBMPFD mutants were transfected to HEK293, HOS cells and primary neurons, and then imaged with an inverted fluorescence microscope. (D) Quantitative analysis of the fluorescence intensity ratio between the nucleus and the cytoplasm (FIRNC) in HEK293 cells expressing IBMPFD mutants was performed using Olympus CellSens Dimension software. The result shows that the FIRNC of all the IBMPFD mutants is WT is significantly lower than WT (P<0.001, n=50).
3.3. Characterization of the nucleocytoplasmic shuttling of VCP
The nucleocytoplasmic shuttling of VCP was further examined by measuring the recovery of the fluorescence of VCP-GFP after photobleaching. Following the photobleach of the nucleus, the recovered fluorescence showed a noticeable increase within cells expressing GFP in 4 s, which was then elevated over time (Fig. 3A). The fluorescence recovery of VCP-GFP was much slower than GFP alone. The recovered fluorescence intensity of VCP-GFP in the nucleus was only 5%, as compared to 50% for GFP alone in 60 s (Fig. 3B). GFP reached an equilibrium state between the cytoplasm and nucleus at 6 min while the nuclear concentration of VCP-GFP only recovered approximately 20%. It suggests that VCP-GFP possesses a mechanism for nuclear entry, which is distinct from GFP.
Fig. 3.
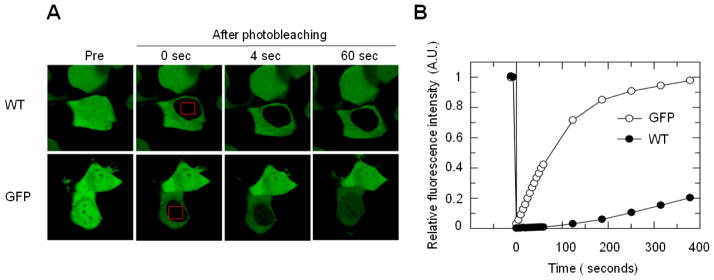
FRAP analysis of the nuclear entry of VCP–GFP in HEK293 cells. (A) The nucleus of the cells expressing VCP–GFP or GFP were photobleached for 4 s (area indicated by red boxes), and the fluorescence images were obtained at 4 s intervals over 400 s by confocal microscopy. (B) Quantitative analysis from results in (A) for the fluorescence recovery in the nucleus using single exponential FRAP of MIPAV software, where differences in fluorescence recovery rate were observed between VCP-GFP and GFP.
3.4. Effect of ATPase activity on the nucleocytoplasmic shuttling of VCP
The IBMPFD mutations have varied effect on the ATPase activity of VCP. While most of these mutants show slightly elevated ATPase activity, R155C and A232E mutants exhibit approximately two fold increase [16–18], suggesting that the altered nucleocytoplasmic shuttling of these mutants may not have been due to the altered ATPase activity. We analyzed the cellular distribution of the site-specific mutant of VCP lacking ATP binding or ATP hydrolysis, A1 or A2, and calculated the FIRNC from the confocal microscopy obtained images and found that both A1 and A2 were present at lower level in the nucleus than in the cytoplasm (Fig. 4A). The FIRNC of these two mutants was about 50% of WT VCP (Fig. 4B). This suggests that the ATPase activity affects the nucleocytoplasmic shuttling of VCP. However, the suppressed nucleocytoplasmic shuttling of N domain mutants is not caused directly through their effect on ATPase activity.
Fig. 4.
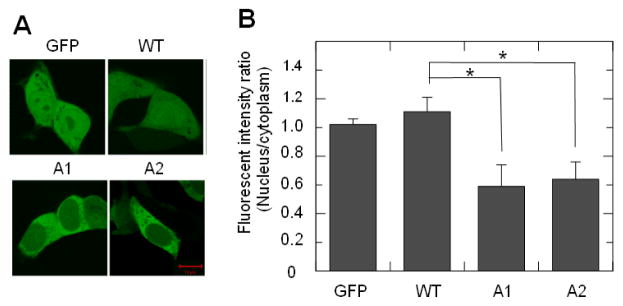
The mutation of ATP binding site in D1 or D2 domain of VCP-GFP decreases the nuclear retention. (A) HEK293 cells transfected with VCP-GFP mutant A1 or A2 were imaged using confocal microscopy. (B) Quantitative analysis of the nuclear retention. Data are presented as the fluorescence intensity ratio between the nucleus and the cytoplasm calculated using MIPAV software (n=36, * P<0.05).
3.5 The C-terminal region regulates the nuclear export of VCP
Since the C-terminal region of the yeast homolog of VCP, Cdc48, regulates nuclear entry, we examined the role of the C-terminal region of VCP on nucleocytoplasmic shuttling. We generated three sequential C-terminal deletion mutants using WT VCP-GFP as a template. Both VCP-Δ780-806 and VCP-Δ786-806 lack the acidic sequence, E798DNDDDLY, in the C-terminal region, but both of them showed similar distribution patterns as WT VCP-GFP (Fig. 5A and Fig. 2A), suggesting this acidic sequence was not involved in regulating the nuclear entry of VCP. Surprisingly, the VCP-Δ780-806 mutant predominately accumulated in the nucleus as indicated by the colocalization of the high intensity green fluorescence of VCP-Δ780-806 within the Hoechst 33342 stained nucleus (Fig. 5A and 5C). FIRNC analysis showed that the nuclear concentration of VCP-Δ780-806 is about 2-fold higher than that of the cytoplasm, and the FIRNC of VCP-Δ780-806 is significantly higher than that of VCP-Δ786-806 and VCP-Δ797-806 (Fig. 5B).
Fig. 5.

The sequence 780-785 in the C-terminal region regulates VCP-GFP retention in the nucleus and nucleoli. HEK293 cells were transiently transfected with C-terminal region deletion mutant VCP-Δ780-806, VCP-Δ786-806 and VCP-Δ797-806 for 16 hr. The images were taken under an inverted fluorescence microscope. (A) The images showed that VCP-Δ780-806, but not VCP-Δ786-806 and VCP-Δ797-806, accumulates in the nucleus and forms foci as indicated by red arrows. (B) Quantitative analysis of the results presented in (A). Data are presented as the fluorescence intensity ratio between the nucleus and the cytoplasm calculated using MIPAV software (n=48, * p<0.01). (C) The VCP-Δ780-806 transfected cells were stained using Hoechst 33342 and imaged with fluorescence microscope and the images of the green fluorescence of VCP-Δ780-806 and the nucleus were merged (scale bar = 5 μM). (D) HEK293 cells expressing VCP-Δ780-806 and VCP-Δ786-806 mutants were fixed with cold methanol and stained with anti-nucleolin antibody and TRITC labeled secondary antibody. Images were taken using confocal microscopy (scale bar=5 μM).
In addition, the results show VCP-Δ780-806 proteins form foci in the nucleus (Fig. 5A, indicated by red arrows). We hypothesize that these foci reside at the nucleoli, sites for ribosome assembly, because VCP can bind misfolded proteins [19] and the nucleoli function as the deposition sites for misfolded proteins, heat shock proteins and proteasomes [20–22]. We fixed these cells with cold methanol and stained cells with an antibody to detect the nucleolin proteins, the marker of nucleoli. Cell imaging showed that the VCP-Δ780-806 and VCP-Δ786-806 proteins were leaked out the nucleus after fixation, but VCP-Δ780-806 proteins were retained in foci and colocalized with the nucleoli (Fig. 5D).
To further determine the effect of the deletion of the G780AGPSQ sequence on the nuclear entry of VCP, we performed FRAP analysis to determine the shuttle efficiency of VCP-Δ780-806 in crossing the nuclear membrane. The results showed that the fluorescence recovery of VCP-Δ780-806 in the nucleus was more rapid than in the cytoplasm, while WT VCP showed similar recovery efficiency in both the nucleus and the cytoplasm (Fig. 6A). In addition, the recovery of VCP-Δ780-806 in the nucleus is approximately twice as fast as that of WT (Fig. 6B). Thus, the G780AGPSQ sequence plays a critical role in regulating the export of VCP from the nucleus and nucleoli to the cytoplasm, and may serve as a nuclear export signal.
Fig. 6.

VCP-Δ780-806 proteins exhibit a higher nuclear import rate than WT VCP-GFP. HEK293 cells were transiently transfected with VCP-Δ780-806 or WT for 16 hr. (A) Large ROIs indicated as red regions in the nucleus or cytoplasm were photobleached. Images were then consecutively taken at 1 min intervals. (A) Representative images show increased fluorescence intensity in the nucleus in cells expressing VCP-Δ780-806, but not WT, noticeable after 5 and 9 min recovery. (B) Fluorescence photobleaching and recovery curves are plotted using single exponential of MIPAV software. Photobleaching in the nucleus or cytoplasm indicated as WT-N and VCP-Δ780-806-N or WT-C and VCP-Δ780-806-C.
3.6. VCP-Δ780-806 increases the nuclear distribution of N domain mutants
To increase the nuclear distribution of IBMPFD mutants of VCP, we hypothesize that overexpression of WT VCP cannot overcome this deficiency, but the G780AGPSQ deletant could promote the nuclear retention of the N domain mutants. The reason is that VCP-GFP proteins are hexamerized with endogenous VCP proteins in cells (Fig. 1). The deficiency in the nuclear distribution manifested by these hybrid hexamers suggests that the intact hexameric N domains are required for the nuclear entry of VCP. Therefore, overexpression of WT VCP cannot enable these IBMPFD mutants to form intact hexameric N domains for mediating their nuclear entry. However, VCP-Δ780-806 could form hexamers with VCP-Δ1-206 and IBMPFD mutants through the D1 domain. In this way, VCP-Δ780-806 could increase the nuclear retention of these mutants. We transfected VCP-Δ780-806 or WT with IBMPFD mutants or VCP-Δ1-206 in HEK293 cells, and as expected, the fluorescence intensity of R155H and VCP-Δ780-806 co-transfected in the nucleus was clearly higher than R155H and WT co-expressing cells (Fig. 7A). Further cell imaging analysis showed that the FIRNC of the WT and IBMPFD co-tranfectant did not show significant increases. In the other hand, the FIRNC of VCP-Δ780-806 and R155H was elevated approximately 2-fold (Fig. 7B). Moreover, western blotting analysis showed that the protein amount of R155H was lower in the nuclear fraction than the cytoplasmic fraction (Fig. 7C lane 1 vs 2). The expression of VCP-Δ780-806, but not VCP-1-206 or WT, with R155H led to an increase of R155H in the nuclear fraction (Fig. 7C lane 5 vs 6, 3 vs 4 and 7 vs 8). These data suggest that VCP-Δ780-806 elevated the nuclear distribution of the R155H mutant.
Fig. 7.

Effect of VCP-Δ780-806 on the nuclear distribution of VCP-GFP bearing R155H mutation. HEK293 cells were co-transfected with R155H and control vector, VCP-Δ1-206, VCP-Δ780-806 or WT for 16 hr. (A) The cells were imaged with an inverted fluorescence microscope. (B) Fluorescence intensity ratio between nucleus and cytoplasm (FIRNC) were calculated using Olympus CellSens Dimension software. The result showed that the FIRNC of R155H and VCP-Δ780-806 is significant higher than other cotransfectants (p<0.05, n=50). (C) Distribution of the coexpressed VCP variants in the nuclear and cytoplasmic fractions. HEK293 cells were transiently transfected with VCP-GFP variants as indicated and then fractionated into the nuclear fraction (N) and cytoplasmic fraction (C). VCP-GFP variants were detected by anti-GFP antibody. GAPDH and lamin A/C were used as cytoplasmic and nuclear markers, respectively.
We further examined the effect of VCP-Δ780-806 on the nuclear distribution of other IBMPFD mutants in HEK293 cells, and found that VCP-Δ780-806 had similar effects for all of them. The FIRNC of cells co-expressing VCP-Δ780-806 with R95G, R155C, R155P, Q191A or A232E are 1.29±0.42, 1.18±0.39, 1.13±0.22, 1.15±0.26 and 1.01±0.33 (mean±SD, n=50). These results suggest that VCP-Δ780-806 can promote the nuclear distribution of IBMPFD mutants.
Since the C-terminal region may affect the N domain mutants on other subunits or the same subunit in the hexamer, we examined the in trans and in cis effects using cells expressing both of the VCP-Δ780-806 and VCP-Δ1-206 proteins or VCP-Δ1-206-Δ780-806 proteins (Fig. 8). The nuclear fluorescence intensity were increased around 2.1-fold in the cells with co-transfection of VCP-Δ780-806 and VCP-Δ1-206 compared to VCP-Δ1-206 only (Fig. 8A and B). The western blotting analysis showed that the protein amount of VCP-Δ780-806 was higher in the nuclear fraction than the cytoplasmic fraction while VCP-Δ1-206 was predominately in the cytoplasmic fraction (Fig. 8C, lane 1 vs 2 and 5 vs 6). The expression of VCP-Δ780-806 caused the increase of VCP-Δ1-206 in the nucleus (Fig. 8C lane 3 vs 4 and 5 vs 6). These results suggest that the C-terminal region can increase the retention of the N domain deletion in trans. In addition, the nuclear fluorescence of VCP-Δ1-206-Δ780-806 was also higher than VCP-Δ1-206. The FIRNC of these cells was increased by approximately 1.5-fold (Fig. 8A and B). The western blotting analysis showed that the protein amount of VCP-Δ1-206-Δ780-806 in the nuclear fraction is slightly higher than VCP-Δ1-206 (Fig. 8C, lane 7 vs 8 and 5 vs 6), suggesting that the C-terminal region can also increase the nuclear distribution in cis, but with lower efficiency than the effect in trans. Thus, the C-terminal region of VCP can regulate the nuclear entry of VCP both in trans and in cis.
Fig. 8.

in cis and in trans effect of the C-terminal sequence 780-806 on VCP-Δ1-206. Cells were transfected with VCP-Δ780-806 with control vector or VCP-Δ1-206, and VCP-Δ1-206 with control vector, or VCP-Δ1-206-Δ780-806 for 16 hr. (A) The cells were imaged with an inverted fluorescence microscope. (B) Fluorescence intensity ratio between the nucleus and cytoplasm (FIRNC) were calculated using Olympus CellSens Dimension software. The comparison of FIRNC between VCP-Δ780-806+VCP-Δ1-206 and VCP-Δ1-206 using ANOVA showed a significant difference (p<0.05, n=50). (C) Distribution of VCP deletion mutants in the nuclear and cytoplasmic fractions. HEK293 cells transiently transfected with VCP-GFP deletion mutants as indicated, and then fractionated into the nuclear fraction (N) and cytoplasmic fraction (C). VCP-GFP variants were detected by anti-GFP antibody. GAPDH and lamin A/C were used as cytoplasmic and nuclear markers, respectively.
4. Discussion
In the past 20 years, studies have established that VCP plays critical roles in a number of cellular activities. VCP not only maintains proteostasis through mediating protein degradation via the ubiquitin proteasome system, autophagy and collecting protein aggregates to aggresomes, but is also involved in nuclear formation and maintenance of DNA integration [1–3]. Nucleocytoplasmic shuttling is essential for VCP to function spatially in the nucleus and cytoplasm. Previous studies have suggested that the N domain is involved in the regulation of nuclear entry based on N domain deletion of VCP. However, whether the site-specific mutation of VCP affects the nuclear distribution is unclear. In addition, whether the C-terminal region of VCP retains similar regulatory function as its homolog Cdc48 remains unknown. As summarized in Fig. 9, here we show that the IBMPFD mutants in the N domain, or N-D1 domain interface, suppress the nuclear entry of VCP. In contrast, the C-terminal region deletion causes VCP to accumulate in the nucleus. In addition, the C-terminal deletion can promote the nuclear concentration of N domain mutants. These findings further clarify the role of N domain in regulating nuclear entry and reveal that the G780AGPSQ sequence in the C-terminus regulates the nuclear export of VCP which may serve as a nuclear export signal.
Fig. 9.

Schematic representation of VCP-GFP variants and summary of the nuclear retention. The N domain or C-terminal deletion mutants are depicted. The nuclear retention (from Fig. 2, Fig. 4 and Fig. 5) are summarized as +/−, +, ++ and +++, which indicate low to high fluorescence intensities.
In our studies, we utilize the VCP-GFP fusion protein to assess the kinetic distribution of VCP. It has previously been used to analyze the intracellular distribution of VCP [4, 13], and our earlier studies showed that VCP-GFP demonstrated similar function as WT VCP in the formation of aggresomes [13]. However, the biological characterization of the fusion protein has not been evaluated. Here, we determined the key biological features of VCP-GFP including the distribution pattern, oligomerization properties and the ATPase activity, and found that all of these features of VCP-GFP are similar to WT VCP. This indicates that the fusion of GFP does not affect the biological function of VCP.
Previous studies showed that the N domain is essential for the nuclear entry of VCP [4], and the deletion of the N domain suppresses the nuclear entry. The IBMPFD mutations are scattered within the N domain or N-D1 interface (A232E) [10], but whether these mutations affect nucleocytoplasmic shuttling of VCP was unknown. By imaging analysis of the transfected HEK293 cells, we found that all of these mutants exhibit decreased nuclear distribution compared with WT VCP. Since these mutations are widely distributed in the N domain, the traditional NLS cannot explain why they all affect the nuclear distribution of VCP. Since these mutations alter the conformation of the N domain [23], this may suggest that VCP interacts with a docking protein on the nuclear membrane via the N domain. The defective N domain of these mutants may suppress the binding of VCP to the docking proteins required for nuclear entry.
In addition, we found that the C-terminal region of VCP exhibits a different regulatory mechanism when compared with Cdc48. Although the amino acid sequence E799NDDDLY in VCP is homologous to the regulatory sequence E828EDDDLY in Cdc48, it is not required for regulating the nucleocytoplasmic shuttling of VCP because C-terminal deletion mutants of VCP (e.g. VCP-Δ786-806 and VCP-Δ797-806) lacking this sequence still show similar nuclear distribution as WT VCP. Instead, the sequence, G780AGPSQ, in the C-terminal region, regulates the nuclear export of VCP. The deletion of this sequence in VCP-Δ780-806, but not VCP-Δ786-806 and VCP-Δ797-806, causes high accumulation of VCP in the nucleus. A blast search of Cdc48 sequence (GenBank: CAA98694.1, http://blast.ncbi.nlm.nih.gov) does not reveal any homolog of G780AGPSQ. Thus, although Cdc48 and VCP are evolutionally conserved in protein degradation, they still use different mechanisms to regulate their nuclear retention.
Interestingly, a homolog of VCP, nuclear valosin-containing protein-like isoform 2 (NVL2), is mainly distributed in the nucleus and nucleoli [24]. Blast analysis of human VCP (accession P55072) and NVL2 (accession NP_002524) shows these two proteins have no similarity in the N domain, the other regions share 59% homology but the G780AGPSQ sequence is absent in NVL2. This implies that lacking this nuclear export sequence could contribute to the accumulation of NVL2 in the nucleus and nucleoli.
It remains unclear whether the nuclear export signal (NES) of VCP interacts with export receptors. CRM1 (chromosome region maintenance 1 or exportin1) is a member of the importin β superfamily of nuclear transport receptors, and recognizes proteins bearing a leucine-rich nuclear export sequence [25, 26]. This sequence is present in a number of proteins, such as human T-cell leukemia type I Rex [27], cAMP-dependent protein kinase inhibitor [28], and mitogen-activated protein kinase kinase [29]. However, G780AGPSQ functions as NES without any leucine residues, suggesting it may function through different nuclear transport receptors.
Even though the ATPase activity of VCP plays an important role in various cellular functions, we found that it is not sufficient for the nucleocytoplamic shuttling of VCP. The N domain deletion mutant exhibits normal ATPase activity of VCP [30], while it lacks the capacity for nuclear entry [4, 14]. Similarly, the IBMPFD mutants exhibit increased ATPase activity [16–18], yet all of them also showed decreased nuclear distribution. However, mutation of either the D1 or D2 domain leads to a decrease of nuclear distribution. This seemingly contradictory observation suggests that although IBMPFD mutants affect both the interaction of VCP with other proteins and the ATPase activity, the enhanced ATPase activity of these mutants is likely due to less of an impact by a potential N domain conformational change [23]. Previous studies showed that restraining the conformational change of the N domain by binding to cofactors such as p47 suppresses the ATPase activity of VCP [30, 31]. This suggests that the binding of the N domain to docking proteins is a primary gateway for VCP to enter the nucleus, while the ATPase activity may be required to regulate the N domain conformational change during the nuclear entry process.
Although mutation can induce protein misfolding, protein aggregation is not a cause of the abnormal localization of the IBMPFD mutants. Because VCP is a molecular chaperone, even the deletion mutants of VCP still can prevent protein aggregation [19]. In addition, these IBMPFD mutants manifest normal or elevated ATPase activity [16–18], suggesting that the altered structures of IBMPFD mutants remain enzymatic functional. In our experiments, we observed that these IBMPFD proteins are mainly in the soluble fractions (Fig. 7C and data not shown). Thus, the dysregulated nucleocytoplasmic shuttling of IBMPFD mutants is not caused by protein aggregation.
Finally, we showed that the C-terminal region can promote the nuclear entry of N domain mutants. Co-transfection of VCP-Δ780-806 increases the nuclear distribution of VCP-Δ1-206 and IBMPFD mutants. Since hexamerization is mainly achieved through the D1 domain and GFP does not affect the hexamerization of VCP, VCP-Δ780-806 should form hybrids with the N domain mutants. In this way, VCP-Δ780-806 promotes the nuclear entry of N domain deletion and IBMPFD mutants in trans. Consistently, co-transfection of WT-GFP does not significantly increase the nuclear distribution of VCP, suggesting most of WT-GFP proteins hybridize with N domain mutants and exhibit suppressed nuclear entry. In addition, VCP-Δ1-206-Δ780-806 with the double deletion of both N domain and the C-terminal region showed higher nuclear distribution than VCP-Δ1-206, suggesting that G780AGPSQ also increases the nuclear distribution in cis. Thus, the C-terminal region may be a potential molecular target for screening chemical entities to increase the nuclear distribution of IBMPFD VCP proteins.
Highlights.
IBMPFD mutations decrease the nuclear distribution of VCP
The peptide G780AGPSQ of VCP regulates its retention in the nucleus and nucleoli
C-terminal region deletion elevates the nuclear retention of VCP IBMPFD mutants
Acknowledgments
We thank Drs. Carols Barrero, Ying Wang and Ren-Ming Dai for technical support and Dr. Madesh Muniswamy for providing the confocal imaging facility. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. The publisher or recipient acknowledges the right of the US Government to retain a nonexclusive, royalty-free license in and to any copyright covering the manuscript. This research was funded by the NIEHS 1R21ES023051 for C.S. Other supports include: DA-14230, DA-25532, and DA 13429 for T.J.R.; NCI Contract No. NO1CO56000 and HHSN261200800001E for S.L; Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research for N.C., C.C.L. and J.M.W.; National Natural Science Foundation of China No. 30672433, 30271215 and Natural Science Foundation of Shandong Province No. 1872010ZRE27212 for S.C.Z..
Abbreviations
- VCP or p97
valosin containing protein
- AAA
ATPases associated with diverse cellular activities
- ER
endoplasmic reticulum
- ATP
adenosine triphosphate
- IBMPFD
inclusion body myopathy associated with Paget’s disease of bone and frontotemporal dementia
- HEK293
human embryonic kidney 293
- FRAP
fluorescence recovery after photobleaching
- FIRNC
fluorescence intensity ratio between the nucleus and cytoplasm
- NES
nuclear export signal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang Q, Song C, Li CC. Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological functions. J Struct Biol. 2004;146:44–57. doi: 10.1016/j.jsb.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 2.Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14:117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- 3.Yamanaka K, Sasagawa Y, Ogura T. Recent advances in p97/VCP/Cdc48 cellular functions. Biochim Biophys Acta. 2012;1823:130–137. doi: 10.1016/j.bbamcr.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Partridge JJ, Lopreiato JO, Jr, Latterich M, Indig FE. DNA Damage Modulates Nucleolar Interaction of the Werner Protein with the AAA ATPase p97/VCP. Mol Biol Cell. 2003;14:4221–4229. doi: 10.1091/mbc.E03-02-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Madeo F, Schlauer J, Zischka H, Mecke D, Frohlich KU. Tyrosine phosphorylation regulates cell cycle-dependent nuclear localization of Cdc48p. Mol Biol Cell. 1998;9:131–141. doi: 10.1091/mbc.9.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, Song C, Yang X, Li CC. D1 ring is stable and nucleotide-independent, whereas D2 ring undergoes major conformational changes during the ATPase cycle of p97-VCP. J Biol Chem. 2003;278:32784–32793. doi: 10.1074/jbc.M303869200. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, Gorman MA, Kondo H, Dokurno P, Lally J, Leonard G, Meyer H, van Heel M, Freemont PS. Structure of the AAA ATPase p97. Mol Cell. 2000;6:1473–1484. doi: 10.1016/s1097-2765(00)00143-x. [DOI] [PubMed] [Google Scholar]
- 8.Song C, Wang Q, Li CC. ATPase activity of p97-valosin-containing protein (VCP). D2 mediates the major enzyme activity, and D1 contributes to the heat-induced activity. J Biol Chem. 2003;278:3648–3655. doi: 10.1074/jbc.M208422200. [DOI] [PubMed] [Google Scholar]
- 9.Yeung HO, Kloppsteck P, Niwa H, Isaacson RL, Matthews S, Zhang X, Freemont PS. Insights into adaptor binding to the AAA protein p97. Biochem Soc Trans. 2008;36:62–67. doi: 10.1042/BST0360062. [DOI] [PubMed] [Google Scholar]
- 10.Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 11.Badadani M, Nalbandian A, Watts GD, Vesa J, Kitazawa M, Su H, Tanaja J, Dec E, Wallace DC, Mukherjee J, Caiozzo V, Warman M, Kimonis VE. VCP associated inclusion body myopathy and paget disease of bone knock-in mouse model exhibits tissue pathology typical of human disease. PLoS One. 2010;5:e13183. doi: 10.1371/journal.pone.0013183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeLaBarre B, Brunger AT. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol. 2003;10:856–863. doi: 10.1038/nsb972. [DOI] [PubMed] [Google Scholar]
- 13.Song C, Xiao Z, Nagashima K, Li CC, Lockett SJ, Dai RM, Cho EH, Conrads TP, Veenstra TD, Colburn NH, Wang Q, Wang JM. The heavy metal cadmium induces valosin-containing protein (VCP)-mediated aggresome formation. Toxicol Appl Pharmacol. 2008;228:351–363. doi: 10.1016/j.taap.2007.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Indig FE, Partridge JJ, Kobbe CCv, Aladjem MI, Latterich M, Bohr VA. Werner syndrome protein directly binds to the AAA ATPase p97/VCP in an ATP-dependent fashion. J Struct Biol. 2004;146:251–259. doi: 10.1016/j.jsb.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Song C, Li CC. Hexamerization of p97-VCP is promoted by ATP binding to the D1 domain and required for ATPase and biological activities. Biochem Biophys Res Commun. 2003;300:253–260. doi: 10.1016/s0006-291x(02)02840-1. [DOI] [PubMed] [Google Scholar]
- 16.Niwa H, Ewens CA, Tsang C, Yeung HO, Zhang X, Freemont PS. The role of the N-domain in the ATPase activity of the mammalian AAA ATPase p97/VCP. J Biol Chem. 2012;287:8561–8570. doi: 10.1074/jbc.M111.302778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halawani D, LeBlanc AC, Rouiller I, Michnick SW, Servant MJ, Latterich M. Hereditary inclusion body myopathy-linked p97/VCP mutations in the NH2 domain and the D1 ring modulate p97/VCP ATPase activity and D2 ring conformation. Mol Cell Biol. 2009;29:4484–4494. doi: 10.1128/MCB.00252-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manno A, Noguchi M, Fukushi J, Motohashi Y, Kakizuka A. Enhanced ATPase activities as a primary defect of mutant valosin-containing proteins that cause inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Genes Cells. 2010;15:911–922. doi: 10.1111/j.1365-2443.2010.01428.x. [DOI] [PubMed] [Google Scholar]
- 19.Song C, Wang Q, Li CC. Characterization of the aggregation-prevention activity of p97/valosin-containing protein. Biochemistry. 2007;46:14889–14898. doi: 10.1021/bi700499j. [DOI] [PubMed] [Google Scholar]
- 20.Subjeck JR, Shyy T, Shen J, Johnson RJ. Association between the mammalian 110,000-dalton heat-shock protein and nucleoli. J Cell Biol. 1983;97:1389–1395. doi: 10.1083/jcb.97.5.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Latonen L, Moore HM, Bai B, Jaamaa S, Laiho M. Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene. 2011;30:790–805. doi: 10.1038/onc.2010.469. [DOI] [PubMed] [Google Scholar]
- 22.Palanca A, Casafont I, Berciano MT, Lafarga M. Reactive nucleolar and Cajal body responses to proteasome inhibition in sensory ganglion neurons. Biochim Biophys Acta. 2014;1842:848–859. doi: 10.1016/j.bbadis.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 23.Tang WK, Li D, Li CC, Esser L, Dai R, Guo L, Xia D. A novel ATP-dependent conformation in p97 N-D1 fragment revealed by crystal structures of disease-related mutants. EMBO J. 2010;29:2217–2229. doi: 10.1038/emboj.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujiwara Y, Fujiwara K, Goda N, Iwaya N, Tenno T, Shirakawa M, Hiroaki H. Structure and function of the N-terminal nucleolin binding domain of nuclear valosin-containing protein-like 2 (NVL2) harboring a nucleolar localization signal. J Biol Chem. 2011;286:21732–21741. doi: 10.1074/jbc.M110.174680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutten S, Kehlenbach RH. CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol. 2007;17:193–201. doi: 10.1016/j.tcb.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278:141–144. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- 27.Palmeri D, Malim MH. The human T-cell leukemia virus type 1 posttranscriptional trans-activator Rex contains a nuclear export signal. J Virol. 1996;70:6442–6445. doi: 10.1128/jvi.70.9.6442-6445.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- 29.Fukuda M, Gotoh I, Gotoh Y, Nishida E. Cytoplasmic localization of mitogen-activated protein kinase kinase directed by its NH2-terminal, leucine-rich short amino acid sequence, which acts as a nuclear export signal. J Biol Chem. 1996;271:20024–20028. doi: 10.1074/jbc.271.33.20024. [DOI] [PubMed] [Google Scholar]
- 30.Wang Q, Song C, Irizarry L, Dai R, Zhang X, Li CC. Multifunctional roles of the conserved Arg residues in the second region of homology of p97/valosin-containing protein. J Biol Chem. 2005;280:40515–40523. doi: 10.1074/jbc.M509636200. [DOI] [PubMed] [Google Scholar]
- 31.Meyer HH, Kondo H, Warren G. The p47 co-factor regulates the ATPase activity of the membrane fusion protein, p97. FEBS Lett. 1998;437:255–257. doi: 10.1016/s0014-5793(98)01232-0. [DOI] [PubMed] [Google Scholar]