

Abstract
Human-associated microbes are the source of many bioactive microbial products (proteins and metabolites) that play key functions both in human host pathways and in microbe-microbe interactions. Culture-independent studies now provide an accelerated means of exploring novel bioactives in the human microbiome; however, intriguingly, a substantial fraction of the microbial metagenome cannot be mapped to annotated genes or isolate genomes and is thus of unknown function. Meta'omic approaches, including metagenomic sequencing, metatranscriptomics, metabolomics, and integration of multiple assay types, represent an opportunity to efficiently explore this large pool of potential therapeutics. In combination with appropriate follow-up validation, high-throughput culture-independent assays can be combined with computational approaches to identify and characterize novel and biologically interesting microbial products. Here, we briefly review the state of microbial product identification and characterization and discuss possible next steps to catalog and leverage the large uncharted fraction of the microbial metagenome.
2 Introduction: the great microbial unknown
The human microbiome comprises trillions of bacteria, archaea, fungi, protozoa, and viruses. Disruptions in host-microbe balance are associated with a wide range of diseases, including obesity (Ley et al., 2006; Turnbaugh et al., 2006), malnutrition (Smith et al., 2013a), inflammatory bowel disease (IBD) (Dicksved et al., 2008; Morgan et al., 2012), liver disease (Wong et al., 2013; Zhu et al., 2013), and cancer (Castellarin et al., 2012; Iida et al., 2013; Kostic et al., 2012). The microbiota of healthy populations, which have been cataloged by efforts such as the Human Microbiome Project (HMP) (Human Microbiome Project Consortium, 2012b) and MetaHIT (Qin et al., 2010), encode at least 100-fold more genes than do their human hosts, and it is estimated that ∼10% of all circulating metabolites in the human body are microbially derived (Wikoff et al., 2009). Pharmaceuticals as fundamental as tetracycline are microbial products, and human-associated microbial isolates have historically yielded therapeutic compounds ranging from antibiotics to antitumor therapies (Berdy, 2005). Culture-independent assays now provide a route to identify new classes of biologically active molecules, which may represent novel therapeutics themselves or, alternatively, targets for further drug discovery (e.g. by inhibition or competitive binding) (Lemon et al., 2012). Processes such as host-microbe cross-talk, immune activation and inflammation, microbe-microbe signaling, microbial metabolism, and antimicrobial activity are all, by definition, bioactive in ecosystems such as the human gut (Holmes et al., 2012). Likewise, a wide range of molecules are candidate mediators of these processes, including small molecule microbial products of primary metabolism (e.g. short-chain fatty acids) as well as a diverse array of secondary metabolites including both secreted and cell surface peptides or sugars (Fischbach and Sonnenburg, 2011; Lopez et al., 2014).
Only in recent years has it become routine to perform culture-independent assessments of microbial communities. Due to the falling costs of sequencing, metagenomes are one of the fastest-growing sources of new microbial community data. As of 2013, a total of over 20,000 microbial community profiles had been deposited in the sequence read archive (SRA), comprising an estimated 10 million genes (Li et al., 2014; NCBI Resource Coordinators, 2014). Even prior to this flood, however, it was well-known that functional characterization of microbial genes lags behind the ability of the field to generate new microbial sequence data (Galperin and Koonin, 2010). Between 30 and 40% (and often as much as 60 to 70%) of the genes from newly-sequenced microbial isolates are functionally uncharacterized despite a growing database of available reference information (Fodor et al., 2012; Galperin and Koonin, 2004).
The problem of uncharacterized novel microbial gene sequences is further exacerbated in microbial communities, in which a large proportion of genes community-wide remains uncharacterized after annotation. Roughly 50% of genes in the gut microbiomes of HMP participants, for example, could not be characterized using standard annotation methods (Human Microbiome Project Consortium, 2012a) (Fig. 1a). Even among genes with putative function (e.g. EC number, corresponding to the specific chemical reactions catalyzed), the majority remained broadly annotated (e.g. the enzyme was classified as a hydrolase, but its substrates were unclear). Furthermore, specific annotations, when provided, were highly unevenly distributed across functional groups (e.g. enzyme families, Fig. 1b). This large fraction of uncharacterized genes dramatically inhibits our ability to understand the functional activity and systems biology of microbial communities. It represents an enormous opportunity, however, to identify novel and biologically interesting microbial products. It is therefore critical to develop efficient hypothesis-generating pipelines and validation approaches in order to successfully mine bioactive products from the vast genetic reservoir of the microbiome.
Figure 1. Uncharacterized microbial genes represent over half of the human gut metagenome.
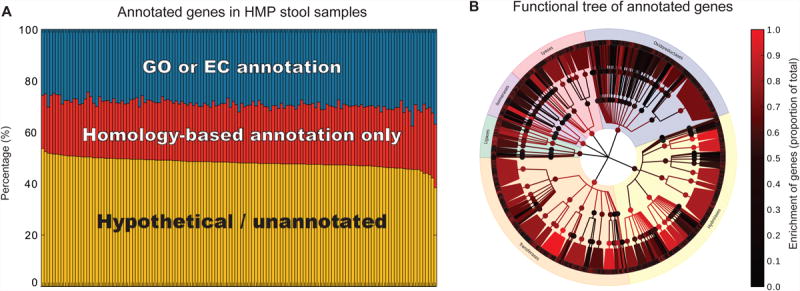
Data from 139 Human Microbiome Project (HMP) stool sample shotgun metagenomes (Human Microbiome Project Consortium, 2012b). The HMP Data Analysis and Coordinating Center (http://hmpdacc.org) annotated microbial community genes with a GO term (Ashburner et al., 2000), EC number (Bairoch, 2000), and/or gene name when possible. (A) Relative abundances of GO and/or EC annotated genes, uncharacterized genes with a homology-based gene name, and completely uncharacterized genes. (B) Distribution of EC-annotated genes in the Enzyme Commission functional hierarchy. Tree shows log-scaled percent coverages of direct EC annotations at each level as observed in the stool samples. With the exception of transferases and hydrolases, even when microbial genes receive EC annotations, they are often at non-specific higher levels within the hierarchy rather than to specific EC subclasses, highlighting the need for deeper microbial gene product characterization in the human microbiome.
Most current bioinformatic methods for gene function prediction provide gene annotations by alignment to homologous genes with existing annotations (e.g. BLAST) (Finn et al., 2013; Gish and States, 1993; Powell et al., 2014) (Fig. 2). Such reference-based strategies are of course limited by the availability of appropriate curated reference genomes. Although thousands of microbial genomes are now being sequenced each year, remarkably, the vast majority of bacterial genomes sequenced to date come from only four phyla (Rinke et al., 2013). Furthermore, automatic reliance on homology-based functional annotation approaches has led to the accrual of uncharacterized and poorly characterized genes, a known problem in microbial genomes that has now emerged in even more dramatic form for community metagenomes (Richardson and Watson, 2013; Wood et al., 2012).
Figure 2. Identification and validation of microbe-derived gene product functions.
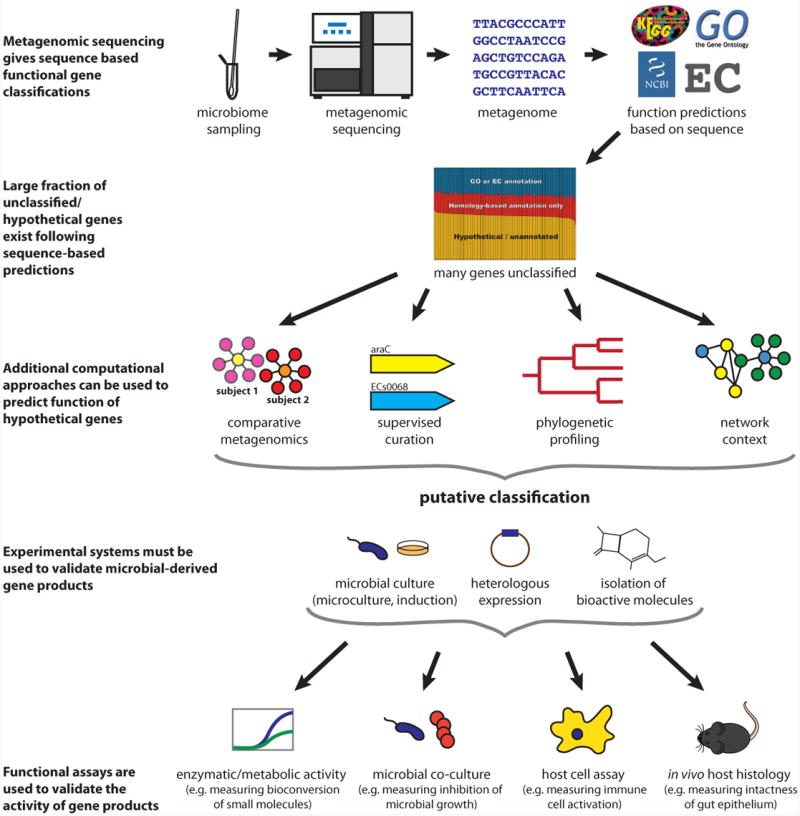
An overview of the process of microbial gene functional annotation and validation. In microbial isolate genomes and metagenomes alike, gene function is typically first assigned using standard sequence analysis methods (homology-based assignment (Loewenstein et al., 2009) and domain profiling (Finn et al., 2013)). These predictions can be further refined by additional bioinformatic approaches, such as comparative metagenomics (“guilt by association” of uncharacterized microbial products with characterized genes across samples through the use of data integration), supervised curation (manual determination of a consensus among multiple complementary automated annotations (Richardson and Watson, 2013)), phylogenic profiling (analysis of co-occurrence of genes across isolates (Eisen and Fraser, 2003)), and network context (“guilt by association” in isolate coexpression, interaction, or functional linkage networks (Sharan et al., 2007)). Following putative classification, bioactivity must be validated and further characterized by experimental methods. When standard culture is challenging (as is common for the microbiome), microculture and induction culture, as well as heterologous expression of genes and direct isolation of products are particularly useful. Functional assays for investigating the activity of microbial products include enzymatic/metabolic activity assays (Craciun and Balskus, 2012), microbial co-culture (Yan et al., 2013), host cell profiling (Wieland Brown et al., 2013), and in vivo host phenotype assessments (Olle, 2013).
Other computational methodologies, e.g. comparative metagenomics, phylogenetic profiling, and network context-based approaches (Bornigen et al., 2012; Goncalves et al., 2012; Hwang et al., 2011; Park et al., 2010; Park et al., 2013; Radivojac et al., 2013; Wang et al., 2012; Zuberi et al., 2013), integrate additional information to generate hypotheses regarding gene function, while manual curation of automated sequence-based function predictions can provide additional insight (Fig. 2). These methods can be combined with more standard homology-based methods to elucidate putative gene product functions for subsequent experimental validation. Though these methods have been available for years, they are currently underused in many fields of genomic research. They have not been extensively applied toward better characterizing products from culture-independent microbial communities in general or from the human microbiome in particular. Efforts to better characterize these functionally-uncharacterized microbial genes, especially those from the human microbiome, represent a promising area of research as a result. Not only will this research lead to an improved understanding of basic microbial biology, but novel human-associated microbial products are likely to have important functions in human health. Thousands of metagenomes - human-associated and otherwise - have already been sequenced, representing an extensive database for mining biologically active microbial products. Even now, re-analyses of these data using a systematic, integrated approach to microbial product characterization could easily yield, for example, a “Most Wanted Genes” list (analogous to the HMP's “Most Wanted Taxa” (Fodor et al., 2012)), comprising abundant or otherwise “important” uncharacterized genes in the human microbiome.
The aims of this perspective are thus (i) to review major categories of bioactive microbial products already isolated from the human microbiome, (ii) to discuss methods used to experimentally validate these functions, (iii) to suggest complementary computational approaches that can be used to improve characterization of the uncharacterized fraction of the metagenome, and (iv) to describe methods for integrating data across multiple platforms and studies to validate hypothesized gene functions.
3 Microbe-derived metabolites affect both host and community
The human gut harbors a multi-million gene microbial metagenome that outnumbers the human gene complement by at least 150 fold and produces an extraordinary array of structural components, cell surface molecules, and metabolic enzymes and byproducts (Qin et al., 2010). Although approximately half of these genes have unknown or poorly-characterized functions (Human Microbiome Project Consortium, 2012b; Qin et al., 2010), studies to date have revealed a vibrant, dynamic ecosystem in which microbial community members influence host functions as well as affect the survival of other microbes in the ecosystem.
3.1 Microbial products involved in host-microbe interactions
Microbial proteins and metabolites are essential in host digestion and biochemistry. The gut microbiota synthesizes valuable nutrients such as vitamins B12 and K (LeBlanc et al., 2013) and produces enzymes that enable the breakdown of complex carbohydrates such as cellulose that would otherwise be indigestible by the host (Xu et al., 2003). Microbial alteration of bile acids plays a key role in the digestion and absorption of lipids, as well as in glucose homeostasis (Jones et al., 2014). Gut microbiota have also been shown to modify drugs consumed by the host, potentially altering drug response (Haiser et al., 2013; Wallace et al., 2010). Microbial communities in other sites of the body also play a role in host biochemistry. In the vagina, the Lactobacillus species produces lactic acid, which maintains a low vaginal pH and inhibits colonization by non-commensal organisms (Ravel et al., 2011).
In addition to host homeostatic and metabolic functions, microbial products are involved in maintaining epithelial barrier function and immune homeostasis. For example, the bacterial fermentation products short chain fatty acids (SCFAs: i.e. acetate, propionate, butyrate) promote normal gut epithelial function, as reviewed in Maslowski et al 2011 (Maslowski and Mackay, 2011). Another major role of microbial products is their role in immune regulation. SCFAs, along with other microbial products and metabolites such as bile acids, peptidoglycan, and sphingolipids have all been described to modulate the immune system, as reviewed in Brestoff et al 2013 (Brestoff and Artis, 2013). While the list of known microbial products is comparatively small in comparison to the microbial metagenome, these characterized products have already been shown to have far-reaching implications for human hosts.
3.2 Microbial products involved in microbe-microbe interactions
The ability of a human-associated microbe to survive and thrive is contingent upon active maintenance of a balanced relationship not only with the host, but also with the microbial community. Consequently, microbes produce many products dedicated to preserving ecosystem homeostasis via microbe-microbe interactions. To monitor their own species abundance in the community, microbes produce and detect self-made metabolites in the process of quorum sensing (Bassler and Losick, 2006); they also secrete inhibitory metabolites such as bacteriocins to directly kill or inhibit growth of competing organisms (Cotter et al., 2005; McCaughey et al., 2014). Currently-characterized mediators of microbe-microbe interactions fall into two broad classes: within-clade positive signaling, and between-clade antagonism. These are extensively reviewed elsewhere (Kuramitsu et al., 2007; Little et al., 2008; Marx, 2009; Phelan et al., 2012). A recent example from the nasal microbial community, in which a species of Corynebacterium inhibited the growth of Staphylococcus aureus in vitro through an as-yet-unidentified mediator (Yan et al., 2013), underscores that a potentially vast array of ecological interactions and mediating molecules have yet to be discovered.
3.3 Functional characterization of microbial products in vitro and in vivo
A wide range of functional assays can be used to evaluate the biological activity of microbial gene products, particularly when culturable isolates are available for the microbe(s) of interest. Testing the effects of microbial products on host cell function requires three components: a host cell system to perturb, a microbially-derived perturbation, and a readout of host cell state. The first component may be a model organism, gnotobiotic animal, cell culture, organoid (Sato and Clevers, 2013), or a structured culture such as a transwell (Moon et al., 2014) or gut-on-chip (Huh et al., 2013). The second component may include live or inactivated microbial cells, whole lysate, media, or an isolated, purified, or synthesized product. The third component includes a range of functional assays to assess the bioactivity of microbial products. These may include assays for enzymatic activity, immune cell activation, or pathology in an animal model. As many human-associated microbes are challenging to culture, combinatorial genetics provides an alternative route for investigating genes of interest identified meta'omically. Genes may be cloned and expressed heterologously in a model organism such as E. coli; or alternatively, knock-out approaches in phylogenetically-related isolates or homologous pathways may validate the activity of a bioactive gene product. A combination of experimental approaches have been used to demonstrate the immunomodulatory activity of some of the well-known microbial products, such as the SCFAs (Arpaia et al., 2013; Atarashi et al., 2013; Maslowski et al., 2009; Smith et al., 2013b), bacterial polysaccharide (PSA) (Mazmanian et al., 2005) and sphingolipids (An et al.).
4 Computational approaches to hypothesize function for human-associated microbial gene products
Uncharacterized novel gene sequences abound in microbial genomes, and the ever-increasing rate of sequence data generation drives a great need for a renewed focus on effectively using existing computational methods for putative gene characterization and functional prediction. In addition to supervised curation (i.e. manual correction of annotated genomes), which can greatly aid in assigning appropriate gene function (Richardson and Watson, 2013), a number of computational approaches can improve upon the simplest homology-based strategies (e.g. best BLAST hit) for assigning gene function. These include (i) advanced sequence-guided methods, (ii) structure-based approaches, (iii) functional prediction methods based on evolutionary conservation and phylogeny, and (iv) approaches that use gene context within networks (e.g. coexpression or metabolic networks) in order to guide functional assignment (Fig. 2).
4.1 Advanced sequence-based approaches
The simplest form of homology-based sequence annotation begins with an unannotated nucleotide sequence, searches its translated amino acid sequence against one or more annotated microbial protein catalogs, and assigns an annotation to the new sequence if nucleotide or amino acid identity exceeds a predefined threshold. Threshold recommendations for confidently transferring function (i.e. assigning function based on sequence similarity) range from 40% to 80% amino acid identity (Rost, 2002; Tian and Skolnick, 2003; Todd et al., 2001). This process requires a number of subjective assumptions regarding nucleotide and amino acid conservation, the relationships between primary sequence and protein function, and evolutionary mutation rates. More sophisticated methods supplement simple best-hit approaches with comparative genomics, comparing sequences to databases of gene products, functions, and pathways to place them into functional context; these databases may include Gene Ontology (GO) (Ashburner et al., 2000), Clusters of Orthologous Genes (COG) (Tatusov et al., 2003), Enzyme Commission (EC) (Bairoch, 2000), or Kyoto Encyclopedia of Genes and Genomes (KEGG)(Kanehisa et al., 2004). Many comparative genomics-based automated pipelines have been built for bacterial genome annotation (Stothard and Wishart, 2006); more recently, annotation pipelines such as MG-RAST (Meyer et al., 2008) and IMG/M (Markowitz et al., 2012) were created for metagenomes.
Unfortunately, overall similarity in protein sequence does not guarantee similarity of function (Bork and Koonin, 1998; Rost, 2002; Rost et al., 2003; Skolnick and Fetrow, 2000; Whisstock and Lesk, 2003). This issue can be partially mitigated by extending the best-hit annotation strategy to include comparisons with sequence-diverse protein families or recurring sequence motifs. For example, the SMART (Schultz et al., 1998) and Pfam (Punta et al., 2012) databases catalog recurring protein domains and binding motifs that can be directly compared with an unannotated protein sequence. Position-specific iterative BLAST (PSI-BLAST) (Altschul et al., 1997) takes a related approach to this problem: an unannotated protein is first searched against a broad sequence database; the hits from this initial search are then used to guide subsequent searches of the database, gradually building a sequence-diverse family centered on the unannotated protein. Notably, PSI-BLAST was used to identify a large group of highly-abundant protein families in the human gut microbiome that were missed by Pfam (Ellrott et al., 2010).
When extending homology-based annotation techniques beyond the best-hit approach, additional statistical techniques are necessary to detect remote homology and combine annotations from multiple homologous hits. Nearly any machine learning technique can be (and has been) employed for this purpose. For example, hidden Markov models (HMM) are used in SMART and Pfam to model site-specific amino acid distributions across a protein family, while k-nearest neighbor classifers (kNN) have been used to predict GO function based on interrelationships between functional classes (Pandey et al., 2009). GOPET uses support vector machines (SVM) to incorporate data from GO and BLAST and assign confidence levels to each GO prediction (Vinayagam et al., 2006). SVMs are also used in FFPred to predict function in eukaryotes by comparing unannotated sequences to a set of sequence-based features from well-characterized proteins, including amino acid composition, secondary structure, posttranslational modification sites, and localization signals (Lobley et al., 2008). Many of these described techniques have been developed in eukaryotic model organisms, with as-yet limited applications in microbial isolates or communities.
4.2 Structure-guided approaches
As discussed above, various methods exist for transferring annotations from well-characterized proteins to an unannotated protein based on sequence-level homology. These methods are based on the observations that (i) proteins of similar sequence tend to adopt similar 3D structures and (ii) that proteins of similar 3D structure tend to perform similar functions. Naturally, this suggests that one could directly compare an unannotated protein to a library of 3D structures to aid in annotation transfer, and indeed several methods have been proposed to do exactly this. For example, ProFunc, SuMo, and RASMOT-3D, are analogous to functional assignment based on sequence homology, but instead identify compatibility between an unnannotated protein and well-characterized three-dimensional protein structures (Debret et al., 2009; Jambon et al., 2005; Laskowski et al., 2005). Local-global alignment (LGA) (Zemla, 2003) is a further example that behaves as a structural analog to PSI-BLAST. This method iteratively identifies local similarity at the protein structure level, and is one of several methods that increase the specificity of annotation transfer by focusing on local structural motifs for which function is likely to be conserved. Indeed, in cases of remote homology, the ability of structure-guided methods to assess the functional impact of conserved versus non-conserved regions represents a key advantage over sequence-focused methods.
4.3 Phylogenetic and evolutionary approaches
Another approach in computational gene product characterization is to assess the patterns by which genes are evolutionarily conserved, mutated, or lost throughout the microbial phylogeny, a process referred to as phylogenomic profiling (Eisen and Fraser, 2003). Most straightforwardly, if over evolutionary time an uncharacterized gene is gained or lost only in tandem with genes in a characterized pathway, the uncharacterized gene may be placed within that pathway or regulon. This approach can be extended from the broad level of gene gain and loss to the more specific level of joint site-specific mutation rates. For example, proteins that directly bind specific substrates, such as ATP, have been identified by local evolutionary conservation (or substitution) of individual binding site residues (Fang et al., 2014). Tools for this type of evolutionarily-informed analysis include SIFTER (Engelhardt et al., 2005), RIO (Zmasek and Eddy, 2002), OrthoGUI (Hollich et al., 2002), and FlowerPower (Krishnamurthy et al., 2007). Other approaches such as evolutionary tracing combine residue conservation with tertiary structure analysis to identify groups of functionally significant residues, such as exposed active sites (Amin et al., 2013). Particularly in the microbial tree of life, where thousands of isolate genomes are available and genome content is particularly plastic, phylogenomic approaches have been shown to be extremely powerful.
4.4 Gap-filling approaches based on metabolic network reconstruction
An interesting and highly complementary approach for inferring the biochemical functions of some gene products is referred to as “sins of omission” in microbial metabolic network analysis. If a microbial genome carries the vast majority of genes necessary to synthesize an important compound, but a few necessary enzymes remain uncharacterized, the rest of the genome can be more closely scrutinized for gene products able to carry out those roles. One recent example of such an investigation was the remote sequence homology-based identification and experimental validation of the enzyme necessary for processing choline to trimethylamine in the human gut microbiome (Craciun and Balskus, 2012). This approach can be formalized by the process of metabolic network reconstruction using methods such as flux-balance analysis (FBA) (Lewis et al., 2012), which enables genome-wide modeling of the metabolism of an organism. For example, MetaFlux (Latendresse et al., 2012) can be used to identify gaps in an organism's network of biotransformations. Such gaps include “dead-end” metabolites that are known to be consumed or produced but do not appear in the network, and “orphan” reactions that are known to occur, but the genes encoding the necessary enzymes are not present (Orth and Palsson, 2010). While metabolic networks are typically constructed for single organisms, further research is likely to render them feasible for multi-organism communities (Levy and Borenstein, 2013; Zengler and Palsson, 2012).
4.5 Integrative approaches to predict protein function
The most recent and arguably most successful methods for protein function prediction integrate information from many sources, such as primary sequence, tertiary structure, and evolutionary conservation. Hess et al (Hess et al., 2009), for example, used an ensemble method combining three different Bayesian approaches that integrated sequence features, coexpression, protein-protein interactions, binding sites, and subcellular localization to improve protein function prediction. Other studies have combined tertiary structure with primary structural homology and secondary structure biochemistry (Wang et al., 2014); sequence data with gene expression, protein-protein interactions, and evolutionary conservation (Cozzetto et al., 2013); sequence with interaction profiles and domain co-occurrence (Wang et al., 2013); and sequence features with active site motifs and structural alignment for determination of protein fold activities (Zakeri et al., 2014). Purely technical issues have impeded the application of these methods to microbial genomes and metagenomes. These include problems with computational efficiency, consistent identification schemes for microbial taxonomy and gene products, and the assembly of appropriate training data from diverse sources. All of these are surmountable given further work on deep characterization of human-associated microbial gene products.
5 Integration within and among studies in the meta'omic era
5.1 Statistical techniques for data integration
Different types of ‘omics data can be combined in many biologically-informed ways to identify robust “hits”, or novel microbial products that are of interest for follow up characterization. A range of well-studied statistical techniques can be used to integrate high-dimensional data, particularly when it is multivariate (i.e. incorporates multiple features of interest) and heterogeneous (i.e. consists of measurements of multiple types; Fig. 3). These techniques include (i) repeated univariate associations, or networks; (ii) ordination; and (iii) hierarchical regression-based modeling. Each of these methods can be used to integrate either across different meta'omic data types or across different studies containing the same data types. Any type of interaction (or, more commonly, covariation) can be usefully represented as a network: correlation or distance of features such as gene or microbial abundances, or any type of expression data (transcript, protein, metabolite, etc.). Individual (pairwise, univariate, etc.) associations can be tested for statistical significance, or the network structure overall can be analyzed to demonstrate how genes cluster in terms of expression pattern, or how pathways consume and produce metabolites.
Figure 3. Integration methods for multiple data types or datasets.
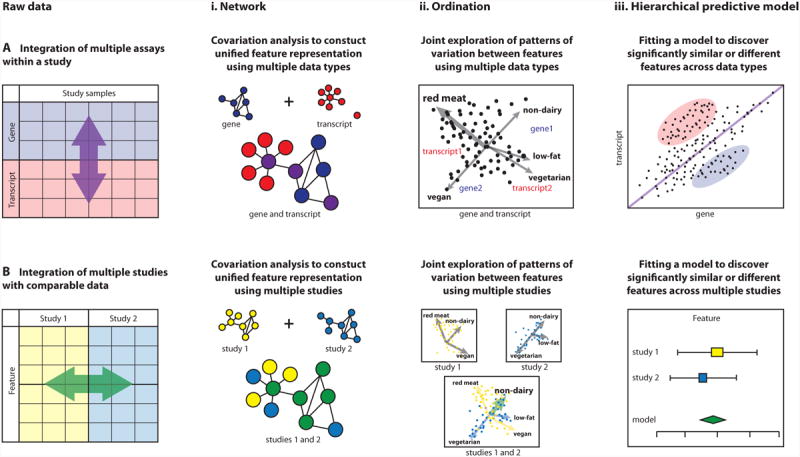
Schematic of approaches for data integration either (A) among different data types within the same study or (B) across different studies assaying the same data type. Integration methods include (i) network analyses capturing similarity of genes/gene products/microbes (correlation, co-abundance, co-expression, etc); (ii) ordination projections, showing overall patterns of clustering or co-variation (shown here applying to samples, can also apply to gene/microbial features); and (iii) hierarchical statistical models such as regression that quantify the degree of association among genes/microbes and sample phenotypes. Each of these methods can be applied to one or more assay types (and phenotype metadata) within study, or they can be applied to a combination of multiple studies. (Ai) Networks of covarying features can be generated separately for different data types (e.g. gene and transcript) or using both data types in one unified network by correlating multiple feature types. (Bi) Networks of covarying features can also be generated separately for different studies or can be summarized in one network to relate features that covary in both studies. (Aii) A combined ordination (or biplot) of multiple data types (e.g. gene and transcript) can reveal patterns of variation that enrich one or more data types or metadata (e.g. red meat consumption) in particular subsets of samples. In this example, samples are ordinated jointly with metadata, genes, and transcripts. (Bii) Ordination can be used to understand patterns of variation either independently in different studies, or a joint ordination can reveal patterns of sample co-variation across studies, possibly as linked to common metadata (e.g. consumption of non-dairy diet). (Aiii) Statistically significantly (un)related features can be identified by formal models such as linear regression. Regression among linked data types (e.g. genes and transcripts) can quantify the degree to which features or metadata associate across data types. In this example, we show feature levels that are similar between data types (close to the diagonal) as well as those that are significantly up- or down-regulated. (Biii) Statistical models can be meta-analyzed by applying them within each study, determining the significance and variability of a result within each study individually, and then comparing the resulting significance and effect sizes across studies. Meta-analysis can be used to detect signals too weak to see in any one study or to assess the reproducibility of a result across studies.
Network representations of feature associations can incorporate data from multiple meta'omics platforms in the same study to form a single integrative network, or their contents and structure can be compared across multiple studies to determine similarities or differences (Fig. 3Ai, Bi). Alternatively, ordinations (such as principal coordinates analysis) are a family of projection methods that provides low-dimensional visualization of high-dimensional data. These can be used to qualitatively determine data clustering patterns, with regard to either metadata (e.g. consumption of red meat, non-dairy, low-fat, vegetarian or vegan diet) or meta'omic features (microbial taxa, genes, transcripts, etc.). This enables assessment of the metadata that covary with the greatest variation among samples, or determination of which samples are enriched for features of interest (e.g. which samples have the greatest abundance of one or more microbes of interest, Fig. 3Aii, Bii). Ordination typically shows these relationships only qualitatively. Although this is useful for visualization, a more quantitative test of statistical significance using hierarchical modeling (such as linear regression) should be performed. This will determine which features (microbial taxa, genes, transcripts, etc.) are associated with one another and with metadata in a multivariate manner. Regression analyses provide this type of significance test for relationships among features of different types within datasets (Fig. 3Aiii), while meta-analyses provide similar results for features of the same type across datasets (Fig. 3Biii).
5.2 Integrating across different data types from the same study
While metagenomic sequencing assays define the total genetic potential of a microbial community, they describe neither which genes are actively being transcribed and translated into proteins, nor the relative quantities of each gene product that each cell is expected to produce. Metatranscriptomics and metaproteomics are therefore excellent complementary approaches to metagenomics, as they can be used to evaluate the environments in which gene products are produced or differentially expressed (e.g. body site, disease state, nutritional intake). For most microbial community analyses, it is critical to pair the measured gene expression (e.g. transcripts, proteins) of each sample with a measure of gene abundance (e.g. metagenomics). It is otherwise impossible to determine whether changes in gene product abundances arise due to differential underlying cell count (e.g. microbial growth) or to differential expression or activity.
Regression analysis can be used as an integrative approach for combining gene and transcript data (as depicted in Fig. 3Aiii) in microbiome studies, demonstrating the upregulation of a subset of pathways under specific conditions. For example, in one study, iron acquisition and lipopolysaccharide synthesis pathways were highly upregulated in subjects with periodontal disease as compared with healthy subjects (Duran-Pinedo et al., 2014). In another study, although Methanobrevibacter smithii was present only at low abundance (<1%), in the human gut, genes in the methanogenesis pathway were highly transcribed, suggesting the importance of that pathway in gut function (Franzosa et al., 2014). The additional context of transcriptional activity may also prove informative in understanding gene functionality with regard to host condition (e.g. identifying/characterizing novel virulence factors based on upregulation during host disease); thus, integrative approaches will be useful in the characterization of novel genes.
In addition to transcriptomics, the integration of metagenomic data with metabolomics can aid in identifying the specific microbial gene products that underlie associations found between metagenomic taxonomy data and clinical/phenotypic metadata. For example, a combination of 16S sequence profiling and metabolomics was recently used to identify 4EPS as one of several metabolites with altered levels in the maternal immune activation mouse model of autism (Hsiao et al., 2013). 16S sequencing revealed that Bacteroidia was one of the major classes that discriminated between disease and health in this model system, which led to experiments demonstrating that Bacteroides fragilis restored healthy levels of 4EPS levels in this model system. While metabolomics and metagenomics were not formally integrated in this study, the use of covariation network analyses, joint ordinations between microbial taxa and metabolites with disease/health state, and modeling of taxa versus metabolite abundance are well-suited to support to such studies.
While methods for integrating data from multiple assays have not yet been widely adopted in microbial community meta'omics, these methods have been applied for nearly a decade in human and model organism ‘omics. For example, a combination of metabolomics, proteomics, and metabolic flux analysis was used to generate metabolic networks of multiple knock-out strains of the model microbe E. coli (Yizhak et al., 2010). Likewise, human transcriptomic, proteomic, and metabolomic data were integrated to form functional linkage networks (Chen et al., 2012), which were subsequently explored to identify pathways that changed in expression over time. The ability to jointly observe multiple levels of regulation of the same underlying biological entities – genes and their products – revealed a clear upregulation of infection and stress response pathways at the onset of a viral respiratory infection in the subject. Only some of these pathways were detectable in analysis of the individual data types, either due to decreased power or biological reasons (e.g. post-transcriptional regulation). As these integrative methods have proven useful in identifying associations in human and model systems, their use should be extended to studies of microbial communities.
5.3 Integrating similar data types across different studies
Jointly analyzing information from one particular assay (e.g. metagenomics) across multiple studies (e.g. cohorts) can likewise serve as a means of integration to increase the specificity and power of hypothesis generation. This type of approach, typically referred to as a meta-analysis, has also been well-studied in low-dimensional (e.g. clinical trials) and ‘omics (e.g. gene expression, genome-wide association studies) contexts (Tonelli et al., 2009). Although any one dataset is typically collected to answer a few particular questions, comparative meta-analyses are one way to leverage existing data – improving power and reproducibility – and to compare a new study to the existing body of previous data. Given the large body of existing meta'omics data sets, meta-analyses of meta'omics can therefore be a powerful approach for discovering novel associations between taxonomy/genes and metadata, as well as for investigating the putative functions of uncharacterized genes.
A meta-analysis of one data type across studies typically includes at least two steps. First, to the extent that it is possible, systematic differences in measurements are normalized between data sets. Such differences are inevitable in any high-dimensional biology; they are also referred to as batch effects (Leek et al., 2010), They can be minimized by careful adherence to standardized protocols but rarely completely abrogated. Instead, integrative analyses should plan to explicitly account for their effects, either by pre-normalizing each dataset (e.g. by rank, median, or z-score normalization if appropriate (Bolstad et al., 2003)), or by using a statistical model incorporating multiple datasets (e.g. ComBat (Johnson et al., 2007) or fRMA (McCall et al., 2010) from gene expression analysis). When cross-study normalization is either not feasible or insufficient to correct systematic biases, meta-analyses can be performed in terms of standardized statistics such as effect sizes. This ensures that raw data are only compared within and not between studies (Crowther et al., 2010). For example, this is a standard approach in differential gene expression analysis, in which a sophisticated variant of a t-test might be performed within each study, and the resulting t-statistic values (rather than raw expression data) are compared across studies to identify genes with reproducibly variable expression.
Second, the target measurements or statistics calculated within each dataset are compared across the meta-analyzed studies. This not only identifies signals that reproduce across studies (depicted by the green nodes and arrows in Fig. 3Bii-iii), but detects weak but consistent signals with poor detection power in any single dataset. Although it is early for such techniques to be widespread in studies of microbial community bioactives, one example is in the assessment of cluster structure (e.g. enterotypes) among human microbiomes in several different body sites and cohorts (Koren et al., 2013). In this study, the measured effect was the degree to which groups of individuals formed robust, discrete clusters (prediction strength (Tibshirani and Walther, 2005)), which reproduced in some body sites (e.g. vaginal microbiome) but not others (e.g. gut). Note that simply merging multiple datasets or applying the same analytical approaches to multiple datasets is not a meta-analysis and in fact will often be prone to the biases that meta-analytical techniques were designed to prevent. Opportunities for robust within- and between-study integration will continue to arise as more functional profiles of the human microbiome become available.
6 Summary
In this perspective, we discussed some of the hallmark microbial products of the human microbiome, how they affect the host and other microbes, and how further microbial products of biological significance may be identified in culture-independent studies. These include compounds such as secreted peptides, non-ribosomal peptides, small molecules and metabolites, any of which can influence microbe-microbe or host-microbe interactions. Computational methods for predicting the biological roles of gene products have been widely used for microbial isolates and include sequence-based, structural, and integrative approaches; but these have as yet to be extensively applied to meta'omic studies.
As new studies and tools are created to identify microbial products of interest, it is worth noting that the toolbox of existing bioinformatic and statistical methods has not been exhaustively applied toward this end, nor has the portfolio of existing meta'omic datasets. With at least 50% of the genes in the human microbiome still uncharacterized, it is a worthwhile venture to focus on better characterization of this enormous portion of genetic material. Existing computational approaches for putative function assignment are one underexplored route, as is better integration of information from across meta'omics platforms and studies. When coupled with appropriate – and vital – experimental validation, these have the potential to accelerate the discovery of bioactives in the human microbiome.
Acknowledgments
We would like to thank Eric Franzosa, Galeb Abu-Ali and Aleksander Kostic for helpful feedback and Ramnik Xavier and Dirk Gevers for close collaboration while preparing this manuscript. This work was funded in part by grants PLF-5972-GD (Danone Research) to Wendy Garrett; R01HG005969 (NIH), DBI-1053486, (NSF), and 2012-5-39 MBRP (Sloan Foundation) to CH; and by the Robert F. Naka Fellowship to KY.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Xochitl C. Morgan, Email: xmorgan@hsph.harvard.edu.
Curtis Huttenhower, Email: chuttenh@hsph.harvard.edu.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin SR, Erdin S, Ward RM, Lua RC, Lichtarge O. Prediction and experimental validation of enzyme substrate specificity in protein structures. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E4195–4202. doi: 10.1073/pnas.1305162110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An D, Oh SF, Olszak T, Neves JF, Avci FY, Erturk-Hasdemir D, Lu X, Zeissig S, Blumberg RS, Kasper DL. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell. 2014;156:123–133. doi: 10.1016/j.cell.2013.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- Bairoch A. The ENZYME database in 2000. Nucleic acids research. 2000;28:304–305. doi: 10.1093/nar/28.1.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler BL, Losick R. Bacterially speaking. Cell. 2006;125:237–246. doi: 10.1016/j.cell.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Berdy J. Bioactive microbial metabolites. J Antibiot (Tokyo) 2005;58:1–26. doi: 10.1038/ja.2005.1. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Bork P, Koonin EV. Predicting functions from protein sequences--where are the bottlenecks? Nature genetics. 1998;18:313–318. doi: 10.1038/ng0498-313. [DOI] [PubMed] [Google Scholar]
- Bornigen D, Tranchevent LC, Bonachela-Capdevila F, Devriendt K, De Moor B, De Causmaecker P, Moreau Y. An unbiased evaluation of gene prioritization tools. Bioinformatics. 2012;28:3081–3088. doi: 10.1093/bioinformatics/bts581. [DOI] [PubMed] [Google Scholar]
- Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nature immunology. 2013;14:676–684. doi: 10.1038/ni.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome research. 2012;22:299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Chen R, Miriami E, Karczewski KJ, Hariharan M, Dewey FE, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148:1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter PD, Hill C, Ross RP. Bacteriocins: developing innate immunity for food. Nature reviews Microbiology. 2005;3:777–788. doi: 10.1038/nrmicro1273. [DOI] [PubMed] [Google Scholar]
- Cozzetto D, Buchan DW, Bryson K, Jones DT. Protein function prediction by massive integration of evolutionary analyses and multiple data sources. BMC Bioinformatics. 2013;14(Suppl 3):S1. doi: 10.1186/1471-2105-14-S3-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther M, Lim W, Crowther MA. Systematic review and meta-analysis methodology. Blood. 2010;116:3140–3146. doi: 10.1182/blood-2010-05-280883. [DOI] [PubMed] [Google Scholar]
- Debret G, Martel A, Cuniasse P. RASMOT-3D PRO: a 3D motif search webserver. Nucleic acids research. 2009;37:W459–464. doi: 10.1093/nar/gkp304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicksved J, Halfvarson J, Rosenquist M, Jarnerot G, Tysk C, Apajalahti J, Engstrand L, Jansson JK. Molecular analysis of the gut microbiota of identical twins with Crohn's disease. Isme J. 2008;2:716–727. doi: 10.1038/ismej.2008.37. [DOI] [PubMed] [Google Scholar]
- Duran-Pinedo AE, Chen T, Teles R, Starr JR, Wang X, Krishnan K, Frias-Lopez J. Community-wide transcriptome of the oral microbiome in subjects with and without periodontitis. Isme J. 2014 doi: 10.1038/ismej.2014.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA, Fraser CM. Phylogenomics: intersection of evolution and genomics. Science. 2003;300:1706–1707. doi: 10.1126/science.1086292. [DOI] [PubMed] [Google Scholar]
- Ellrott K, Jaroszewski L, Li W, Wooley JC, Godzik A. Expansion of the protein repertoire in newly explored environments: human gut microbiome specific protein families. PLoS computational biology. 2010;6:e1000798. doi: 10.1371/journal.pcbi.1000798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt BE, Jordan MI, Muratore KE, Brenner SE. Protein molecular function prediction by Bayesian phylogenomics. PLoS computational biology. 2005;1:e45. doi: 10.1371/journal.pcbi.0010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C, Noguchi T, Yamana H. Simplified sequence-based method for ATP-binding prediction using contextual local evolutionary conservation. Algorithms for molecular biology : AMB. 2014;9:7. doi: 10.1186/1748-7188-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. Pfam: the protein families database. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, Sonnenburg JL. Eating for two: how metabolism establishes interspecies interactions in the gut. Cell host & microbe. 2011;10:336–347. doi: 10.1016/j.chom.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor AA, DeSantis TZ, Wylie KM, Badger JH, Ye Y, Hepburn T, Hu P, Sodergren E, Liolios K, Huot-Creasy H, et al. The “most wanted” taxa from the human microbiome for whole genome sequencing. PLoS ONE. 2012;7:e41294. doi: 10.1371/journal.pone.0041294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, Giannoukos G, Boylan MR, Ciulla D, Gevers D, et al. Relating the metatranscriptome and metagenome of the human gut. Proceedings of the National Academy of Sciences of the United States of America. 2014 doi: 10.1073/pnas.1319284111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin MY, Koonin EV. ‘Conserved hypothetical’ proteins: prioritization of targets for experimental study. Nucleic acids research. 2004;32:5452–5463. doi: 10.1093/nar/gkh885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin MY, Koonin EV. From complete genome sequence to ‘complete’ understanding? Trends in biotechnology. 2010;28:398–406. doi: 10.1016/j.tibtech.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gish W, States DJ. Identification of protein coding regions by database similarity search. Nature genetics. 1993;3:266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- Goncalves JP, Francisco AP, Moreau Y, Madeira SC. Interactogeneous: disease gene prioritization using heterogeneous networks and full topology scores. PLoS One. 2012;7:e49634. doi: 10.1371/journal.pone.0049634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science. 2013;341:295–298. doi: 10.1126/science.1235872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DC, Myers CL, Huttenhower C, Hibbs MA, Hayes AP, Paw J, Clore JJ, Mendoza RM, Luis BS, Nislow C, et al. Computationally driven, quantitative experiments discover genes required for mitochondrial biogenesis. PLoS genetics. 2009;5:e1000407. doi: 10.1371/journal.pgen.1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollich V, Storm CE, Sonnhammer EL. OrthoGUI: graphical presentation of Orthostrapper results. Bioinformatics. 2002;18:1272–1273. doi: 10.1093/bioinformatics/18.9.1272. [DOI] [PubMed] [Google Scholar]
- Holmes E, Kinross J, Gibson GR, Burcelin R, Jia W, Pettersson S, Nicholson JK. Therapeutic modulation of microbiota-host metabolic interactions. Sci Transl Med. 2012;4:137rv136. doi: 10.1126/scitranslmed.3004244. [DOI] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155:1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh D, Kim HJ, Fraser JP, Shea DE, Khan M, Bahinski A, Hamilton GA, Ingber DE. Microfabrication of human organs-on-chips. Nat Protoc. 2013;8:2135–2157. doi: 10.1038/nprot.2013.137. [DOI] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature. 2012a;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012b;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Rhee SY, Marcotte EM, Lee I. Systematic prediction of gene function in Arabidopsis thaliana using a probabilistic functional gene network. Nat Protoc. 2011;6:1429–1442. doi: 10.1038/nprot.2011.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–970. doi: 10.1126/science.1240527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambon M, Andrieu O, Combet C, Deleage G, Delfaud F, Geourjon C. The SuMo server: 3D search for protein functional sites. Bioinformatics. 2005;21:3929–3930. doi: 10.1093/bioinformatics/bti645. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Jones ML, Martoni CJ, Ganopolsky JG, Labbe A, Prakash S. The human microbiome and bile acid metabolism: dysbiosis, dysmetabolism, disease and intervention. Expert opinion on biological therapy. 2014;14:467–482. doi: 10.1517/14712598.2014.880420. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic acids research. 2004;32:D277–280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren O, Knights D, Gonzalez A, Waldron L, Segata N, Knight R, Huttenhower C, Ley RE. A guide to enterotypes across the human body: meta-analysis of microbial community structures in human microbiome datasets. PLoS computational biology. 2013;9:e1002863. doi: 10.1371/journal.pcbi.1002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome research. 2012;22:292–298. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy N, Brown D, Sjolander K. FlowerPower: clustering proteins into domain architecture classes for phylogenomic inference of protein function. BMC evolutionary biology. 2007;7(Suppl 1):S12. doi: 10.1186/1471-2148-7-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramitsu HK, He X, Lux R, Anderson MH, Shi W. Interspecies interactions within oral microbial communities. Microbiology and molecular biology reviews : MMBR. 2007;71:653–670. doi: 10.1128/MMBR.00024-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Watson JD, Thornton JM. ProFunc: a server for predicting protein function from 3D structure. Nucleic acids research. 2005;33:W89–93. doi: 10.1093/nar/gki414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latendresse M, Krummenacker M, Trupp M, Karp PD. Construction and completion of flux balance models from pathway databases. Bioinformatics. 2012;28:388–396. doi: 10.1093/bioinformatics/btr681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Current opinion in biotechnology. 2013;24:160–168. doi: 10.1016/j.copbio.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K, Irizarry RA. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11:733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon KP, Armitage GC, Relman DA, Fischbach MA. Microbiota-targeted therapies an ecological perspective. Science translational medicine. 2012;4:137rv135. doi: 10.1126/scitranslmed.3004183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R, Borenstein E. Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:12804–12809. doi: 10.1073/pnas.1300926110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis NE, Nagarajan H, Palsson BO. Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nature reviews. Microbiology. 2012;10:291–305. doi: 10.1038/nrmicro2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- Little AE, Robinson CJ, Peterson SB, Raffa KF, Handelsman J. Rules of engagement: interspecies interactions that regulate microbial communities. Annual review of microbiology. 2008;62:375–401. doi: 10.1146/annurev.micro.030608.101423. [DOI] [PubMed] [Google Scholar]
- Lobley AE, Nugent T, Orengo CA, Jones DT. FFPred: an integrated feature-based function prediction server for vertebrate proteomes. Nucleic acids research. 2008;36:W297–302. doi: 10.1093/nar/gkn193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewenstein Y, Raimondo D, Redfern OC, Watson J, Frishman D, Linial M, Orengo C, Thornton J, Tramontano A. Protein function annotation by homology-based inference. Genome Biol. 2009;10:207. doi: 10.1186/gb-2009-10-2-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez CA, Kingsbury DD, Velazquez EM, Baumler AJ. Collateral Damage: Microbiota-Derived Metabolites and Immune Function in the Antibiotic Era. Cell host & microbe. 2014;16:156–163. doi: 10.1016/j.chom.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, Chen IM, Chu K, Szeto E, Palaniappan K, Grechkin Y, Ratner A, Jacob B, Pati A, Huntemann M, et al. IMG/M: the integrated metagenome data management and comparative analysis system. Nucleic acids research. 2012;40:D123–129. doi: 10.1093/nar/gkr975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx CJ. Microbiology. Getting in touch with your friends. Science. 2009;324:1150–1151. doi: 10.1126/science.1173088. [DOI] [PubMed] [Google Scholar]
- Maslowski KM, Mackay CR. Diet, gut microbiota and immune responses. Nature immunology. 2011;12:5–9. doi: 10.1038/ni0111-5. [DOI] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA) Biostatistics. 2010;11:242–253. doi: 10.1093/biostatistics/kxp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaughey LC, Grinter R, Josts I, Roszak AW, Waloen KI, Cogdell RJ, Milner J, Evans T, Kelly S, Tucker NP, et al. Lectin-like bacteriocins from Pseudomonas spp. utilise D-rhamnose containing lipopolysaccharide as a cellular receptor. PLoS pathogens. 2014;10:e1003898. doi: 10.1371/journal.ppat.1003898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon C, VanDussen KL, Miyoshi H, Stappenbeck TS. Development of a primary mouse intestinal epithelial cell monolayer culture system to evaluate factors that modulate IgA transcytosis. Mucosal Immunol. 2014;7:818–828. doi: 10.1038/mi.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic acids research. 2014;42:D7–17. doi: 10.1093/nar/gkt1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olle B. Medicines from microbiota. Nat Biotechnol. 2013;31:309–315. doi: 10.1038/nbt.2548. [DOI] [PubMed] [Google Scholar]
- Orth JD, Palsson BO. Systematizing the generation of missing metabolic knowledge. Biotechnology and bioengineering. 2010;107:403–412. doi: 10.1002/bit.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey G, Myers CL, Kumar V. Incorporating functional inter-relationships into protein function prediction algorithms. BMC Bioinformatics. 2009;10:142. doi: 10.1186/1471-2105-10-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Hess DC, Huttenhower C, Troyanskaya OG. Simultaneous genome-wide inference of physical, genetic, regulatory, and functional pathway components. PLoS computational biology. 2010;6:e1001009. doi: 10.1371/journal.pcbi.1001009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Wong AK, Greene CS, Rowland J, Guan Y, Bongo LA, Burdine RD, Troyanskaya OG. Functional knowledge transfer for high-accuracy prediction of under-studied biological processes. PLoS Comput Biol. 2013;9:e1002957. doi: 10.1371/journal.pcbi.1002957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan VV, Liu WT, Pogliano K, Dorrestein PC. Microbial metabolic exchange—the chemotype-to-phenotype link. Nature chemical biology. 2012;8:26–35. doi: 10.1038/nchembio.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell S, Forslund K, Szklarczyk D, Trachana K, Roth A, Huerta-Cepas J, Gabaldon T, Rattei T, Creevey C, Kuhn M, et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Research. 2014;42:D231–239. doi: 10.1093/nar/gkt1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic acids research. 2012;40:D290–301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radivojac P, Clark WT, Oron TR, Schnoes AM, Wittkop T, Sokolov A, Graim K, Funk C, Verspoor K, Ben-Hur A, et al. A large-scale evaluation of computational protein function prediction. Nature methods. 2013;10:221–227. doi: 10.1038/nmeth.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson EJ, Watson M. The automatic annotation of bacterial genomes. Briefings in bioinformatics. 2013;14:1–12. doi: 10.1093/bib/bbs007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- Rost B. Enzyme function less conserved than anticipated. Journal of molecular biology. 2002;318:595–608. doi: 10.1016/S0022-2836(02)00016-5. [DOI] [PubMed] [Google Scholar]
- Rost B, Liu J, Nair R, Wrzeszczynski KO, Ofran Y. Automatic prediction of protein function. Cellular and molecular life sciences : CMLS. 2003;60:2637–2650. doi: 10.1007/s00018-003-3114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340:1190–1194. doi: 10.1126/science.1234852. [DOI] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan R, Ulitsky I, Shamir R. Network-based prediction of protein function. Mol Syst Biol. 2007;3:88. doi: 10.1038/msb4100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skolnick J, Fetrow JS. From genes to protein structure and function: novel applications of computational approaches in the genomic era. Trends in biotechnology. 2000;18:34–39. doi: 10.1016/s0167-7799(99)01398-0. [DOI] [PubMed] [Google Scholar]
- Skolnick J, Fetrow JS, Kolinski A. Structural genomics and its importance for gene function analysis. Nat Biotechnol. 2000;18:283–287. doi: 10.1038/73723. [DOI] [PubMed] [Google Scholar]
- Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, Kau AL, Rich SS, Concannon P, Mychaleckyj JC, et al. Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science. 2013a;339:548–554. doi: 10.1126/science.1229000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013b;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothard P, Wishart DS. Automated bacterial genome analysis and annotation. Current opinion in microbiology. 2006;9:505–510. doi: 10.1016/j.mib.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, et al. The COG database: an updated version includes eukaryotes. BMC bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W, Skolnick J. How well is enzyme function conserved as a function of pairwise sequence identity? Journal of molecular biology. 2003;333:863–882. doi: 10.1016/j.jmb.2003.08.057. [DOI] [PubMed] [Google Scholar]
- Tibshirani R, Walther G. Cluster validation by prediction strength. Journal of Computational and Graphical Statistics. 2005;14:511–528. [Google Scholar]
- Todd AE, Orengo CA, Thornton JM. Evolution of function in protein superfamilies, from a structural perspective. Journal of molecular biology. 2001;307:1113–1143. doi: 10.1006/jmbi.2001.4513. [DOI] [PubMed] [Google Scholar]
- Tonelli M, Hackam D, Garg AX. Primer on systematic review and meta-analysis. Methods Mol Biol. 2009;473:217–233. doi: 10.1007/978-1-59745-385-1_13. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Vinayagam A, del Val C, Schubert F, Eils R, Glatting KH, Suhai S, Konig R. GOPET: a tool for automated predictions of Gene Ontology terms. BMC Bioinformatics. 2006;7:161. doi: 10.1186/1471-2105-7-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh LA, Mani S, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831–835. doi: 10.1126/science.1191175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Li Y, Liu X, Dai Q, Yao Y, He P. High-accuracy prediction of protein structural classes using PseAA structural properties and secondary structural patterns. Biochimie. 2014;101:104–112. doi: 10.1016/j.biochi.2013.12.021. [DOI] [PubMed] [Google Scholar]
- Wang PI, Hwang S, Kincaid RP, Sullivan CS, Lee I, Marcotte EM. RIDDLE: reflective diffusion and local extension reveal functional associations for unannotated gene sets via proximity in a gene network. Genome Biol. 2012;13:R125. doi: 10.1186/gb-2012-13-12-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Cao R, Cheng J. Three-level prediction of protein function by combining profile-sequence search, profile-profile search, and domain co-occurrence networks. BMC Bioinformatics. 2013;14(Suppl 3):S3. doi: 10.1186/1471-2105-14-S3-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whisstock JC, Lesk AM. Prediction of protein function from protein sequence and structure. Quarterly reviews of biophysics. 2003;36:307–340. doi: 10.1017/s0033583503003901. [DOI] [PubMed] [Google Scholar]
- Wieland Brown LC, Penaranda C, Kashyap PC, Williams BB, Clardy J, Kronenberg M, Sonnenburg JL, Comstock LE, Bluestone JA, Fischbach MA. Production of alpha-galactosylceramide by a prominent member of the human gut microbiota. PLoS biology. 2013;11:e1001610. doi: 10.1371/journal.pbio.1001610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3698–3703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong VW, Tse CH, Lam TT, Wong GL, Chim AM, Chu WC, Yeung DK, Law PT, Kwan HS, Yu J, et al. Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis--a longitudinal study. PLoS ONE. 2013;8:e62885. doi: 10.1371/journal.pone.0062885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DE, Lin H, Levy-Moonshine A, Swaminathan R, Chang YC, Anton BP, Osmani L, Steffen M, Kasif S, Salzberg SL. Thousands of missed genes found in bacterial genomes and their analysis with COMBREX. Biology direct. 2012;7:37. doi: 10.1186/1745-6150-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 2003;299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- Yan M, Pamp SJ, Fukuyama J, Hwang PH, Cho DY, Holmes S, Relman DA. Nasal microenvironments and interspecific interactions influence nasal microbiota complexity and S. aureus carriage. Cell host & microbe. 2013;14:631–640. doi: 10.1016/j.chom.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak K, Benyamini T, Liebermeister W, Ruppin E, Shlomi T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics. 2010;26:i255–260. doi: 10.1093/bioinformatics/btq183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakeri P, Jeuris B, Vandebril R, Moreau Y. Protein fold recognition using geometric kernel data fusion. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemla A. LGA: A method for finding 3D similarities in protein structures. Nucleic acids research. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengler K, Palsson BO. A road map for the development of community systems (CoSy) biology. Nature reviews Microbiology. 2012;10:366–372. doi: 10.1038/nrmicro2763. [DOI] [PubMed] [Google Scholar]
- Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–609. doi: 10.1002/hep.26093. [DOI] [PubMed] [Google Scholar]
- Zmasek CM, Eddy SR. RIO: analyzing proteomes by automated phylogenomics using resampled inference of orthologs. BMC Bioinformatics. 2002;3:14. doi: 10.1186/1471-2105-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuberi K, Franz M, Rodriguez H, Montojo J, Lopes CT, Bader GD, Morris Q. GeneMANIA prediction server 2013 update. Nucleic Acids Res. 2013;41:W115–122. doi: 10.1093/nar/gkt533. [DOI] [PMC free article] [PubMed] [Google Scholar]