

SUMMARY
Systemic amyloidoses result from the aberrant secretion of destabilized, amyloidogenic proteins to the serum where they aggregate into proteotoxic soluble aggregates and amyloid fibrils. Few therapeutic approaches exist to attenuate extracellular pathologic aggregation of amyloidogenic proteins, necessitating the development of new strategies to intervene in these devastating disorders. We show that stress-independent activation of the Unfolded Protein Response-associated transcription factor ATF6 increases ER quality control stringency for the amyloidogenic protein transthyretin (TTR), preferentially reducing secretion of disease-associated TTR variants to an extent corresponding to the variants’ destabilization of the TTR tetramer. This decrease in destabilized TTR variant secretion attenuates extracellular, concentration-dependent aggregation of amyloidogenic TTRs into soluble aggregates commonly associated with proteotoxicity in disease. Collectively, our results indicate that increasing ER quality control stringency through ATF6 activation is a strategy to attenuate pathologic aggregation of a destabilized, amyloidogenic protein, revealing a potential approach to intervene in systemic amyloid disease pathology.
INTRODUCTION
The ER is the primary organelle responsible for the folding and trafficking of proteins destined for downstream environments of the secretory pathway, including the extracellular space. Newly synthesized proteins are cotranslationally translocated into the ER lumen, where ER chaperones and folding enzymes facilitate their proper folding into native 3D conformations (Gidalevitz et al., 2013; Kleizen and Braakman, 2004). Once folded, these proteins are directed to coat protein complex II vesicles for trafficking to downstream secretory environments (D’Arcangelo et al., 2013). Proteins unable to properly fold in the ER lumen are targeted to ER degradation pathways including ER-associated degradation (ERAD) and autophagy (Brodsky and Skach, 2011). This partitioning of ER targeted polypeptides between ER protein folding/trafficking and degradation pathways, also referred to as ER quality control, prevents the ER accumulation and secretion of misfolding-prone proteins that could aggregate into pathologic proteotoxic conformations (Brodsky and Skach, 2011; Gidalevitz et al., 2013; Powers et al., 2009).
While ER quality control is generally efficient at preventing the secretion of aggregation-prone proteins, the aberrant secretion of destabilized, amyloidogenic proteins is pathologically associated with >13 protein misfolding diseases including the systemic amyloidoses (Blancas-Mejía and Ramirez-Alvarado, 2013). In these diseases, efficient secretion of destabilized, amyloidogenic proteins leads to their extracellular, concentration-dependent assembly into proteotoxic soluble aggregates and amyloid fibrils that deposit on tissues distal from the site of protein synthesis, leading to organ malfunction and eventual death (Gillmore and Hawkins, 2013). The dependence of pathologic extra-cellular protein aggregation on the secretion of destabilized, amyloidogenic proteins indicates that inadequate ER quality control stringency directly contributes to disease pathogenesis (Kleizen and Braakman, 2004; Sekijima et al., 2005; Wiseman et al., 2007).
The importance of ER quality control in the pathology of systemic amyloid diseases is most evident in the transthyretin (TTR) amyloidoses, a set of familial systemic amyloid disorders causally linked to the aberrant hepatic secretion of >100 destabilized TTR variants (Johnson et al., 2012). Highly destabilized, highly aggregation-prone TTR variants such as TTRD18G and TTRA25T are recognized by the liver’s ER quality control pathways and targeted to degradation in the ER (Hammarström et al., 2003; Sato et al., 2007; Sekijima et al., 2003, 2005). The increased partitioning of destabilized TTR variants to ER degradation pathways decreases their secretion and reduces their serum concentrations, attenuating pathologic concentration-dependent TTR aggregation in the serum. As such, patients expressing highly destabilized TTR mutations present with moderate disease pathologies inconsistent with the high amyloidogenicity of these variants (Hammarström et al., 2003; Sekijima et al., 2003). Alternatively, moderately destabilized, but still highly amyloidogenic, TTR variants such as TTRL55P are efficiently secreted from the liver, resulting in high serum concentrations that facilitate pathologic aggregation into proteotoxic soluble aggregates (Hammarström et al., 2002; Sekijima et al., 2005). Owing to the failure of ER quality control to prevent the secretion of these more moderately destabilizing TTR variants, individuals harboring mutations of this type present with the most severe and earliest-onset systemic amyloid disease pathology (Hammarström et al., 2002; Jacobson et al., 1992). A similar relationship between ER quality control stringency and amyloidogenic protein secretion has been proposed to explain the variable disease pathologies associated with other systemic amyloidoses (Kumita et al., 2006; Marchesi et al., 2011). The relationship between ER quality control of amyloidogenic proteins and amyloid disease pathology suggests that increasing ER quality control stringency to reduce the secretion of destabilized protein variants is a potential strategy to attenuate the extracellular protein aggregation responsible for disease pathogenesis.
Many of the protein factors responsible for maintaining ER quality control stringency are transcriptional targets of the ER unfolded protein response (UPR) (Walter and Ron, 2011). The UPR consists of three integrated signaling pathways activated downstream of the ER transmembrane proteins IRE1, activating transcription factor 6 (ATF6), and PERK. These pathways are activated in response to the accumulation of misfolded proteins within the ER lumen (i.e., ER stress), resulting in transient translational attenuation (downstream of PERK) and the transcriptional remodeling of ER protein folding, trafficking, and degradation pathways. UPR-dependent remodeling of these ER proteostasis pathways is primarily achieved through the two UPR-associated transcription factors spliced XBP1 (XBP1s, activated downstream of IRE1) and ATF6 (a cleaved product of full length ATF6) that induce overlapping, but distinct, sets of ER proteostasis factors (Adachi et al., 2008; Shoulders et al., 2013; Yamamoto et al., 2004). This indicates that activation of XBP1s and/or ATF6 offers a unique opportunity to differentially influence ER quality control of destabilized amyloidogenic protein variants.
Previously, we showed that stress-independent ATF6 activation reduces secretion of destabilized amyloidogenic TTRA25T (Shoulders et al., 2013). Here, we show that ATF6 activation similarly attenuates secretion of other destabilized, amyloidogenic TTR variants through a mechanism involving the increased partitioning of destabilized TTR monomers to degradation. Intriguingly, we show a sigmoidal relationship between the impact of ATF6 activation on TTR variant secretion and the destabilization of the TTR variant tetramer, demonstrating that ATF6-dependent remodeling of the ER proteostasis environment preferentially reduces secretion of destabilized, aggregation-prone TTRs. Furthermore, we show that the capacity for ATF6 activation to reduce secretion and hence extracellular concentrations of aggregation-prone TTR variants also decreases their extracellular aggregation into proteotoxic soluble aggregates. Collectively, our results demonstrate that increasing ER quality control stringency through stress-independent ATF6 activation is a viable strategy to decrease secretion and consequent extracellular aggregation of destabilized TTR variants responsible for the pathogenesis of familial TTR amyloidoses.
RESULTS
ATF6 Activation Preferentially Reduces Secretion of Disease-Associated TTR Variants
We previously showed that ATF6 activation reduces the secretion of the disease-associated TTRA25T (Shoulders et al., 2013). To evaluate whether ATF6 activation similarly affects the secretion of other disease-associated, amyloidogenic TTR variants, we chose five TTR variants that predispose patients to varying levels of amyloid disease pathology: TTRD18G and TTRA25T (poorly secreted TTR variants that present with moderate disease pathology, see Hammarström et al., 2003; Sekijima et al., 2003; Vidal et al., 1996), TTRL55P (the TTR variant that presents with the most severe TTR amyloid disease in patients, see Hammarström et al., 2002; Jacobson et al., 1992), TTRV30M (the most prominent TTR variant in patients that presents with varying disease pathologies within endemic populations, see Buxbaum et al., 2010), and TTRV122I (a TTR variant found in 5% of the African American population that presents with a late onset cardiac disease pathology, see Buxbaum et al., 2006). We also chose two TTR variants that do not cause familial TTR amyloidoses: wild-type TTR (TTRWT) and TTRT119M (a highly stabilized TTR variant that transsuppresses TTR amyloid disease in heterozygote patients, see Hammarström et al., 2001).
We used [35S] metabolic labeling to measure the secretion of FLAG tagged TTR (FTTTR) variants from human embryonic kidney (HEK)293DAX cells (Figure S1A available online). These cells stably and constitutively express a protein fusion between destabilized dihydrofolate reductase (DHFR) and the active ATF6 transcription factor (DHFR.ATF6) that is rapidly degraded by the proteasome (Figure 1A) (Shoulders et al., 2013). The addition of the small molecule DHFR pharmacologic chaperone trimethoprim (TMP) stabilizes the DHFR.ATF6 fusion protein, prevents proteasomal degradation, and promotes transcriptional activity of DHFR.ATF6 (Figure 1A). Thus, in HEK293DAX, the ATF6 transcriptional program can be activated in the absence of ER stress through TMP-dependent DHFR.ATF6 stabilization (Shoulders et al., 2013).
Figure 1. Stress-Independent ATF6 Preactivation Preferentially Decreases Secretion of Disease-Associated TTR Variants.

(A) Schematic illustrating the TMP-mediated, posttranslational regulation of DHFR.ATF6 in HEK293DAX cells (Shoulders et al., 2013).
(B–H) Quantification of fraction secreted [35S]-labeled FTTTR variants from HEK293DAX cells in the absence (black) or presence (blue) of ATF6 preactivation (TMP, 10 μM; 16 hr). Representative autoradiograms and the experimental protocol are shown in Figure S1A. Fraction secreted was calculated from autoradiograms using the equation: Fraction Secreted = {Total FTTTR Signal in Media at Time = t } divided by {Total FTTTR Signal in Lysate at Time = 0} + {Total FTTTR Signal in Media at Time = 0} (Sekijima et al., 2005; Shoulders et al., 2013). Error bars represent SEM from biological replicates (n ≥ 3). Identical plots of fraction secreted for FTTTRD18G and FTTTRA25T with the same scales used for the other TTR variants are shown in Figures S1B and S1C.
(I) Bar graph comparing fraction secreted at 4 hr for FTTTR variants in the absence (gray) or presence of ATF6 preactivation (blue), as shown in Figures 1B–1H. Shading shows familial disease-associated FTTTR variants (pink) and FTTTR variants not involved in familial disease (brown). Error bars represent SEM from biological replicates (n ≥ 3).
**p < 0.01; ***p < 0.001.
We found that FTTTR variants are secreted from HEK293DAX with identical efficiencies to those previously reported for BHK cells (Figures 1B–1H and S1A–S1C) (Sekijima et al., 2005). FTTTRD18G and FTTTRA25T are poorly secreted from HEK293DAX cells, demonstrating secretion efficiencies of 6% and 27%, respectively, following a 4 hr chase in nonradioactive media (Figures 1B and 1C and S1B and S1C). Conversely, other FTTTR variants were secreted to levels nearly identical to those observed for FTTTRWT, demonstrating ~60% secretion efficiency following a 4 hr chase (Figures 1D–1H). ATF6 preactivation results in a significant ~40% decrease in the secretion of the disease-associated variants FTTTRD18G, FTTTRA25T, FTTTRL55P, and FTTTRV30M and a 30% decrease in FTTTRV122I secretion. Alternatively, the secretion of FTTTRWT and the highly stable FTTTRT119M were only modestly reduced by ~15% following ATF6 preactivation. These results show that ATF6 preactivation preferentially reduces the secretion of amyloidogenic TTR variants associated with familial TTR amyloid diseases relative to FTTTRWT and the nonpathogenic FTTTRT119M (Figure 1I).
Reduced Secretion of TTR Variants upon ATF6 Activation Follows a Sigmoidal Relationship with Energetic Stability
The partitioning of TTR between ER protein folding/trafficking and degradation is influenced by the thermodynamic (the equilibrium propensity to attain a folded conformation) and kinetic (the rate of interconversion between folded and unfolded conformations) stabilities of the TTR tetramer (Sekijima et al., 2005). Previously, the secretion of TTR variants from mammalian cells was shown to depend on their stability as expressed by the combined stability score (CSS), an energetic parameter determined by a linear regression against in vitro-derived thermodynamic (Cm of urea denaturation) and kinetic (t1/2 of tetramer dissociation/unfolding) measures of TTR variant stability (Sekijima et al., 2005) (Table S1). We find a similar dependence of FTTTR variant secretion on CSS in the absence or presence of ATF6 activation (Figure 2A). The FoldEx mathematical model for protein secretion predicts a sigmoidal relationship between the energetic stability and fraction secreted for a given series of destabilized variants of a single protein (Wiseman et al., 2007). This sigmoidal relationship has been previously indicated for destabilized variants of monomeric TTR (M-TTR), bovine pancreatic trypsin inhibitor, and lysozyme (Kumita et al., 2006; Wiseman et al., 2007) and reflects the energetically defined partitioning of protein variants between ER protein folding, trafficking, and degradation pathways (Wiseman et al., 2007). We find that the relationship between FTTTR variant energetic stability (defined by the CSS) and fraction secreted is also consistent with a sigmoid (Figure 2A). Interestingly, ATF6 activation appears to shift the sigmoidal transition to higher CSS values, indicating that ATF6 activation preferentially decreases the partitioning of destabilized FTTTR variants to ER trafficking pathways and increases the energetic requirements for TTR variant secretion (Powers et al., 2009; Wiseman et al., 2007).
Figure 2. The ATF6-Dependent Decrease in Destabilized TTR Variant Secretion Correlates with the Energetic Stability of the TTR Tetramer.
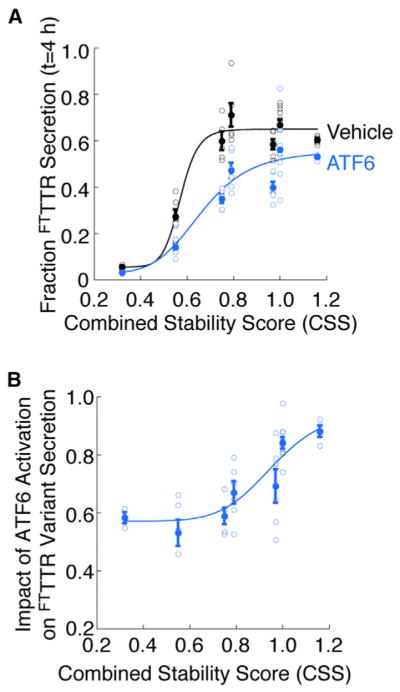
(A) Plot relating the TTR variant CSS as calculated in (Sekijima et al., 2005) (Table S1) to the fraction FTTTR variant secreted at 4 hr in the absence (black) or presence (blue) of ATF6 preactivation. The solid circles show the average for all replicates and the open circles show all individual replicates. The error bars show SEM for n ≥ 3. The curves describe the least-squares fit of the individual replicates to the sigmoid function (defined in Experimental Procedures).
(B) Plot relating the TTR variant CSS as calculated in (Sekijima et al., 2005) (Table S1) to the impact of ATF6 activation on FTTTR variant secretion. The impact of ATF6 activation on FTTTR variant secretion is defined as {fraction FTTTR secretion at 4 hr in the presence of ATF6 preactivation} divided by {fraction FTTTR secretion at 4 hr in the absence of ATF6 preactivation}. The solid circles show the average impact of ATF6 activation for all replicates and the open circles show all individual replicates. The error bars show SEM for n ≥ 3. The line describes the least-squares fit of the individual replicate data to the sigmoid function defined in Experimental Procedures.
To better visualize the effect of ATF6 activation on the secretion of destabilized FTTTR variants, we plotted the impact of ATF6 activation (defined as [fraction TTR variant secretion at 4 hr in the presence of ATF6 preactivation]/[fraction TTR variant secretion at 4 hr in the absence of ATF6 activation]) against the CSS for TTR variants (Figure 2B). As a ratio of two sigmoids, this plot is also sigmoidal. This relationship shows that ATF6 activation influences FTTTR variant secretion to extents defined by the inherent instability of the protein fold (described by the CSS).
ATF6 Activation Increases Targeting of Destabilized TTR Monomers to ER Degradation Pathways
ER quality control is defined by the partitioning of destabilized, misfolding-prone proteins between secretion and degradation in the ER lumen. The results shown in Figure 2 indicate that ATF6 activation reduces FTTTR variant secretion to levels dictated by the energetic destabilization of the protein fold, showing that ATF6 activation increases ER quality control stringency for TTR (i.e., the energetic requirements for TTR variant secretion). We next tested whether ATF6 preactivation also influenced the partitioning of destabilized TTR variants to degradation.
We measured ATF6-dependent increases in TTR degradation by monitoring the fraction of total [35S]-labeled TTR (media + lysates) remaining following a 4 hr chase in the experiments shown in Figures 1 and S1. ATF6 preactivation reduced the recovery of destabilized FTTTRD18G, FTTTRA25T, FTTTRL55P, and FTTTRV30M (Figure 3A), without increasing intracellular levels of these variants (Figure S2A). The recovery of the more stable FTTTRV122I, FTTTRWT, and FTTTRT119M were not affected by ATF6 preactivation. Interestingly, the relationship between the TTR variant CSS and the impact of ATF6 activation on FTTTR variant recovery shows that ATF6 activation preferentially reduces the recovery of the most destabilized TTR variants (Figure 3B). This suggests that ATF6 preactivation preferentially increases partitioning of destabilized FTTTR variants toward degradation, preventing the ER accumulation of these aggregation prone proteins that could disrupt ER proteostasis and function. Consistent with this prediction, we previously showed that ATF6 activation does not increase the accumulation of destabilized FTTTRA25T in insoluble aggregates (Shoulders et al., 2013).
Figure 3. Stress-Independent ATF6 Pre-activation Increases Partitioning of Destabilized TTR Monomers to ER Degradation Pathways.

(A) Bar graph comparing the fraction remaining of [35S]-labeled FTTTR variants at 4 hr in the absence (gray) or presence (blue) of ATF6 preactivation. Fraction remaining was calculated from autoradiograms as shown in Figure S1A, using the equation: Fraction Remaining = {Total FTTTR Signal in Lysate at Time = t } + {Total FTTTR Signal in Media at Time = t } divided by {Total FTTTR Signal in Lysate at Time = 0 } + {Total FTTTR Signal in Media at Time = 0 } (Sekijima et al., 2005; Shoulders et al., 2013). Shading shows familial disease-associated FTTTR variants (pink) and FTTTR variants not involved in familial disease (brown). Error bars represent SEM from biological replicates (n ≥ 3).
(B) Plot relating the impact of ATF6 activation on the fraction remaining for FTTTR variants, as shown in Figure 3A, to each respective TTR variants’ CSS (Table S1). The impact of ATF6 activation on FTTTR variant fraction remaining is defined as {fraction FTTTR remaining t = 4 hr in the presence of ATF6} divided by {fraction FTTTR remaining t = 4 hr in the absence of ATF6}. Error bars represent SEM from biological replicates (n ≥ 3).
(C) Quantification of fraction remaining for [35S]-labeled FTTTRD18G in HEK293DAX following ATF6 preactivation (10 μM TMP; 16 hr) when chased in the presence of vehicle (black), MG132 (10 μM; orange), NH4Cl (25 mM; green), or both MG132 and NH4Cl (purple). A representative autoradiogram and experimental protocol employed are shown in Figure S2A. Error bars represent SEM from biological replicates (n ≥ 3).
(D) Quantification of fraction remaining of [35S]-labeled FTM-TTR in HEK293DAX in the absence (black) or presence (blue) of ATF6 preactivation (10 μM TMP; 16 hr). A representative autoradiogram is shown in Figure S2D. Error bars represent SEM from biological replicates (n = 6).
*p < 0.05, **p < 0.01, ***p < 0.001.
ER degradation can occur through ERAD or autophagic pathways (Brodsky and Wojcikiewicz, 2009; Houck and Cyr, 2012). We evaluated the contributions of these two pathways to the ATF6-dependent increase in destabilized TTR degradation using FTTTRD18G, a highly destabilized TTR variant that does not form stable TTR tetramers (Hammarström et al., 2003). TTRD18G is a known ERAD substrate that is degraded >50% in mammalian cells (Sato et al., 2007; Sekijima et al., 2005; Shoulders et al., 2013). Thus, as expected, the recovery of [35S]-metabolically labeled FTTTRD18G in the absence of ATF6 preactivation is modestly increased by the addition of the proteasome inhibitor MG132 (Figures S2B and S2C). Interestingly, FTTTRD18G recovery was similarly increased upon addition of NH4Cl, an inhibitor of lysosomal degradation, suggesting that FTTTRD18G also can be degraded through autophagic pathways (Figures S2B and S2C). These results suggest that ATF6 activation could increase FTTTRD18G degradation by increasing the partitioning of destabilized TTRs to ERAD and/or autophagic pathways.
Surprisingly, the recovery of FTTTRD18G following ATF6 preactivation does not appear to be affected by the addition of either MG132 or NH4Cl (Figures 3C and S2B). Alternatively, coadministration of MG132 and NH4Cl increased the recovery of FTTTRD18G by 30% following ATF6 preactivation. This result suggests that ATF6 activation can increase the partitioning of FTTTRD18G to ERAD and autophagic pathways, and that inhibition of either pathway independently results in increased flux through the alternative pathway (Lamark and Johansen, 2010).
Since TTRD18G exists as a destabilized monomer (Hammarström et al., 2003), we evaluated whether a stable monomeric TTR (M-TTR, see Jiang et al., 2001) is also affected by ATF6 activation. M-TTR tetramerization is inhibited by two mutations at the dimer interface, resulting in a monomeric TTR whose stability is identical to unassembled TTRWT subunits. Although ATF6 pre-activation reduces FTM-TTR secretion by 30% (Figure S2D), FTM-TTR is not significantly degraded upon ATF6 preactivation (Figure 3D). This suggests that ATF6-dependent increases in destabilized TTR degradation require destabilization of the monomer. Collectively, the above results suggest a model whereby enhanced ER quality control stringency for TTR upon ATF6 activation proceeds, at least in part, through increased partitioning of destabilized FTTTR monomers to ERAD and/or autophagic ER degradation pathways.
ATF6 Activation Reduces Secretion of Destabilized FTTTRA25T from Liver-Derived HepG2 Cells
In heterozygote patients, TTR can be secreted as heterotetramers, comprised of TTR variant and TTRWT subunits (Figure 4A), the stability of which correlates with the number of TTRWT subunits (Sekijima et al., 2005). This heterotetramerization with TTRWT subunits stabilizes and promotes the secretion of destabilized TTR variants such as TTRA25T (Sekijima et al., 2005). Since our results indicate that ATF6 activation increases degradation of destabilized TTR monomers prior to tetramerization, we predicted that ATF6 activation would decrease secretion of destabilized TTR variant subunits in TTRWT-containing heterotetramers.
Figure 4. ATF6 Preactivation Decreases Secretion of the Destabilized FTTTRA25T Variant from Liver-Derived HepG2DA Cells Coexpressing Equal Amounts of Endogenous TTRWT.

(A) Schematic of TTR heterotetramer formation and secretion from HepG2DA cells expressing endogenous TTRWT and FTTTR variants.
(B) Quantitative PCR (qPCR) analysis of TTR in HepG2DA cells transduced with adenovirus encoding FTTTR variants. qPCR data is normalized relative to mock-transfected HepG2DA cells. Error bars represent 95% confidence interval for technical replicates.
(C and D) Plots showing fraction secreted (C) or fraction remaining (D) of FTTTRA25T in HepG2DA in the absence (black) or presence (blue) of ATF6 preactivation (10 μM TMP; 16 hr). A representative autoradiogram is shown in Figure S3A. Error bars represent SEM from biological replicates (n = 6).
(E and F) Plots showing the fraction secreted (E) or fraction remaining (F) of FTTTRWT in HepG2DA in the absence (black) or presence (blue) of ATF6 preactivation (10 μM TMP; 16 hr). A representative autoradiogram is shown in Figure S3A. Error bars represent SEM from biological replicates (n = 6).
(G) Graph showing the normalized fraction secreted of FTTTRWT (gray) or FTTTRA25T (red) at 4 hr from HepG2DA in the absence or presence of ATF6 preactivation, as shown in Figures 4C and 4E. Error bars represent SEM from biological replicates (n = 6).
*p < 0.05, **p < 0.01, ***p < 0.001.
We employed a human, liver-derived HepG2 cell line stably expressing DHFR.ATF6 (HepG2DA) to test this prediction (Shoulders et al., 2013). HepG2 cells secrete high levels of endogenous TTRWT, as liver cells are the primary secretory tissue for TTR in vivo (Johnson et al., 2012). We previously showed that ATF6 activation does not significantly influence secretion of endogenous TTRWT from HepG2DA (Shoulders et al., 2013). We transduced these cells with adenovirus encoding FTTTRWT or FTTTRA25T, titered to allow identical expression of FTTTR variants and endogenous TTRWT to mimic TTR expression in heterozygote patient livers (Figure 4B). Endogenous TTRWT and exogenous FTTTR separate on SDS-PAGE, allowing quantification of the FTTTR variants independent of the endogenous protein (Sekijima et al., 2005; Shoulders et al., 2013). We measured the impact of ATF6 activation on the secretion of FTTTR variants using a [35S] metabolic labeling approach, immunopurifying the FTTTR protein with a FLAG tag antibody. The efficient formation of heterotetramers containing TTRWT and FTTTR subunits in these cells is evident by the coimmunopurification of endogenous TTRWT in our FLAG immunopurifications (Figure S3A).
The preactivation of ATF6 in HepG2DA reduces secretion of FTTTRA25T by 50% (Figure 4C). This decrease in FTTTRA25T secretion corresponds with increased degradation of FTTTRA25T (Figure 4D). The secretion and degradation of FTTTRWT was only modestly affected by ATF6 preactivation in HepG2DA cells (Figures 4E–4G). The degradation of FTTTRA25T in HepG2DA cells in the presence or absence of ATF6 preactivation is attenuated upon the addition of both MG132 and NH4Cl, but not upon the addition of either inhibitor independently (Figures S3B and S3C). This suggests that FTTTRA25T degradation in liver-derived HepG2DA cells is mediated through the activities of ERAD and autophagic pathways. This also suggests that ATF6-dependent reductions in FTTTRA25T secretion from HepG2DA cells involves increased partitioning of destabilized, FTTTRA25T monomers to ER degradation pathways. With less FTTTRA25T available, we expected that ATF6 activation would reduce secretion of TTR heterotetramers that contain three or four FTTTRA25T subunits relative to those that contain one or two FTTTRA25T subunits (Figure 4A), effectively increasing the kinetic stability of the secreted TTR population (Johnson et al., 2012). Consistent with this prediction, we observe an ATF6-dependent increase in the recovery of [35S]-labeled TTRWT in FLAG-immunopurification of FTTTRA25T from the experiments described in Figure 4D, which suggests an ATF6-dependent increase in extracellular populations of tetramers containing one or two FTTTRA25T subunits (Figure S3D). These findings support our model, whereby ATF6 activation reduces TTR variant secretion through a mechanism involving the increased targeting of destabilized TTR monomers to ER degradation pathways prior to tetramer assembly.
Kinetic Stabilization of the TTR Tetramer Does Not Inhibit ATF6-Dependent Reductions in Destabilized Transthyretin Secretion
Small molecule TTR kinetic stabilizers such as Tafamidis (Taf) bind to the native TTR tetramer and increase the energetic barrier for TTR tetramer disassembly, thus preventing a requisite step in the TTR amyloid cascade (Bulawa et al., 2012). We applied Taf to further evaluate the capacity for ATF6 activation to reduce FTTTR secretion by targeting destabilized FTTTR monomers to degradation prior to tetramer assembly. Since Taf stabilizes the TTR tetramer, but does not bind to the monomer prior to assembly, we predicted that, in the presence of Taf, ATF6 activation would maintain its capacity to decrease secretion of destabilized FTTTR variants.
We tested this prediction in HEK293DAX cells overexpressing destabilized FTTTRL55P. The tetramer dissociation rate of TTRL55P is slow (t1/2 = 4.4 hr) relative to the rate of secretion (Sekijima et al., 2005). This implies that stabilization of TTRL55P tetramers by Taf should not influence the distribution of tetramers and monomers in the steady-state environment of the ER, as any protein that forms a tetramer will be secreted prior to dissociation (Figure 5A). The addition of Taf does not influence FTTTRL55P secretion in the absence of ATF6 preactivation (Figures 5B, 5C, and S4A). Furthermore, the addition of Taf does not affect the ATF6-dependent decrease in FTTTRL55P secretion. These results indicate that kinetic stabilization of the FTTTRL55P tetramer does not influence ATF6-dependent reductions in TTR variant secretion, supporting our model, whereby ATF6 activation influences FTTTR secretion prior to tetramerization.
Figure 5. ATF6 Preactivation Decreases Secretion of Destabilized FTTTR Variants in the Presence of the Small Molecule TTR Tetramer Kinetic Stabilizer Tafamidis.

(A) Illustration showing the folding, assembly, secretion, and Taf binding for TTRL55P in the ER lumen. Note that the rate of TTRL55P tetramer dissociation is slow relative to the rate of secretion, indicating that the effect of Taf binding on the ER population of TTR tetramers will be negligible.
(B) Plot showing fraction FTTTRL55P secretion from HEK293DAX cells in the absence of ATF6 preactivation (black), the presence of ATF6 preactivation (10 μM TMP; 16 hr; blue), in the presence of Taf included in the pretreatment and the chase media (10 μM 16 hr; green), or in the presence of ATF6 pre-activation where Taf was added during the preincubation and the chase media (brown). A representative autoradiogram and the experimental protocol employed are shown in Figure S4A. Error bars represent SEM from biological replicates (n = 6).
(C) Bar graph depicting the normalized fraction secreted for FTTTRL55P at 4 hr as shown in Figure 5B. Error bars represent SEM from biological replicates (n = 6).
(D) Illustration showing the folding, assembly, secretion, and Taf binding for TTRA25T in the ER lumen. Note that the rate of TTRA25T tetramer dissociation is fast relative to the rate of secretion, indicating that Taf can bind to and stabilize TTRA25T tetramers in the ER lumen increasing TTRA25T secretion.
(E) Plot showing fraction FTTTRA25T secretion from HEK293DAX cells in the absence of ATF6 preactivation (black), the presence of ATF6 preactivation (10 μM TMP; 16 hr; blue), in the presence of Taf included in the pretreatment and the chase media (10 μM 16 hr; green), or in the presence of ATF6 pre-activation where Taf was added during the preincubation and the chase media (brown). Error bars represent SEM from biological replicates (n = 3).
(F) Bar graph depicting the normalized fraction secreted for FTTTRA25T at 4 hr as shown in Figure 5E. Error bars represent SE from biological replicates (n = 3).
**p < 0.01.
We next evaluated the capacity for Taf and/or ATF6 activation to influence the secretion of FTTTRA25T, a TTR tetramer that has a fast tetramer dissociation rate relative to secretion (t1/2 = 0.035 hr) (Sekijima et al., 2005). The rapid tetramer dissociation rate of TTRA25T allows equilibration between TTR tetramers and monomers in the ER prior to secretion (Figure 5D). As shown previously (Shoulders et al., 2013), the addition of Taf increases FTTTRA25T secretion from HEK293DAX cells in the absence of ATF6 preactivation, demonstrating that Taf-dependent stabilization of FTTTRA25T tetramers in the ER increases secretion of this variant from mammalian cells (Figures 5E, 5F, and S4B). The capacity for Taf to influence FTTTRA25T secretion demonstrates that FTTTRA25T tetramer dissociation influences the conformational distribution of FTTTRA25T tetramers and monomers in the ER. The preactivation of ATF6 attenuates the Taf-dependent increase in FTTTRA25T secretion, although Taf does increase FTTTRA25T secretion relative to cells treated with ATF6 preactivation alone (Figures 5E and 5F). Thus, even though Taf stabilizes FTTTRA25T tetramers in the ER lumen, ATF6 activation still reduces FTTTRA25T secretion, further supporting our model wherein ATF6 activation influences secretion of destabilized TTR prior to tetramerization.
ATF6-Dependent Reduction in Destabilized TTR Secretion Attenuates Extracellular Aggregation of TTR
Extracellular aggregation of TTR into proteotoxic soluble aggregates and amyloid fibrils is dependent on two parameters: the destabilization of the TTR tetramer afforded by the mutation and the extracellular concentration of destabilized TTR available for concentration-dependent aggregation (Hammarström et al., 2002) (Figure 6A). The capacity for ATF6 activation to preferentially reduce secretion of destabilized TTR variants directly addresses both of these parameters. The aggregation propensity for TTR was previously shown to inversely correlate with the CSS of the TTR variant tetramer (Sekijima et al., 2005). Since ATF6-dependent reductions in FTTTR variant secretion also relates to the CSS, ATF6 activation should preferentially impact secretion of amyloidogenic TTR variants. This effect is observed in our experimental results as the impact of ATF6 activation on FTTTR variant secretion inversely correlates with the optimal pH of TTR fibril formation (Figure 6B), an in vitro-derived measure of TTR variant aggregation propensity (Sekijima et al., 2005). The correlation shown in Figure 6B demonstrates that ATF6 activation preferentially reduces extracellular concentrations of the most amyloidogenic TTR variants. Since no mouse model of TTR familial amyloidosis recapitulates disease pathology (Buxbaum et al., 2003), we evaluated the potential for ATF6 activation to attenuate extracellular aggregation of secreted, destabilized FTTTR variants into proteotoxic soluble aggregates, which are predicted to be the proximal cause of TTR amyloid disease pathology (Reixach et al., 2004).
Figure 6. ATF6 Activation Attenuates the Extracellular Concentration of Soluble FTTTR Aggregates in Conditioned Media.

(A) Illustration showing that the partitioning of destabilized TTRs between degradation and secretion in the ER impacts the extracellular concentration of aggregation-prone protein available for concentration-dependent aggregation into proteotoxic soluble aggregates.
(B) Plot comparing the impact of ATF6 activation on FTTTR variant fraction secreted to the optimal pH for TTR variant fibril formation determined in (Sekijima et al., 2005) and shown in Table S1.
(C) CN-PAGE/immunoblot (IB) of media conditioned for 24 hr on HEK293T cells overexpressing FTTTRWT or FTTTRA25T following transfection with the indicated equivalents of plasmid DNA. An SDS-PAGE/IB of identical samples is shown as a control for total FTTTR levels in the conditioned media for each treatment.
(D) CN-PAGE/IB of media conditioned for 24 hr on HEK293DAX cells transfected with one equivalent of FTTTRA25T and treated with vehicle or 10 μM Taf during conditioning, as indicated. An SDS-PAGE/ IB of identical samples is shown as a control for total FTTTR levels in the conditioned media for each treatment.
(E) Representative CN-PAGE/IB and quantification of FTTTRA25T soluble aggregates in media conditioned for 24 hr on HEK293DAX cells transfected with one equivalent of FTTTRA25T in the absence or presence of ATF6 activation (TMP; 10 μM) as indicated. An SDS-PAGE/IB of identical samples is shown as a control for total FTTTR levels in the conditioned media for each treatment. Error bars represent SEM for n = 3.
**p < 0.01
We measured the aggregation of FTTTR variants in conditioned media using Clear-Native PAGE (CN-PAGE) (Upadhaya et al., 2012). Destabilized FTTTRA25T secreted from HEK293T cells spontaneously aggregates into soluble aggregates with molecular weights between 200–900 kDa (Figure 6C). This aggregation is dependent on the amount of FTTTRA25T secreted from the cells, as evidenced by the reduced aggregate concentrations in media conditioned on cells transfected with decreasing amounts of FTTTRA25T DNA. FTTTRA25T soluble aggregates were also identified by gel filtration chromatography (Figure S5A). Cell secreted FTTTRWT does not efficiently form soluble aggregates (Figures 6C and S5A). Identical results were observed using the anti-TTR antibody in CN-PAGE/immunoblotting assays (Figure S5B). The addition of the TTR tetramer kinetic stabilizer Taf to cells secreting FTTTRA25T inhibits TTR aggregation (Figure 6D), demonstrating that the observed aggregation requires rate-limiting dissociation of the TTR tetramer (Johnson et al., 2012). Taf similarly reduced the extracellular aggregation of other destabilized, aggregation-prone TTR variants (Figure S5B). The activation of ATF6 during the conditioning of media prepared on HEK293DAX cells expressing FTTTRA25T reduces FTTTRA25T soluble aggregates by 50%, consistent with the ATF6-dependent reduction in total FTTTRA25T secreted into the media (Figure 6E). ATF6 activation similarly reduces extracellular soluble aggregates of FTTTRD18G, FTTTRL55P, and FTTTRV30M (Figure S5C). These results indicate that reducing secretion of destabilized, amyloidogenic FTTTR variants through stress-independent ATF6 activation can decrease extracellular populations of soluble TTR aggregates predicted to induce distal proteotoxicity in TTR amyloid disease pathology (Reixach et al., 2004).
DISCUSSION
The extracellular aggregation of destabilized, amyloidogenic protein variants is pathologically linked to the onset and pathology of multiple familial systemic amyloid diseases (Blancas-Mejía and Ramirez-Alvarado, 2013). Because protein aggregation is concentration-dependent, high serum concentrations of destabilized amyloidogenic proteins facilitate their aggregation into proteotoxic soluble aggregates and amyloid fibrils, directly contributing to disease pathogenesis. Thus, reducing serum concentrations of destabilized proteins will attenuate pathologic extracellular protein aggregation involved in systemic amyloid disease pathogenesis (Ryno et al., 2013).
A strategy to reduce serum concentrations of amyloidogenic proteins is to increase ER quality control in cells secreting destabilized, disease-associated variants of amyloidogenic proteins. ER protein secretion models predict that increasing ER quality control stringency through adaptation of ER proteostasis pathways can preferentially attenuate secretion of destabilized protein variants without significantly affecting secretion of the stable, WT protein (Wiseman et al., 2007). This increase in ER quality control stringency can be reflected by a shift in the sigmoidal relationship between secretion efficiency and energetic stability for a series of destabilized variants of a single protein (Figure 7A). Since amyloidogenicity often correlates with protein destabilization (Blancas-Mejía and Ramirez-Alvarado, 2013; Jahn and Radford, 2005; Johnson et al., 2012), reducing the secretion of destabilized protein variants by increasing ER quality control stringency will decrease their extracellular populations available for concentration-dependent aggregation and attenuate extracellular aggregate load (Figure 7B).
Figure 7. Increasing ER Quality Control Stringency Is a Viable Therapeutic Approach to Reduce Extracellular Concentrations of Proteotoxic Aggregates.

(A) Illustration showing that adapting the ER proteostasis environment to enhance ER quality control stringency can reduce secretion of destabilized amyloidogenic protein variants (red) without significantly impacting secretion of stable, WT protein (black).
(B) Illustration showing that increasing ER quality control stringency through the adaption of the ER proteostasis environment provides a therapeutic approach to preferentially reduce the extracellular aggregation of destabilized, aggregation-prone protein variants most commonly associated with disease pathogenesis (red). Highly destabilized protein variants are recognized by ER quality control pathways, increasing their degradation in the ER, decreasing their secretion to the extracellular space, and reducing extracellular concentrations available for concentration-dependent aggregation (left arrow). Alternatively, stable protein variants prevent the initial misfolding steps required for protein aggregation (right arrow).
Here, we demonstrate that stress-independent ATF6 activation increases ER quality control stringency for the amyloidogenic protein TTR, preferentially reducing the secretion of destabilized, familial disease-associated TTR variants. ATF6 activation decreases TTR variant secretion to extents defined by the inherent destabilization of the TTR tetramer, reflecting the shift in ER quality control stringency described in Figure 7A. The capacity for ATF6 activation to preferentially reduce secretion of destabilized TTR variants based on energetic stability demonstrates that this approach is broadly applicable to reduce secretion of the remaining >100 destabilized, amyloidogenic TTR variants involved in familial TTR amyloid disease pathology (Johnson et al., 2012). Furthermore, we show that ATF6-dependent reduction in the secretion of destabilized FTTTRs decreases extracellular concentrations of soluble TTR aggregates, reflecting the capacity for increased ER quality stringency to influence extracellular aggregate load predicted in Figure 7B.
The increased ER quality control stringency for TTR observed upon ATF6 activation corresponds with an increased partitioning of destabilized TTRs to ERAD and autophagic degradation pathways. Using monomeric TTR, heterotetramerization with TTRWT, and a small molecule TTR tetramer stabilizer, we demonstrate that ATF6-dependent increases in TTR degradation are mediated prior to tetramerization, primarily through the increased partitioning of destabilized TTR monomers to ER degradation pathways. This indicates that ATF6-dependent increases in ER quality control do not result in the intracellular accumulation of destabilized, aggregation-prone TTR monomers that could aggregate into toxic conformations, disrupt ER proteostasis, and/or reduce cellular viability (Powers et al., 2009). Furthermore, ATF6 activation increases degradation and reduces secretion of destabilized TTRs in liver-derived HepG2DA cells, indicating that this approach is likely applicable in heterozygote patient livers, where destabilized TTRs can be stabilized and trafficked to the extracellular space in heterotetramers otherwise composed of TTRWT subunits.
The ATF6 transcriptional program has evolved to increase ER quality control during ER stress and prevent the secretion of misfolded protein conformations to the extracellular space where they could aggregate into proteotoxic conformations. Our results demonstrate that we can exploit this evolved function of ATF6 to alter the partitioning of proteins between ER protein folding/secretion and degradation based primarily on protein stability. Consistent with this prediction, ATF6 activation reduces secretion and/or intracellular aggregation of aggregation-prone variants of α1-antitrypsin and rhodopsin that are associated with nonamyloidogenic protein aggregation diseases without affecting trafficking of the corresponding stable, WT proteins (Chiang et al., 2012; Smith et al., 2011). Furthermore, ATF6 activation does not globally impact secretion of the endogenous secreted proteome (Shoulders et al., 2013). These results suggest that stress-independent ATF6 activation offers a unique opportunity to increase ER quality control stringency for destabilized, misfolding-prone proteins without significant consequences on the secretion and/or extracellular function of the endogenous secreted proteome. Critically, overexpression of the prominent ATF6 target gene BiP, the central HSP70 chaperone in the ER, attenuates degradation of destabilized TTR variants without influencing TTR secretion (Susuki et al., 2009). This suggests that emergent properties of global ER proteostasis remodeling afforded by ATF6 activation are distinct from those accessible through targeting specific ER proteostasis factors, further highlighting the advantage of accessing the endogenous ATF6 transcriptional program to influence ER proteostasis of aggregation-prone proteins.
The capacity for stress-independent activation of ATF6 to increase ER quality control stringency for TTR suggests that similar approaches can be employed to reduce secretion and extracellular aggregation of destabilized, amyloidogenic protein variants involved in other systemic amyloid diseases, many of which have no current therapeutic options (Ryno et al., 2013). For example, stress-free ATF6 activation selectively reduces the secretion and extracellular aggregation of destabilized, amyloidogenic variants of immunoglobulin light chain (LC) (Cooley et al., 2014), indicating that this approach has potential to ameliorate the LC proteotoxicity associated with Light Chain Amyloidosis, a systemic amyloid disease affecting ten people per million per year (Blancas-Mejía and Ramirez-Alvarado, 2013). The capacity for stress-independent ATF6 activation to reduce secretion and subsequent extracellular aggregation of destabilized, amyloidogenic TTR and LC variants, and potentially disease-associated variants of other amyloidogenic proteins (Ryno et al., 2013), bypasses all the challenges associated with establishing protein specific therapies for each systemic amyloid disease, potentially allowing a single therapeutic approach to be broadly applied to these etiologically diverse diseases of protein aggregation.
SIGNIFICANCE
The results described in this manuscript show that stress-independent activation of the UPR-associated transcription factor ATF6 preferentially reduces the secretion and extra-cellular aggregation of destabilized, aggregation-prone variants of the amyloidogenic protein TTR, as compared to stable WT TTR and nonamyloidogenic TTR variants. The impact of ATF6 activation on TTR variant secretion corresponds with the destabilization of the TTR tetramer afforded by mutation, indicating that ATF6 activation enhances the stringency of ER quality control for TTR secretion. The capacity for ATF6 to preferentially reduce extracellular aggregation of destabilized, disease-associated TTR variants based on the extent of destabilization suggests that similar approaches can be applied to attenuate the secretion and extracellular aggregation of destabilized, amyloidogenic variants of other amyloidogenic proteins involved in multiple systemic amyloid diseases, many of which lack available treatment options. Thus, ultimately, our results highlight the therapeutic potential for targeting ATF6 signaling as a broadly applicable therapeutic strategy to attenuate the secretion and extracellular proteotoxic aggregation of amyloidogenic proteins involved in systemic amyloid diseases.
EXPERIMENTAL PROCEDURES
Cell Culture, Plasmids, and Transfections
HEK293T, HEK293DAX, and HepG2DA cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with glutamine, penicillin/ streptomycin, and 10% fetal bovine serum, as previously described (Shoulders et al., 2013). All TTR point mutations were incorporated into the FLAG2.TTR.pcDNA3.1 vector (Shoulders et al., 2013) through site-directed mutagenesis. Transient transfections of FTTTR variants were performed by calcium phosphate transfection. Adenoviruses for FLAG2.TTR variants were prepared in the pAD-V5-DEST vector (Life Technologies) and grown and propagated in HEK293A cells (Life Technologies), as previously described (Shoulders et al., 2013). Adenoviruses encoding FTTTR variants were transduced into HepG2 cells at multiplicities of infection experimentally determined to express FTTTR mRNA to levels identical to those of the endogenous TTR.
Immunoblotting, Clear-Native-PAGE, and SDS-PAGE
Immunoblotting was performed as previously described and analyzed using the Li-COR Biosciences Odyssey System (Shoulders et al., 2013). CN-PAGE was performed on conditioned media prepared on HEK293T or HEK293DAX cells expressing FTTTR variants as described previously (Wittig and Schägger, 2008) using 4%–16% gradient NativePage gels (Life Technologies), 50 mM Bis-Tris pH 7.0 anodic buffer, 50 mM Tricine, 15 mM Bis-Tris pH 7.0 cathodic buffer, and loading buffer containing 5% glycerol, 0.01% Ponceau S, 50 mM 6-aminohexanoic acid, and 10 mM Bis-Tris pH 7.0. NativeMark Unstained Protein Standard (Life Technologies) was used for molecular weight calibration. SDS-PAGE was performed using freshly prepared 15% tris-glycine gels. Monoclonal mouse M2 anti-FLAG antibody was purchased from Sigma and polyclonal rabbit anti-TTR antibody was purchased from Dako.
[35S] Metabolic Labeling Experiments
HEK293DAX or HepG2DA cells plated on poly-D-lysine coated plates were metabolically labeled in, DMEM-Cys/-Met (CellGro) supplemented with glutamine, penicillin/streptomycin, dialyzed fetal bovine serum, and EasyTag EXPRESS35S Protein Labeling Mix (Perkin Elmer) for 30 min. Proteasome and lysosome inhibitors were added at the indicated concentrations 1 hr prior to metabolic labeling and were included in both the metabolic labeling and chase. Cells were washed twice with complete media and incubated in pre-warmed DMEM for the indicated times. Media or lysates were harvested at the indicated times. Lysates were prepared in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris pH 7.5, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS) with fresh protease inhibitor cocktail (Roche). FLAG tagged TTR variants were immunopurified using M1 anti-FLAG agarose beads (Sigma-Aldrich) and washed with RIPA buffer. The immunoisolates were then eluted by boiling in Laemmli buffer and separated on SDS-PAGE. The gels were then dried, exposed to phosphorimager plates (GE Healthcare), and imaged with a Typhoon imager. Band intensities were quantified by densitometry in ImageQuant.
Sigmoidal Fitting of Activating Transcription Factor 6-Dependent Reductions in Transthyretin Secretion
Figure 2A was prepared by fitting the raw data of TTR variant fraction secreted at t = 4 hr versus the variant CSS in Kaleidograph using the following equation: Y(X) = m1 + (m2−m1)/(1+(X/m3)^m4), where m1 is the pretransition baseline, m2 is the posttransition baseline, m3 is the midpoint of transition, and m4 is the sharpness of transition. The same equation was used to fit the raw data of impact of ATF6 activation on TTR variant secretion versus the variant CSS, as shown in Figure 2B.
Additional experimental procedures are included in the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
The authors are grateful to Jeffery Kelly and Evan Powers for helpful discussion. R.L.W. thanks NIH (AG036634, NS079882, DK075295, DK102635, and AG046495), Arlene and Arnold Goldstein, the Ellison Medical Foundation, and the Scripps Research Institute for financial support. J.C.G. was supported by the NIH (HL099245). M.D.S. was supported by the American Cancer Society.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.chembiol.2014.09.009.
AUTHOR CONTRIBUTIONS
J.J.C., J.C.G., and R.L.W. designed experiments. J.J.C., J.C.G., and S.Q. performed experiments. J.D.H. and M.D.S. provided reagents and tools. J.J.C., J.C.G., and R.L.W. interpreted experimental results and prepared the manuscript with comments from all authors.
References
- Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct. 2008;33:75–89. doi: 10.1247/csf.07044. [DOI] [PubMed] [Google Scholar]
- Blancas-Mejía LM, Ramirez-Alvarado M. Systemic amyloidoses. Annu Rev Biochem. 2013;82:745–774. doi: 10.1146/annurev-biochem-072611-130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL, Wojcikiewicz RJ. Substrate-specific mediators of ER associated degradation (ERAD) Curr Opin Cell Biol. 2009;21:516–521. doi: 10.1016/j.ceb.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL, Skach WR. Protein folding and quality control in the endoplasmic reticulum: Recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol. 2011;23:464–475. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA. 2012;109:9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum J, Tagoe C, Gallo G, Reixach N, French D. The pathogenesis of transthyretin tissue deposition: lessons from transgenic mice. Amyloid. 2003;10(Suppl 1):2–6. [PubMed] [Google Scholar]
- Buxbaum J, Jacobson DR, Tagoe C, Alexander A, Kitzman DW, Greenberg B, Thaneemit-Chen S, Lavori P. Transthyretin V122I in African Americans with congestive heart failure. J Am Coll Cardiol. 2006;47:1724–1725. doi: 10.1016/j.jacc.2006.01.042. [DOI] [PubMed] [Google Scholar]
- Buxbaum J, Anan I, Suhr O. Serum transthyretin levels in Swedish TTR V30M carriers. Amyloid. 2010;17:83–85. doi: 10.3109/13506129.2010.483118. [DOI] [PubMed] [Google Scholar]
- Chiang WC, Hiramatsu N, Messah C, Kroeger H, Lin JH. Selective activation of ATF6 and PERK endoplasmic reticulum stress signaling pathways prevent mutant rhodopsin accumulation. Invest Ophthalmol Vis Sci. 2012;53:7159–7166. doi: 10.1167/iovs.12-10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley CB, Ryno LM, Plate L, Morgan GJ, Hulleman JD, Kelly JW, Wiseman RL. Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain. Proc Natl Acad Sci USA. 2014;36:13046–13051. doi: 10.1073/pnas.1406050111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Arcangelo JG, Stahmer KR, Miller EA. Vesicle-mediated export from the ER: COPII coat function and regulation. Biochim Biophys Acta. 2013;1833:2464–2472. doi: 10.1016/j.bbamcr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidalevitz T, Stevens F, Argon Y. Orchestration of secretory protein folding by ER chaperones. Biochim Biophys Acta. 2013;1833:2410–2424. doi: 10.1016/j.bbamcr.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillmore JD, Hawkins PN. Pathophysiology and treatment of systemic amyloidosis. Nat Rev Nephrol. 2013;9:574–586. doi: 10.1038/nrneph.2013.171. [DOI] [PubMed] [Google Scholar]
- Hammarström P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293:2459–2462. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- Hammarström P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci USA. 2002;99(Suppl 4):16427–16432. doi: 10.1073/pnas.202495199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarström P, Sekijima Y, White JT, Wiseman RL, Lim A, Costello CE, Altland K, Garzuly F, Budka H, Kelly JW. D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: a prescription for central nervous system amyloidosis? Biochemistry. 2003;42:6656–6663. doi: 10.1021/bi027319b. [DOI] [PubMed] [Google Scholar]
- Houck SA, Cyr DM. Mechanisms for quality control of misfolded transmembrane proteins. Biochim Biophys Acta. 2012;1818:1108–1114. doi: 10.1016/j.bbamem.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson DR, McFarlin DE, Kane I, Buxbaum JN. Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet. 1992;89:353–356. doi: 10.1007/BF00220559. [DOI] [PubMed] [Google Scholar]
- Jahn TR, Radford SE. The Yin and Yang of protein folding. FEBS J. 2005;272:5962–5970. doi: 10.1111/j.1742-4658.2005.05021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Smith CS, Petrassi HM, Hammarström P, White JT, Sacchettini JC, Kelly JW. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 2001;40:11442–11452. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012;421:185–203. doi: 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleizen B, Braakman I. Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol. 2004;16:343–349. doi: 10.1016/j.ceb.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Kumita JR, Johnson RJ, Alcocer MJ, Dumoulin M, Holmqvist F, McCammon MG, Robinson CV, Archer DB, Dobson CM. Impact of the native-state stability of human lysozyme variants on protein secretion by Pichia pastoris. FEBS J. 2006;273:711–720. doi: 10.1111/j.1742-4658.2005.05099.x. [DOI] [PubMed] [Google Scholar]
- Lamark T, Johansen T. Autophagy: links with the proteasome. Curr Opin Cell Biol. 2010;22:192–198. doi: 10.1016/j.ceb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Marchesi M, Parolini C, Valetti C, Mangione P, Obici L, Giorgetti S, Raimondi S, Donadei S, Gregorini G, Merlini G, et al. The intracellular quality control system down-regulates the secretion of amyloidogenic apolipoprotein A-I variants: a possible impact on the natural history of the disease. Biochim Biophys Acta. 2011;1812:87–93. doi: 10.1016/j.bbadis.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci USA. 2004;101:2817–2822. doi: 10.1073/pnas.0400062101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryno LM, Wiseman RL, Kelly JW. Targeting unfolded protein response signaling pathways to ameliorate protein misfolding diseases. Curr Opin Chem Biol. 2013;17:346–352. doi: 10.1016/j.cbpa.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Susuki S, Suico MA, Miyata M, Ando Y, Mizuguchi M, Takeuchi M, Dobashi M, Shuto T, Kai H. Endoplasmic reticulum quality control regulates the fate of transthyretin variants in the cell. EMBO J. 2007;26:2501–2512. doi: 10.1038/sj.emboj.7601685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekijima Y, Hammarstrom P, Matsumura M, Shimizu Y, Iwata M, Tokuda T, Ikeda S, Kelly JW. Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab Invest. 2003;83:409–417. doi: 10.1097/01.lab.0000059937.11023.1f. [DOI] [PubMed] [Google Scholar]
- Sekijima Y, Wiseman RL, Matteson J, Hammarström P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR, 3rd, Su AI, Kelly JW, Wiseman RL. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Reports. 2013;3:1279–1292. doi: 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Granell S, Salcedo-Sicilia L, Baldini G, Egea G, Teckman JH, Baldini G. Activating transcription factor 6 limits intracellular accumulation of mutant α(1)-antitrypsin Z and mitochondrial damage in hepatoma cells. J Biol Chem. 2011;286:41563–41577. doi: 10.1074/jbc.M111.280073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susuki S, Sato T, Miyata M, Momohara M, Suico MA, Shuto T, Ando Y, Kai H. The endoplasmic reticulum-associated degradation of transthyretin variants is negatively regulated by BiP in mammalian cells. J Biol Chem. 2009;284:8312–8321. doi: 10.1074/jbc.M809354200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhaya AR, Lungrin I, Yamaguchi H, Fändrich M, Thal DR. High-molecular weight Aβ oligomers and protofibrils are the predominant Aβ species in the native soluble protein fraction of the AD brain. J Cell Mol Med. 2012;16:287–295. doi: 10.1111/j.1582-4934.2011.01306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal R, Garzuly F, Budka H, Lalowski M, Linke RP, Brittig F, Frangione B, Wisniewski T. Meningocerebrovascular amyloidosis associated with a novel transthyretin mis-sense mutation at codon 18 (TTRD 18G) Am J Pathol. 1996;148:361–366. [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wiseman RL, Powers ET, Buxbaum JN, Kelly JW, Balch WE. An adaptable standard for protein export from the endoplasmic reticulum. Cell. 2007;131:809–821. doi: 10.1016/j.cell.2007.10.025. [DOI] [PubMed] [Google Scholar]
- Wittig I, Schägger H. Features and applications of blue-native and clear-native electrophoresis. Proteomics. 2008;8:3974–3990. doi: 10.1002/pmic.200800017. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem. 2004;136:343–350. doi: 10.1093/jb/mvh122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.