

Abstract
Bridged bicyclic rings containing nitrogen heterocycles are important motifs in bioactive small organic molecules. An enantioselective copper-catalyzed alkene carboamination reaction that creates bridged heterocycles is reported herein. Two new rings are formed in this alkene carboamination reaction where N-sulfonyl-2-aryl-4-pentenamines are converted to 6-azabicyclo[3.2.1]octanes using [Ph-Box-Cu](OTf)2 or related catalysts in the presence of MnO2 as stoichiometric oxidant in moderate to good yields and generally excellent enantioselectivities. Two new stereocenters are formed in the reaction, and the C-C bond-forming arene addition is a net C-H functionalization.
Keywords: carboamination, copper-catalyzed, enantioselective, 6-azabicyclo[3.2.1]octane, alkaloids
Bridge-containing organic compounds are attractive molecular scaffolds due to their relative rigidity and ability to precisely display functional groups in three dimensions.[1] The azabicyclo[3.2.1]octane skeleton is at the core of important biologically active natural products such as cocaine, aphanorphine, and securinine.[2] In addition, synthetic analogs have demonstrated promising therapeutic potential for the treatment of drug addiction and pain.[3] Although a number of methods for their synthesis have been developed,[4] few form two rings in one step,[5] and of these latter examples, none provide azabicyclo[3.2.1]octanes enantioselectively through asymmetric catalysis.[5e,f] This report describes the synthesis of 6-azabicyclo[3.2.1]octanes via copper-catalyzed enantioselective alkene carboamination.
We have previously shown that N-tosyl-2,2-diphenyl-4-pentenamine 1a can undergo copper-catalyzed enantioselective alkene carboamination to form the chiral bicyclic sultam 2 with 94% ee (Scheme 1).[6a] The new N-C bond is thought to be set by an enantioselective aminocupration step. Subsequent C-Cu(II) bond homolysis is thought to occur, forming intermediate A. Carbon-carbon bond formation via intramolecular addition of the primary carbon radical of A to the pendant arylsulfonyl group followed by oxidation/rearomatization provides sultam 2. More recently, we reported that in the presence of 1,1-diphenylethylene, the same carbon radical intermediate can undergo an intermolecular C-C bond formation to give chiral pyrrolidine 4 with 95% ee.[7] In both examples, formation of the bridged bicyclic carboamination product 3a via path b (Scheme 1) is possible but was not observed.
Scheme 1.

Possible carboamination pathways.
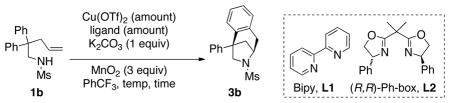
We hypothesized that in the absence of all other radical acceptors (tosyl, 1,1-diphenylethylene), 2-aryl pentenamines 1 could undergo intramolecular carboamination reactions to give 6-azabicyclo[3.2.1]octanes 3. To our delight, we found that this process could indeed occur using N-mesyl-2,2-diphenylpenteneamine 1b as the model substrate (Table 1, entry 1). Furthermore, the reaction could be rendered enantioselective to give 3b with 67% ee using the commercially available (R,R)-Ph-Box ligand complexed with Cu(OTf)2 (Table 1, entry 2). Addition of activated 4 Å molecular sieves led to a higher and more reproducible level of enantioselectivity (91% ee, Table 1, entry 3), presumably due to sequestration of adventitious water, which is deleterious to the reaction’s efficiency and selectivity. We were able to reduce the catalyst loading to 15 mol %, the temperature to 110 °C, and the reaction time to 12 h without loss of efficiency or enantioselectivity (Table 1, entry 4).
Table 1.
Optimization of reaction conditions.[a]
| |||||
|---|---|---|---|---|---|
| Entry | Cu(OTf)2 (amount) | Ligand (amount) | Temp, time | Yield (%)[b] | ee (%)[c] |
| 1 | 30 | L1 (40 | 120 °C | 84 | -- |
| mol % | mol%) | 24 h | |||
| 2 | 20 | L2 (25 | 120 °C | 78 | 67 |
| mol % | mol %) | 24 h | |||
| 3[d] | 20 | L2 (25 | 120 °C | 81 | 91 |
| mol % | mol %) | 24 h | |||
| 4[d] | 15 | L2 (19 | 110 °C | 76 | 95 |
| mol % | mol %) | 12 h | |||
| 5[d,e] | 10 | L2 (13 | 110 °C | 72 | 66 |
| mol % | mol %) | 12 h | |||
Conditions: Cu(OTf)2 was combined with L1 or L2 under argon and heated in dry PhCF3 (0.8 mL) for 2 h in a sealed tube. Substrate 1b (50.0 mg, 0.158 mmol), K2CO3 (0.158 mmol), MnO2 (0.473 mmol) and PhCF3 (0.8 mL) were added and the mixture was heated and stirred.
Isolated yield following flash chromatography on silica gel.
Enantiomeric excess determined by chiral HPLC analysis.
Activated flame-dried molecular sieves (20 mg/mL) were used.
Reaction did not go to completion.
The scope of the enantioselective alkene carboamination reaction was examined (Table 2) using either the conditions from Table 1, entry 3 [120 °C, 20 mol % Cu(OTf)2], or Table 1, entry 4 [110 °C, 15 mol % Cu(OTf)2]. N-2-(trimethylsilylethane)sulfonyl-4-pentenamine 1c gave 78% of the bicyclic adduct 3c with 95% ee. This sulfonyl group is attractive as it is easily removed with fluoride reagents such as TBAF. The N-benzylsulfonyl pentenamine 1d gave 83% of 3d with 80% ee. The N-3,5-tert-butyl-4-methoxybenzenesulfonyl substrate 1e gave 76% of 3e with 95% ee. This arylsulfonyl group’s ortho positions are hindered by its meta-tert-butyl groups, rendering formation of the fused ring sultam analogous to 2 (Scheme 1) less favorable. Electron rich (OMe, SMe) and more electron-deficient (Cl, F) para-substituents on the substrate’s backbone aryl rings had little effect on the reaction efficiency and selectivity (Table 2, entries 5–8). The 2,2-bis(2-methylphenyl)-substituted 4-pentenyl sulfonamide 5e underwent the enantioselective carboamination uneventfully, despite the added steric hindrance on the aryl acceptor, giving adduct 6e in 81% yield and 93% ee (Table 2, entry 9). N-mesyl-4-methyl-4-pentenamine 7, a 1,1-distubstituted alkene, gave adduct 8 bearing two fully substituted chiral carbons in 69% yield with 92% ee (Table 2, entry 10). The internal alkene substrates 9a and 9b, however, reacted poorly, producing intractable mixtures of substrate and unidentified products under the reaction conditions (Table 2, entries 11 and 12).
Table 2.
Scope of the enantioselective carboamination.
| Entry | Substrate | Product | Yield (%) | ee (%) |
|---|---|---|---|---|
| 1[a] |
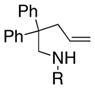 1b, R = Ms |
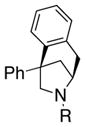 3b |
76 | 95 |
| 2[b] | 1c, R = SES | 3c | 80 | 90 |
| 3[b] | 1d, R = SO2Bn | 3d | 83 | 80 |
| 4[b] |
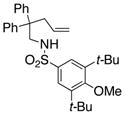 1e |
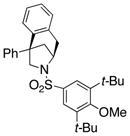 3e |
76 | 95 |
| 5[a] |
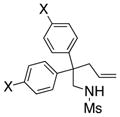 5a, X = OMe |
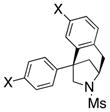 6a |
83 | 95 |
| 6[a] | 5b, X = SMe | 6b | 80 | 93 |
| 7[a] | 5c, X = Cl | 6c | 80 | 90 |
| 8[a] | 5d, X = F | 6d | 68 | 95 |
| 9[b] |
 5e |
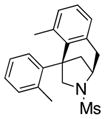 6e |
81 | 93 |
| 10[b] |
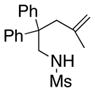 7 |
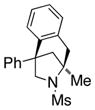 8 |
69 | 92 |
| 11[b] |
 9a, R = Me |
-- | -- | |
| 12[b] | 9b, R = Ph | -- | -- |
An N-mesyl-2,2-diaryl-4-pentenamine bearing meta substitution on its aryl rings produced a 2.5:1 ratio of regioisomers favoring the more hindered ortho isomer 6f (Eq. 1). The level and direction of regioselectivity is consistent with addition of a carbon radical to the arene (e.g. path b, Scheme 1)[6b,c,8] as electrophilic aromatic substitution would be expected to favor the less hindered para product 6g.[9]
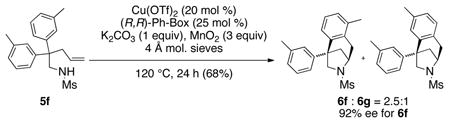 |
(1) |
The absolute configuration of 3b was confirmed by its X-ray structure (Figure 1) and all other enantioenriched products were assigned by analogy to 3b.
Figure 1.

X-ray crystal structure of 3b (CCDC 991873).
We next examined the desymmeterization reactions of bis(2-allyl) substrates 10a and 10b (Scheme 2). The use of our standard conditions (Table 1, entry 3) with the (R,R)-Ph-Box ligand gave 11a in 30% yield with a disappointing 35% ee (not shown), thus we turned to the (4R,5S)-cis-diphenyl bis(oxazoline) ligand, which has demonstrated superior enantioselectivity in challenging cases in copper catalysis.[10] Encouragingly, 11a was formed in 39% yield and 80% ee using this ligand. We were also able to achieve enantioselective desymmeterization using the bis(2-methylallyl) substrate 10b, which gave 11b in 40% yield, with 92% ee. The reduced yield in these reactions is likely due to the additional demand for diastereoselectivity in the N-C bond forming step, although we were unable to isolate and identify minor products from these reactions.
Scheme 2.

Desymmetrization reactions.
The proposed catalytic cycle is given in Scheme 3. The stereoselectivity of the reaction is achieved in the cis-aminocupration step, where the the phenyl rings on the chiral ligand are “trans” to the substrate’s mesyl and alkene groups to avoid steric interaction.[11] The resulting organocopper(II) intermediate undergoes homolysis to form a primary carbon radical which adds across the pyrrolidine ring to the aryl group it is cis to. Oxidation of the resulting aryl radical under the reaction conditions provides the azabicyclic product 3. It should be noted that analogous transannular additions of carbon radicals to alkenes have been reported,[12] but transannular radical additions to arenes are more rare.[8b,13]
Scheme 3.

Proposed catalytic cycle.
In conclusion, the copper(II)-catalyzed enantioselective alkene carboamination reaction is an efficient method to prepare a variety of chiral 6-azabicyclo[3.2.1]octanes. Further investigations into the scope and utility of this method for its application in organic synthesis will be reported in due course.
Experimental Section
For experimental details and spectral data for all new compounds, see Supporting Information.
Representative Procedure for the Enantioselective Carboamination
(1S,4R)-3-(methylsulfonyl)-1-phenyl-2,3,4,5-tetrahydro-1H-1,4-methanobenzo[d]azepine (3b)
Cu(OTf)2 (8.5 mg, 0.024 mmol, 15 mol %), (R,R)-Ph-Box (10.0 mg, 0.030 mmol, 19 mol %) and dry PhCF3 (0.8 mL) were placed in a flame-dried glass reaction tube under argon. The tube was capped and the reaction heated at 60 °C for 2 h. After cooling, sulfonamide 1b (50.0 mg, 0.158 mmol), K2CO3 (21.8 mg, 0.158 mmol, 1 equiv), MnO2 (41.1 mg, 0.473 mmol, 3 equiv) and PhCF3 (0.8 mL) were added. Flame-dried molecular sieves (4 Å, 35 mg/mL) were added to the reaction mixture, the tube was flushed with argon, capped and the mixture was heated at 110 °C for 12 h. Upon cooling, the mixture was diluted with EtOAc and filtered through a pad of silica gel. The filtrate was concentrated in vacuo and the crude residue was purified by flash chromatography on silica gel, providing 38 mg of 6-azabicyclo[3.2.1]octane 3b as a white solid (76% yield). Compound 3b was further purified via HPLC and found to be in 95% enantiomeric excess as determined by analysis on chiral HPLC [Chiralpak AD-RH, 50% CH3CN/H2O, 0.5 mL/min, t(minor) = 15.93 min, t(major) = 18.63 min], [α]D23 = −88.3 ° (c = 0.6 CHCl3); mp 209–211 °C; 1H NMR (300 MHz, CDCl3): δ 7.45-7.28 (m, 5 H), 7.16-7.13 (m, 2 H), 7.01-6.95 (m, 1H), 6.53 (d, J = 8.4 Hz, 1 H), 4.58-4.54 (m, 1 H), 3.88 (ABq, JAB = 9.0 Hz, δν = 17.0 Hz, 2 H), 3.30 (d, J = 17.1 Hz, 1 H), 3.18 (dd, J = 17.1, 2.4 Hz, 1 H), 2.80 (s, 3 H), 2.61 (d, J = 11.1 Hz, 1 H), 2.30 (ddd, J = 12.0, 5.8, 1.5 Hz, 1 H) ppm; 13C NMR (75 MHz, CDCl3): δ 143.8, 142.2, 132.7, 129.5, 127.6, 127.3, 126.8, 126.2, 61.0, 57.0, 51.3, 40.6, 38.5, 36.5 ppm; IR (neat, thin film): 3061, 3031, 2955, 2868, 1485, 1450, 1331, 1157, 1061 cm−1; HRMS (ESI) calcd for [M+H]+ C18H19NO2S: 314.1209, found: 314.1219.
Supplementary Material
Acknowledgments
The National Institute of General Medical Science (GM078383) is gratefully acknowledged for support of this research. We thank Dr. William W. Brennessel and the X-ray Crystallographic Facility at the University of Rochester for the X-ray structure of 3b (CCDC 991873). We thank Timothy W. Liwosz and Michael T. Bovino for the synthesis of bis(oxazoline) ligands as well as Nicole E. Kendal for the preparation of some racemic products.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######. ((Please delete if not appropriate))
References
- 1.Synthetic methods for bridged-ring compounds: Zhao W. Chem Rev. 2010;110:1706. doi: 10.1021/cr9002402.Bonjoch J, Diaba F, Bradshaw B. Synthesis. 2011:993.
- 2.a) Singh S. Chem Rev. 2000;100:925. doi: 10.1021/cr9700538. [DOI] [PubMed] [Google Scholar]; b) Gulavita N, Hori A, Shimizu Y, Laszlo P, Clardy J. Tetrahedron Lett. 1988;29:4381. [Google Scholar]; c) Beutler JA, Karbon EW, Malik AN, Curtis DR, Enna JJ. Brain Res. 1985;330:135. doi: 10.1016/0006-8993(85)90014-9. [DOI] [PubMed] [Google Scholar]
- 3.a) Abraham P, Pinter JB, Lewin AH, Boja JW, Kuhar MJ, Carroll FI. J Med Chem. 1992;35:141. doi: 10.1021/jm00079a018. [DOI] [PubMed] [Google Scholar]; b) Takeda M, Inoue H, Noguchi K, Honma Y, Kawamori M, Tsukamoto G, Yamawaki Y, Saito S, Aoe K, Date T, Nurimoto S, Hayashi G. J Med Chem. 1977;20:221. doi: 10.1021/jm00212a007. [DOI] [PubMed] [Google Scholar]
- 4.a) Rigby JH, Pigge FC. Synlett. 1996:631. [Google Scholar]; b) Rigby JH, Pigge FC. Tetrahedron Lett. 1996;37:2201. [Google Scholar]; c) Han G, LaPorte MG, Folmer JJ, Werner KM, Weinreb SM. J Org Chem. 2000;65:6293. doi: 10.1021/jo000260z. [DOI] [PubMed] [Google Scholar]; d) Tamaru O, Yanagiamachi T, Kobayashi T, Ishibashi H. Org Lett. 2001;3:2427. doi: 10.1021/ol015818j. [DOI] [PubMed] [Google Scholar]; e) Zhai H, Lou S, Ye C, Ma Y. J Org Chem. 2003;68:8268. doi: 10.1021/jo0348726. [DOI] [PubMed] [Google Scholar]; f) Yang X, Zhai H, Li Z. Org Lett. 2008;10:2457. doi: 10.1021/ol800737d. [DOI] [PubMed] [Google Scholar]; g) Fuchs JR, Funk RL. Org Lett. 2001;3:3923. doi: 10.1021/ol016795b. [DOI] [PubMed] [Google Scholar]; h) Mai DN, Rosen BR, Wolfe JP. Org Lett. 2011;13:2932. doi: 10.1021/ol2009895. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Medjahdi M, Gonzalez-Gomez JC, Foubelo F, Yus M. Eur J Org Chem. 2011;12:2230. [Google Scholar]
- 5.de Haro T, Nevado C. Angew Chem Int Ed. 2011;50:906. doi: 10.1002/anie.201005763.Schultz DM, Wolfe JP. Org Lett. 2011;13:2962. doi: 10.1021/ol201051q.Kong A, Blakey SB. Synthesis. 2012;44:1190.Li Q, Jiang X, Fu C, Ma S. Org Lett. 2011;13:466. doi: 10.1021/ol102811x.For related synthesis of [3.3.1] azabicycles using asymmetric catalysis, see: Bradshaw B, Parra C, Bonjoch J. Org Lett. 2013;15:2458. doi: 10.1021/ol400926p.Diaba F, Bonjoch J. Org Biomol Chem. 2009;7:2517. doi: 10.1039/b906835j.
- 6.a) Zeng W, Chemler SR. J Am Chem Soc. 2007;129:12948. doi: 10.1021/ja0762240. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miao L, Haque I, Manzoni MR, Tham WS, Chemler SR. Org Lett. 2010;12:4739. doi: 10.1021/ol102233g. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sherman ES, Fuller PH, Kasi D, Chemler SR. J Org Chem. 2007;72:3896. doi: 10.1021/jo070321u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liwosz TW, Chemler SR. J Am Chem Soc. 2012;134:2020. doi: 10.1021/ja211272v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Conrad SR, Kong J, Laforteza BN, MacMillan DWC. J Am Chem Soc. 2009;131:11640. doi: 10.1021/ja9026902. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miller Y, Miao L, Hosseini AS, Chemler SR. J Am Chem Soc. 2012;134:12149. doi: 10.1021/ja3034075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yip KT, Yang D. Org Lett. 2011;13:2134. doi: 10.1021/ol2006083. [DOI] [PubMed] [Google Scholar]
- 10.a) Sequeira FC, Bovino MT, Chipre AJ, Chemler SR. Synthesis. 2012;44:1481. doi: 10.1055/s-0031-1289762. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bovino MT, Chemler SR. Angew Chem Int Ed. 2012;51:3923. doi: 10.1002/anie.201109044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The pro-S and pro-R transition states for similar reactions have been modeled with density functional theory calculations: Belding L, Chemler SR, Dudding T. J Org Chem. 2013;78:10288. doi: 10.1021/jo401665n.
- 12.These methods also form azabicyclic products: Bennasar ML, Roca T, Garcia-Diaz D. J Org Chem. 2008;73:9033. doi: 10.1021/jo801998h.Quirante J, Vila X, Escolano C, Bonjoch J. J Org Chem. 2002;67:2323. doi: 10.1021/jo0163849.
- 13.Transannular addition to arene: Kostiuk SL, Woodcock T, Dudin LF, Howes PD, Harrowven DC. Chem-Eur J. 2011;17:10906. doi: 10.1002/chem.201101550.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.