

Abstract
Although atrial fibrillation (AF) is clinically and genetically a highly heterogeneous disease, recent studies suggest that the arrhythmia may arise because of interactions between genetic and acquired risk factors – the so called “double-hit” hypothesis. Genome-wide association studies have identified common AF susceptibility loci, and linkage analysis and candidate gene approaches have identified mutations in genes that encode for cardiac ion channels and signaling proteins; however, most of the heritability of AF still remains unexplained. The voltage-dependent cardiac sodium channel, encoded by SCN5A, conducts the main cardiac inward sodium current (INa) and is responsible for the upstroke of the atrial action potential. Mutations in SCN5A, which encodes the α-subunit of the NaV1.5 channel, have been linked with increased susceptibility to not only AF but also ventricular arrhythmias (long QT syndrome, Brugada syndrome), progressive cardiac conduction disease, and overlap syndromes with mixed arrhythmia phenotypes. Over the last decade, functional characterization of SCN5A mutations by expressing the channel in heterologous expression systems and applying cellular electrophysiological techniques has not only advanced our understanding of molecular mechanisms of AF but also potentially identified a mechanism-based approach to treating this common and morbid condition.
Keywords: atrial fibrillation, SCN5A mutations, electrophysiology, SCN5A, gain of function, loss of function, action potential, “two-hit” hypothesis
1. Definition and epidemiology of Atrial Fibrillation
Atrial fibrillation (AF) is the most frequent sustained cardiac arrhythmia encountered in the clinical practice. It is described as a rapid irregular and chaotic electrical activation of atria that results in highly variable ventricular rates.1 The AF electrocardiogram is characterized by the absence of P waves and irregularities in the R-R intervals.
AF is clinically and genetically a highly heterogeneous disease. Both acquired2 and genetic3 risk factors for AF have been identified. About 10% of patients over 80 years of age develop AF. Up to 30% of AF cases occur in individuals with no prior history of cardiac or systemic conditions, a group that has previously been defined as “lone” or early onset AF. Christophersen et al. have estimated this number to be as high as 60% of total AF cases.4 While the number of patients with true “lone” AF is probably very few,5 not all our patients with early onset AF can be categorized as having a monogenic/Mendelian form of AF6 as only ~35% have a positive family history.5 Genome-wide association studies (GWAS) have identified common genetic variants at 9 different chromosomal loci that are significantly associated with AF.7-10 However, most of the heritability of AF still remains unexplained.11 The recognition that both acquired and genetic AF risk factors are associated with increased risk for AF has fundamentally altered our perception of this arrhythmia. Due to its clinical and genetic heterogeneity, AF should be considered a syndrome rather than a homogeneous disease entity, where the arrhythmia represents the culmination of diverse etiologies and pathways.
In 1994, using the Framingham cohort, Benjamin et al. identified independent risk factors for AF.2 The strongest predictor of development of AF was age. AF prevalence increased from 0.5% in the age group 50-60 years old to 8.8% in patients older than 80 years old.12 This study also demonstrated that common risk factors for cardiovascular disease e.g., diabetes, hypertension and cardiac valve disease are associated with increased risk for AF and modification of these risk factors may reduce the incidence of AF as well.2 Importantly however, only some patients with these risk factors develop AF. One possible explanation for this apparent paradox is that both acquired and genetic risk factors are required to develop AF – the so-called “two-hit” hypothesis.13
2. Genetics of AF
Despite AF being the most common arrhythmia in clinical practice requiring pharmacological treatment, response to antiarrhythmic drugs (AADs) is highly unpredictable with ~50% of patients experiencing a symptomatic recurrence of AF within 6-12 months of starting therapy. The limited success of AADs is most likely related to our poor understanding of the molecular pathophysiology of AF, inter-individual differences in underlying mechanisms and our inability to target mechanism-based therapies. There is increasing support for the idea that variability in drug and ablation therapy may in part reflect heterogeneity of the underlying AF mechanisms.13
Over the last decade, we and others have applied diverse genetic approaches to define the genetic architecture of AF and better identify the underlying genetic mechanisms. While a comprehensive review of the genetic basis of AF is beyond the scope of this review, many excellent reviews have recently been published.13,14 Linkage and positional cloning approaches have identified family-specific rare mutations that encode not only cardiac ion channels including SCN5A, but also signaling molecules such as atrial natriuretic peptide (ANP) and nucleoporins (NUP155). Furthermore, a candidate gene approach has uncovered both common and rare genetic variants linked with AF. A more contemporary approach that has been utilized is the identification of common genetic variants (or single nucleotide polymorphisms [SNPs]) by genome-wide association studies (GWAS). These have uncovered novel genes and loci that are important for the initiation and maintenance of AF.13
3. Normal Atrial Electrophysiology and Role of SCN5A/NaV1.5
As cardiac myocytes are excitable cells, they are capable of generating rapid changes in the cell membrane polarity due to changes in their ionic conductance. These cells are characteristically activated by an electrical ‘all-or-none’ response, the action potential (AP). These changes in the electrical potential across the plasma membrane of the cardiomyocytes are a consequence of an orchestrated sequence of openings and closings of cardiac ion channels. The electronegative resting membrane potential in atrial cells (of approximately −80 mV) is critical for the normal propagation of the atrial AP across cells. Cardiomyocytes are heavily dependent mainly on the electrogenic Na/K-ATPase but also inward rectifying potassium currents (mainly, IK1) to maintain the electronegativity of the resting membrane potential. The potassium currents facilitate termination of the AP by repolarizing the depolarized membrane back to resting levels.15
A typical atrial AP can be divided into 5 phases: phase 0 (upstroke), phase 1 (“early repolarization” phase), phase 2 (or “plateau”), phase 3 (or repolarization phase), and phase 4 (resting potential) (Figure 1). The sodium (INa) and the L-type calcium (ICa-L) currents are the two main inward currents that participate in the upstroke (phase 0) of atrial AP.16 One consequence of the dual participation of these two ionic currents during phase 0 is that the [dV/dt]max of atrial AP is less steep than that of ventricular myocytes.15 It is important to note that although these two depolarizing inward currents participate in the generation of phase 0 of the atrial AP, the main determinant of the plateau for both atrial and the ventricular cells is ICa-L.15 In addition to its contribution to phases 0 and 2 of the AP, ICa-L triggers the release of additional Ca2+ from the sarcoplasmic reticulum (SR), which is central for activation of atrial contraction. Moreover, a transient outward 4-aminopyridine-sensitive potassium current (Ito1) contributes to the low membrane potential during the plateau phase. Phase 1 of AP (“early repolarization”) is due to activation of Ito2, a transient outward Ca2+-dependent chloride current that is 4-aminopyridine insensitive and can be suppressed by caffeine and manganese.17 Once Ito1 is inactivated, the ultra-rapid delayed rectifier potassium current (IKur) remains active. IKur is selectively expressed in the atria and is generated by the KV1.5 channel. During the repolarization phase (phase 3 of AP) several other transient inward potassium currents (IKs, IKr, IKAch, IKATP, IK1) ensure return of the membrane potential back to resting levels.13,15
Figure 1.
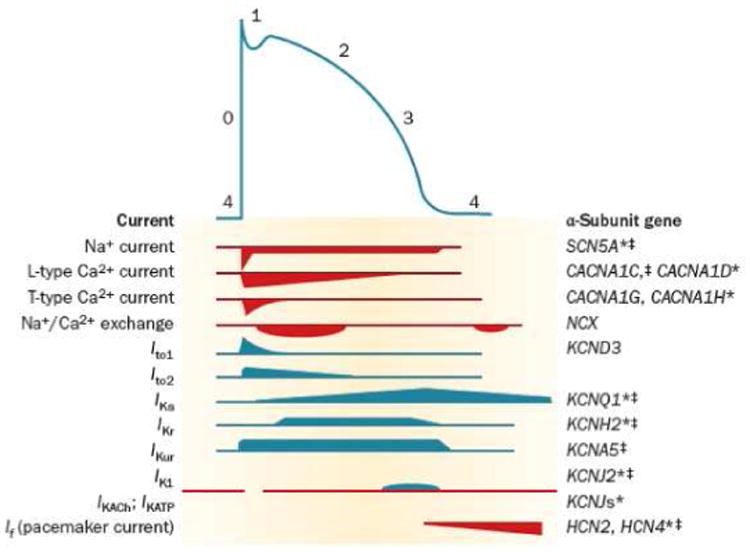
The relationship between ionic currents and the duration of the atrial action potential (AP). The AP is initiated by a rapid influx of Na+ ions (phase 0), followed by an early (phase 1 and 2) and late (phase 3) phases of repolarization, before returning to the resting membrane potential (phase 4). * Function-modifying subunit. ‡ Mutations in this gene were associated with atrial fibrillation. (From Darbar D, Roden DM. Genetic mechanisms of atrial fibrillation: impact on response to treatment. Nat Rev Cardiol 2013;10(6):317-29).
The main sodium channel involved in the upstroke of the atrial AP is the canonical NaV1.5 cardiac channel, which is part of a group of ion channels called voltage-gated sodium channels (VGSCs) (a detailed review of all the VGSCs has recently been published in a book edited by Peter C Ruben. Handbook of Experimental Pharmacology, vol. 221. 2014).18 Similar to other channels VGSCs activate, deactivate, and inactivate in response to changes in the membrane potential. VGSCs conform to a universal ion channel structure consisting of an α-subunit that is organized into 4 homologous domains (DI-DIV). Each domain comprises of 6 transmembrane segments (S1-S6). These 24 α-helical transmembrane segments arranged in 4 domains surround a central aqueous pore that allows sodium ions to flow from the extracellular space into the cytosol of the myocyte causing depolarization of the myocyte. Each of the 4 domains of the sodium channel contains a voltage-sensing domain formed by the first 4 transmembrane segments S1-S4, and a pore domain consisting of segments S5 and S6 and the extracellular linker between these two segments (the p-loop) (Figure 2).19
Figure 2.
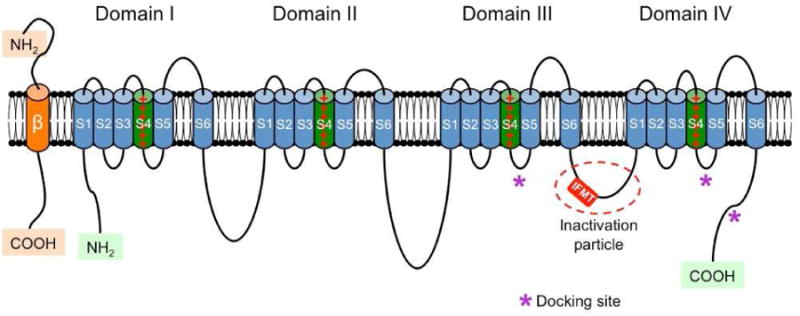
Schematic representation of the α and β subunits of the voltage-gated sodium channel. The four homologous domains (I-IV) of the α subunit are represented; S5 and S6 are the pore-lining segments and S4 is the core of the voltage-sensor. In the cytoplasmic linker between domains III and IV the IFMT (isoleucine, phenylalanine, methionine, and threonine) region is indicated. This is a critical part of the “inactivation particle” (inactivation gate), and substitution of aminoacids in this region can disrupt the inactivation process of the channel. The “docking site” consists of multiple regions that include the cytoplasmic linker between S4-S5 in domains III and IV, and the cytoplasmic end of the S6 segment in domain IV (*). Depending on the subtype of β subunit considered, they can interact (covalently or non-covalently) with the α subunit (From Savio-Galimberti E, Gollob MH, Darbar D. Voltage-gated sodium channels: biophysics, pharmacology, and related channelopathies. Front Pharmacol. 2012 Jul 11;3:124. Doi:10.3389/fphar.2012.00124. eCollection 2012.
Although the α-subunit of the cardiac sodium channel is normally associated with one or more β regulatory subunits, the α-subunit itself conducts sodium.19-21 The β-subunits are integral proteins of the cardiac myocytes sarcolemma. There are 4 different types of β-subunits, β1-β4. Each β-subunit is composed of an N-terminal domain, one transmembrane domain, and one intracellular C-terminal domain. In addition to being cell adhesion molecules (CAMs), the cardiac sodium channel β-subunits also modulate cell surface expression of the α-subunits, enhancing sodium current density and cell excitability.20,21 The α-subunit of the NaV1.5 channel is encoded by the SCN5A gene. β-subunits are encoded by SCN1B through SCN4B. Mutations in both the α- and β-subunits have been linked with increased susceptibility to atrial and ventricular arrhythmias.20,21
Although the main cardiac sodium channel isoform expressed in cardiomyocytes is NaV1.5 (which is tetrodotoxin (TTX)-resistant), there are several other sodium (TTX-sensitive) channel isoforms expressed in the myocardium. These isoforms account for a smaller fraction of the total sodium channel transcripts, and include isoforms NaV1.1-NaV1.4 and NaV1.6. The TTX-sensitive component of the sodium channel contributes to ~8% of the total INa.15,22 TTX-resistant NaV1.8 mRNA has also been detected and quantified in the atria of mice.23 Facer et al. were able to identify the NaV1.8 channel in human atrial samples collected during surgery for valve disease using immunohistochemistry.24
4. Role of Cardiac Potassium Channels Mutations in Increased Susceptibility to AF
The clinical and genetic heterogeneity of AF may in part be related to the poor understanding of the underlying pathophysiology of AF. While most cases of AF are acquired and related to structural remodeling of the atria, in ~30% of patients AF occurs without structural heart disease. It is in this group of patients with early onset AF that common and rare SCN5A variants may not only play an important role in the pathophysiology of AF but also identify underlying mechanisms.25 However, it should be appreciated that the familial monogenic AF is an uncommon disease.
The first mutations linked with AF were identified in genes encoding cardiac potassium channels. A mutation in KCNQ1 (S140G), encoding the α-subunit of the slowly repolarizing potassium channel current IKs, was identified in a large Chinese family with 16 members who developed early onset AF. When the KCNQ1 S140G mutation was expressed in a heterologous expression system, a marked increase in IKs was discovered. It is postulated that this gain-of-function mutation may lead to shortening of the atrial AP duration (APD) and the effective refractory period (ERP) of the atria.26 Several gain-of-function mutations in KCNQ1 have subsequently been reported.14 Identification of KCNQ1 as an AF-causative gene led to screening of other cardiac potassium channels as potential candidate genes for familial AF. Mutations in KCNE1,27 KCNE2,28 KCNE3,29 KCNE5,30 KCND3,31 KCNJ2,32,33 and KCNA534 have now all been associated with increased susceptibility to AF. Most potassium channels' variants associated with AF exhibit a gain-of-function cellular phenotype, and predispose to AF by shortening of the atrial APD and ERP.13,14 However, loss-of-function mutations in KCNA5, encoding the KV1.5 channel, have also been reported.35-37 These loss-of-function mutations are postulated to prolong atrial APD and trigger early afterdepolarizations and thereby provide an electrophysiological substrate for AF. Although mutations in cardiac potassium channels modulate atrial APD and likely generate an electrophysiological substrate for AF, large scale re-sequencing has not identified genes encoding cardiac potassium channels as a common cause for AF.38,39
5. Role of SCN5A Mutations in Mixed Inherited Arrhythmia Syndromes
Both common and rare genetic variants in the cardiac sodium channel have also been associated with the development of AF. SCN5A was considered a strong candidate gene for AF after the publication of two studies which showed that mutations in this gene were linked with a syndrome comprising of dilated cardiomyopathy, AF and cardiac conduction disease.40,41 AF-causing variants have been reported in both the α-subunit (encoded by the SCN5A gene) and associated β-subunits (encoded by the genes SCN1B-SCN4B). Similar to potassium channels genetic variants, both gain- and loss-of-function mutations are capable of creating a pro-arrhythmogenic substrate, further supporting the idea that mutations in cardiac sodium channel genes are also candidate genes for AF.
SCN5A mutations are not only associated with AF but also ventricular inherited syndromes such long-QT syndrome (LQTS), sudden infant death syndrome and Brugada syndrome, as well as progressive cardiac conduction disease, and more complex overlapping inherited arrhythmia syndromes. There is a high incidence (20- 40%) of AF in Brugada patients presenting with SCN5A mutations.42,43 As would be expected with loss of function of the cardiac sodium channel, these patients often also have evidence of progressive conduction disease with prolonged atrioventricular and atrial-His conduction. There is also a high incidence (~2%) of AF in patients with the LQTS.44 Interestingly, Benito et al. identified a moderate sized kindred where an SCN5A gain of function mutation was associated with a mixed phenotype of prolonged QT intervals and AF.45 While the precise electrophysiological mechanisms by which an SCN5A mutation gives rise to a mixed phenotype such as LQTS and AF, Brugada syndrome and progressive conduction disease has not been completely determined, one explanation may relate to differential expression or chamber-specific interactions between the NaV1.5 protein and its partners. These may be β-subunits, other sodium channels such as SCN10A, which encodes Nav1.8 or proteins that have not yet been identified.
6. SCN5A Mutations and AF
The first comprehensive resequencing of the entire coding region of SCN5A in patients with AF was performed in 2008. Here, we screened 375 subjects with early onset AF (n=118) or AF associated with traditional risk factors (n=257) and identified 8 novel SCN5A mutations, not found in a control population, in 10 familial AF probands.39 These variants were in highly conserved residues and co-segregated with AF in 6 familial AF kindreds. Furthermore, when these variants were expressed in a heterologous expression system, they modulated the biophysics of the Nav1.5 channel.46 While 4 of the 8 novel variants were identified in probands with early onset AF (age 36±14 years), AF in the other 4 probands was associated with underlying structural heart disease (cardiomyopathy, hypertension, or ischemic heart disease). We also identified 12 rare SCN5A variants that have previously been reported and 3 common SNPs.39
Since then, other studies have confirmed the association between SCN5A variants and increased susceptibility to AF. In 2008 Ellinor et al. reported the results of a study where they showed that an SCN5A mutation contributes to the development of early onset AF.38 However, this study was limited by its small size; out of a total cohort of 189 patients with early onset AF, only 57 AF probands with family history underwent further evaluation. A single SCN5A mutation (N1986K), absent in 600 control chromosomes, was identified in a small familial AF kindred. The expression of this mutant in Xenopus oocytes demonstrated a hyperpolarizing shift in the steady state inactivation of the N1986K mutant channel, which would be expected to result in an effective loss-of-function of the sodium channel and prolongation of the atrial APD.
While shortening of the atrial APD is the most common postulated mechanism for the initiation of AF, an alternative mechanism may relate to prolongation of the atrial APD.35 This mechanism is supported by a number of studies where SCN5A gain of function mutations have been linked with increased susceptibility to AF. In 2009, Li et al. reported an SCN5A gain-of function mutation in AF patients that can enhance cellular excitability and lowered the AP threshold.13,47 Most recently, Ziyadeh-Isleem et al. reported an SCN5A C-terminal truncating mutation (R1860Gfs*12) identified in a family presenting with a complex clinical phenotype of sick-sinus syndrome and AF or atrial flutter. The mutation was a 1 bp (A) deletion at position 5578 in exon 28. This deletion induced a frame-shift mutation, R1860Gfs*12, which changed the arginine in position 1860 to glycine followed by 10 frameshift amino acids before a premature stop codon. The heterologous expression of the mutant channels alone or in combination with the wild-type channel demonstrated a ~70% reduction in the sodium current density. As this mutation involved the C-terminus, inactivation of the channel was markedly impaired giving rise to persistent late sodium current. This was described as both a combined loss- and gain-of-function mutation, since the mutation not only decreased the peak current but also exhibited a persistent late current. The increased persistent late INa can disrupt repolarization of atrial myocytes, and prolong APD thereby providing another potential mechanism by which the mutation may increase susceptibility to AF.48
7. Challenges Associated with Linking SCN5A Variants with Increased Susceptibility to AF
We and others have identified SCN5A mutations that have been linked with mixed phenotypes and postulated potential mechanisms to explain the diverse arrhythmia syndromes.13,49 However, these studies clearly highlight some of the challenges encountered when trying to determine the pathogenicity of an SCN5A mutation and uncovering the underlying mechanisms.
Multiple approaches have been used to distinguish between benign rare polymorphisms and pathogenic mutations including screening a large ethnically-matched control population; in a large kindred evaluating if the variant co-segregates with AF; determining whether the variant is evolutionary conserved; using in-silico prediction models; and functionally characterizing the variant in-vitro.50 While heterologous expression systems and in-vitro studies can contribute to the characterization of the variants, these systems tend to be oversimplified models that fail to recapitulate the electrical milieu in which the variants function and do not necessarily provide clarification of how both loss- and gain-of-function SCN5A mutations may cause a diverse array of clinical arrhythmia phenotypes.
While other expression modeling systems, such as cardiac myocytes and induced pluripotent stem (iPS) cell-derived myocytes, have also been proposed as suitable models to dissect the underlying mechanisms associated with SCN5A mutations, these approaches also have a number of limitations. One of the techniques used to transfer a gene into mammallian cells (like cardiomyocytes) is using adenoviral or retroviral/lentiviral vectors. As retroviral vectors only accommodate approximately 8kb of DNA, this poses problems when attempting to insert the complete coding region of a gene like SCN5A, whose size is ~ 10,161 kb, and its promoter elements.51,52 An alternate approach is the transfection of sodium channels in neonatal cardiomyocytes instead of adult myocytes. Neonatal cells are less committed and more ‘plastic’ in terms of gene expression. However, the disadvantage of transfecting neonatal cells is that they exhibit an immature electrophysiologic phenotype that does not completely recapitulate the adult atrial AP. The use of iPSc-derived cardiomyocytes (iPSc-CM) is even more technically challenging, with the major obstacle related to the lack of clear atrial structure and electrophysiological phenotype of these early cardiomyocytes. Although criticized, the use of iPSc-CM is a new approach that is actively developing and has been adopted by many scientific groups as the more approachable disease-model that can provide an improved inside in diseases based on genotype specificity.53,54
Genetically modified mice have also been proposed as a complimentary approach to in vitro electrophysiology to explore and improve our understanding of the pathophysiological consequences of SCN5A mutations. While many SCN5A mouse models generated express either a LQT type 3 or Brugada syndrome phenotypes, some do also exhibit mixed phenotypes.55 Furthermore, many mouse models of SCN5A mutations also demonstrate inducible AF.56 The lack of mouse models of SCN5A genetic variants exhibiting purely an AF phenotype highlights some of the challenges and complexities of dissecting the underlying pathophysiology of AF related to cardiac sodium channel mutations.
Key points.
The incidence and prevalence of atrial fibrillation (AF) continues to rise and may in part be explained by the aging of the population.
Recent data suggest that both genetic and acquired risk factors increase susceptibility to AF and their simultaneous occurrence has given rise to the “two-hit” hypothesis for the development of AF.
SCN5A variants have also been linked with an increasing number of cardiac arrhythmia syndromes including long QT syndrome, Brugada syndrome, sick-sinus syndrome, conduction disease, AF, atrial standstill, overlap syndromes with mixed arrhythmia phenotypes, and drug-induced arrhythmias.
Common and rare genetic variants in SCN5A, which encodes the α-subunit of human cardiac sodium channel, have been associated with AF with both gain and loss-of-function variants modulating susceptibility to AF.
SCN5A genetic variants have not only provided important insights into AF mechanisms but have also uncovered novel therapeutic targets for the treatment of this common and morbid condition.
Acknowledgments
This work was supported by the National Institutes of Health (U19 HL65962, HL092217, CTSA award No. UL1TR000445, awarded to DD), and MMPC MicroMouse grant (MMPC/NIH, awarded to ESG).
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brugada R, Kaab S. Chapter 6, pages 69-76: Genetics of AF. In: Natale A, Jalife J, editors. Atrial Fibrillation: From Bench to Bedside. 2008. Humana Press, a part of Springer Science + Bussiness Media, LLC. 999 Riverview Drive, Suite 208, Totowa, NJ 07512 USA.
- 2.Benjamin EJ, Levy D, Vaziri SM, D'Agostino RB, Belanger AJ, Wolf PA. Independent risk factors in a population-based cohort The Framingham Heart Study. JAMA. 1994;271(11):840–844. [PubMed] [Google Scholar]
- 3.Sinner MF, Ellinor PT, Meitinger T, Benjamin EJ, Kaab S. Genome-wide association studies of atrial fibrillation: past, present, and future. Cardiovasc Res. 2011;89(4):701–709. doi: 10.1093/cvr/cvr001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cristophersen IE, Ravn LS, Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH, Christensen K. Familial aggregation of atrial fibrillation:a study in Danish twins. Circ Arrhythm Electrophysiol. 2009;4:378–383. doi: 10.1161/CIRCEP.108.786665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wyse DG, Van Gelder IC, Ellinor PT, Go As, Kalman JM, Narayan SM, Nattel S, Schotten U, Rienstra M. Lone Atrial Fibrillation: Does it exist. J Am Coll Cardiol. 2014;63(17):1715–1723. doi: 10.1016/j.jacc.2014.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirchhof P, et al. Personalized management of atrial fibrillation: Proceedings from the fourth Atrial Fibrillation competence NETwork/European Heart Rhythm Association consensus conference. Europace. 2013;11:1540–1556. doi: 10.1093/europace/eut232. [DOI] [PubMed] [Google Scholar]
- 7.Gudbjartsson DF, Arnar DO, Helgadottir A, Gretarsdottir S, Holm H, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–357. doi: 10.1038/nature06007. [DOI] [PubMed] [Google Scholar]
- 8.Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44(6):670–675. doi: 10.1038/ng.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, Chung MK, Sinner MF, de Bakker PI, Mueller M, et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet. 2010;42(3):240–244. doi: 10.1038/ng.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benjamin EJ, Rice KM, Arking DE, Pfeufer A, van Noord C, Smith AV, Schnabel RB, Bis JC, Boerwinkle E, et al. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet. 2009;41(8):879–881. doi: 10.1038/ng.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parvez B, Darbar D. The “missing” link in atrial fibrillation heritability. J Electrocardiol. 2011;44(6):641–644. doi: 10.1016/j.electrocard.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22(8):983–988. doi: 10.1161/01.str.22.8.983. [DOI] [PubMed] [Google Scholar]
- 13.Darbar D, Roden DM. Genetic mechanisms of atrial fibrillation: impact on response to treatment. Nat Rev Cardiol. 2013;10(6):317–329. doi: 10.1038/nrcardio.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tucker NR, Ellinor PT. Emerging directions in the genetics of atrial fibrillation. Circ Res. 2014;114(9):1469–1482. doi: 10.1161/CIRCRESAHA.114.302225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hatem SN, Coulombe A, Balse E. Specificities of atrial electrophysiology: Clues to a better understanding of cardiac function and the mechanisms of arrhythmias. J Mol Cell Cardiol. 2010;48(1):90–95. doi: 10.1016/j.yjmcc.2009.08.029. [DOI] [PubMed] [Google Scholar]
- 16.Li GR, Nattel S. Properties of human atrial ICa at physiological temperatures and relevance to action potential. Am J Physiol. 1997;272(1 Pt 2):H227–H235. doi: 10.1152/ajpheart.1997.272.1.H227. [DOI] [PubMed] [Google Scholar]
- 17.Escande D, Coulombe A, Faivre JF, Deroubaix E, Coraboeuf E. Two types of transient outward currents in adult human atrial cells. Am J Physiol. 1987;252(1 Pt 2):H142–H148. doi: 10.1152/ajpheart.1987.252.1.H142. [DOI] [PubMed] [Google Scholar]
- 18.Ruben Peter C. Handbook of Experimental Pharmacology. 2014;221. Springer-Verlag Berlin Heidelberg 2014. DOI 10.1007/978-3-642-41588-3.
- 19.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475(7356):353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savio-Galimberti E, Gollob MH, Darbar D. Voltage-gated sodium channels: biophysics, pharmacology, and related channelopathies. Front Pharmacol. 2012 Jul 11;3:124. doi: 10.3389/fphar.201200124. eCollection 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patino GA, Isom LL. Electrophysiology and beyond: multiple roles of Na+ channel beta subunits in development and disease. Neurosci Lett. 2010;486(2):53–59. doi: 10.1016/j.neulet.2010.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haufe V, Chamberland C, Dumaine R. The promiscuous nature of the cardiac sodium channel. J Mol Cell Cardiol. 2007;42(3):469–477. doi: 10.1016/j.yjmcc.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 23.Yang T, Atack T, Stroud DM, Zhang W, Hall L, Roden DM. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res. 2012;111(3):322–332. doi: 10.1161/CIRCRESAHA.112.265173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Facer P, Punjabi PP, Abrari A, Kaba RA, Severs NJ, Chambers J, Kooner JS, Anand P. Localisation of SCN10A gene product Na(v)1.8 and novel pain-related ion channels in human heart. Int Heart J. 2011;52(3):146–152. doi: 10.1536/ihj.52.146. [DOI] [PubMed] [Google Scholar]
- 25.Darbar D, Hardy A, Haines JL, Roden DM. Prolonged signal-averaged P-wave duration as an intermediate phenotype for familial atrial fibrillation. J Am Coll Cardiol. 2008;51(11):1083–1089. doi: 10.1016/j.jacc.2007.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299(5604):251–254. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 27.Olesen MS, Bentzen BH, Nielsen JB, Steffensen AB, David JP, Jabbari J, Jensen HK, Haunso S, Svendsen JH, Schmitt N. Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Med Genet. 2012;13:24. doi: 10.1186/1471-2350-13-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y, Xia M, Jin Q, Bendahhou S, Shi J, Chen Y, Liang B, et al. dentification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet. 2004;75(5):899–905. doi: 10.1086/425342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lundby A, Ravn LS, Svendsen JH, Hauns S, Olesen SP, Schmitt N. KCNE3 mutation V17M identified in a patient with lone atrial fibrillation. Cell Physiol Biochem. 2008;21(1-3):47–54. doi: 10.1159/000113746. [DOI] [PubMed] [Google Scholar]
- 30.Ravn LS, Aizawa Y, Pollevick GD, Hofman-Bang J, Cordeiro JM, Dixen U, et al. Gain of function in IKs secondary to a mutation in KCNE5 associated with atrial fibrillation. Heart Rhythm. 2008;5(3):427–435. doi: 10.1016/j.hrthm.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olesen MS, Refsgaard L, Hoist AG, Larsen AP, Grubb S, Haunso S, Svendsen JH, Olesen SP, Schmitt N, Calloe K. A novel KCND3 gain-of-function mutation associated with early-onset of persistent lone atrial fibrillation. Cardiovasc Res. 2013;98(3):488–495. doi: 10.1093/cvr/cvt028. [DOI] [PubMed] [Google Scholar]
- 32.Deo M, Ruan Y, Padit SV, Shah K, Berenfeld O, Blaufox A, Cerrone M, Noujaim SF, Denegri M, Jalife J, Priori S. KCNJ2 mutation in short QT syndrome 3 results in atrial fibrillation and ventricular proarrhythmia. Proc Natl Acad Sci U S A. 2013;110(11):4291–4296. doi: 10.1073/pnas.1218154110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia M, Jin Q, Bendahhou S, He Y, Larroque MM, Chen Y, et al. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun. 2005;332(4):1012–1019. doi: 10.1016/j.bbrc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 34.Christophensen IE, Olesen MS, Liang B, Andersen MN, Larsen AP, Nielsen JB, Haunso S, Olesen SP, Tveit A, Svendsen JH, Schmitt N. Genetic variation in KCNA5: impact on the atrial-specific potassium current IKur in patients with lone atrial fibrillation. Eur Heart J. 2013;34(20):1517–1525. doi: 10.1093/eurheartj/ehs442. [DOI] [PubMed] [Google Scholar]
- 35.Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15(14):2185–2191. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y, Li J, Lin X, Yang Y, Hong K, Wang L, Liu J, Li L, Yan D, Liang D, Xiao J, Jin H, Wu J, Zhang Y, Chen YH. Novel KCNA5 loss-of-function mutations responsible for atrial fibrillation. J Hum Genet. 2009;54(5):277–283. doi: 10.1038/jhg.2009.26. [DOI] [PubMed] [Google Scholar]
- 37.Yang T, Yang P, Roden DM, Darbar D. Novel KCNA5 mutation implicates tyrosine kinase signaling in human atrial fibrillation. Heart Rhythm. 2010;7(9):1246–1252. doi: 10.1016/j.hrthm.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellinor PT, Nam EG, Shea MA, Milan Dj, Ruskin JN, MacRae CA. Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm. 2008;5(1):99–105. doi: 10.1016/j.hrthm.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 39.Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, George AL, Jr, Roden DM. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008;117(15):1927–1935. doi: 10.1161/CIRCULATIONAHA.107.757955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McNair WP, Ku L, Taylor MR, Fain PR, Dao D, Wolfel E, Mestroni L. Familial Cardiomyopathy Registry Research Group. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;110(15):2163–2167. doi: 10.1161/01.CIR.0000144458.58660.BB. [DOI] [PubMed] [Google Scholar]
- 41.Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293(4):447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morita H, Kusano-Fukushima K, Nagase S, Fujimoto Y, Hisamatsu K, Fujio H, et al. Atrial fibrillation and atrial vulnerability in patients with Brugada Syndrome. J Am Coll Cardiol. 2002;40(8):1437–1444. doi: 10.1016/s0735-1097(02)02167-8. [DOI] [PubMed] [Google Scholar]
- 43.Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111(5):659–670. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- 44.Johnson JN, Tester DJ, Perry J, Salisbury BA, Reed CR, Ackerman MJ. Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm. 2008;5(5):704–709. doi: 10.1016/j.hrthm.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benito B, Brugada R, Perich RM, et al. A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm. 2008;5(10):1434–1440. doi: 10.1016/j.hrthm.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 46.Wang DW, Gillani N, Roden DM, Darbar D, George AL. Sodium channel variants associated with atrial fibrillation exhibit abnormal fast and slow inactivation. Biophys J. 2010;98:S1, P10. [Google Scholar]
- 47.Li Q, Huang H, Liu G, Lam K, Ruttberg J, Green MS, Birnie DH, Lemery R, Chahine M, Gollob MH. Gain-of-function mutation in Nav1.5 in atrial fibrillation enhances cellular excitability and lowers the threshold for action potential firing. Biochem Biophys Res Commun. 2009;380(1):132–7. doi: 10.1016/j.bbrc.2009.01.052. [DOI] [PubMed] [Google Scholar]
- 48.Ziyadeh-Isleem A, Clatot J, Duchatelet S, Gandjbakhch E, Denjoy I, Hidden-Lucet F, Hatem S, Deschenes I, Coulombe A, Neyroud N, Guicheney P. A truncated SCN5A mutation combined with genetic variability causes sick sinus syndrome and early atrial fibrillation. Heart Rhythm. 2014 Febr;Pii:1547-5271(14):00173–8. doi: 10.1016/j.hrthm.2014.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Remme CA, Wilde AAM, Bezzina CR. Cardiac Sodium Channel Overlap Syndromes: Different Faces of SCN5A Mutations. Trends Cardiovasc Med. 2008;18:78–87. doi: 10.1016/j.tcm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Darbar D. IS it time to develop a “pathogenicity” score to distinguish long QT syndrome causing mutations from “background” genetic noise. Heart Rhythm. 2009;6(9):1304–1305. doi: 10.1016/j.hrthm.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dale JW, Von Schantz M, Plant N. From genes to genomes. 3rd. Wiley-Blackwell; 2012. Chapter 11: Modifying organisms. Transgenics. Wiley & Sons, Ltd. Baffins Lane, Chichester, West Sussex, PO19 IUD, England.
- 52.Louch WE, Sheehan KA, Wolska BM. Methods in cardiomyocyte isolation, culture, and gene transfer. J Mol Cel Cardiol. 2011;51:288–298. doi: 10.1016/j.yjmcc.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Josowitz R, Carvajal-Vergara X, Lemischka IR, Gelb BD. Induced pluripotent stem cell-derived cardiomyocytes as models for genetic cardiovascular disorders. Current Opinion in Cardiology. 2011;26:223–229. doi: 10.1097/HCO.0b013e32834598ad. [DOI] [PubMed] [Google Scholar]
- 54.Knollmann BC. Induced pluripotent stem cell-derived cardiomyocytes. Boutique Science or valuable arrhythmia model. Circ Res. 2013;112:969–976. doi: 10.1161/CIRCRESAHA.112.300567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carpentier F, Bourge A, Merot J. Mouse models of SCN5A-related cardiac arrhythmias. Progress in Biophysics and Molec Biol. 2008;98(2-3):230–237. doi: 10.1016/j.pbiomolbio.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 56.Watanabe H, Yang T, Stroud DM, Lowe JS, Harris L, Atack TC, et al. Striking in vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation. 2011;124(9):1001–1011. doi: 10.1161/CIRCULATIONAHA.110.987248. [DOI] [PMC free article] [PubMed] [Google Scholar]