

Abstract
Blood cells derive from hematopoietic stem cells through stepwise fating events. To characterize gene expression programs driving lineage choice we sequenced RNA from eight primary human hematopoietic progenitor populations representing the major myeloid commitment stages and the main lymphoid stage. We identify extensive cell-type specific expression changes: 6,711 genes and 10,724 transcripts, enriched in non-protein coding elements at early stages of differentiation. In addition, we discovered 7,881 novel splice junctions and 2,301 differentially used alternative splicing events, enriched in genes involved in regulatory processes. We demonstrate experimentally cell specific isoform usage, identifying NFIB as a regulator of megakaryocyte maturation – the platelet precursor. Our data highlight the complexity of fating events in closely related progenitor populations, the understanding of which is essential for the advancement of transplantation and regenerative medicine.
Introduction
Hematopoiesis has been extensively studied as a paradigm of stem cell biology and development (1). Hematopoietic stem cells (HSCs) and their progeny have been used to pioneer stem cell therapies for malignant and non-malignant hematological diseases (2) and the successful transplantation of genetically repaired HSCs is at the forefront of regenerative medicine in primary immune deficiency and severe combined immunodeficiency (3, 4). HSCs reside in the bone marrow and can undergo asymmetric cell division (5), thereby generating an identical copy and a multipotent progenitor cell (MPP). MPPs have the ability to generate all hematopoietic cell types, but are incapable of indefinite self-renewal and engraftment (6, 7). This process of expansion, differentiation and maturation culminates in the daily release of up to 1011 newly formed cells into the circulation, mainly red blood cells (RBCs) and platelets (8, 9). The molecular mechanisms driving hematopoiesis have been classically understood as a cascade of gene expression programs propelled by transcription factors (TFs) (10) that direct lineage commitment and maturation by the coordinated regulation of gene transcription. Studies of hematological malignancies and model organisms (1) have identified many of the critical genes and mechanisms regulating hematopoietic development. Owing to species-specific differences, model organisms only contribute partially towards the detailed characterization of transcriptional cascades regulating human hematopoiesis (11-13).
Genome-wide transcriptional profiling of human hematopoietic progenitor populations has identified several transcriptional networks coordinating blood formation (14). However, gene expression datasets using whole-genome expression arrays only produce an incomplete assessment of the full repertoire of transcript isoforms that underpin the fating and expansion of progenitor cells (14-16). Alternative splicing is a widespread post-transcriptional process in eukaryotic organisms where multiple distinct transcripts are produced from a single gene (17). Analysis of RNA sequencing (RNA-seq) has shown that alternative splicing is used in up to 94% of human multi-exonic genes (18, 19), often in a tissue and developmental stage-specific manner (18, 20, 21).
Alternative splicing has an important role in disease with 15% of disease-causing mutations located within splice sites and more than 20% of missense mutations lying within predicted splicing elements (22). Studies have also revealed that somatic mutations of splicing factor genes occur frequently in hematological cancers, including myelodysplasia and chronic lymphocytic leukemia (23-25). Thus, knowledge of cell-type specific alternative splicing and transcript isoform usage is required to interpret the consequences of genetic variation and to inform strategies for therapeutic intervention based on gene repair.
Results
Deep transcriptomes of human hematopoietic progenitors
We used fluorescence-activated cell sorting of umbilical cord blood (CB) mononuclear cells to obtain highly purified populations of HSCs and five progenitor cells (MPP; CLP, common lymphoid progenitor; CMP, common myeloid progenitor; GMP, granulocyte monocyte progenitor; MEP, megakaryocyte erythrocyte precursor). In addition, erythroblasts (EBs) and megakaryocytes (MKs), the nucleated precursors of red blood cells (RBCs) and platelets, were obtained by in vitro differentiation of CB CD34+ cells (Figs. 1A, S1). For simplicity, we address all eight types of cells as progenitors.
Fig. 1.
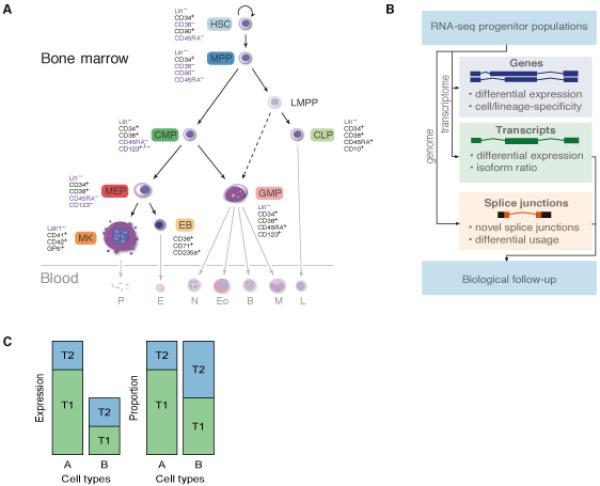
Transcriptional atlas of hematopoietic progenitors and precursors. (A) Schematic representation of the current model of hematopoietic cell ontogeny and samples used in this study. Established ontological relationships are represented as solid lines, emerging ontological relationship are represented as dotted lines. A simplified representation of mature cells is shaded. Antigens used for selecting each population are also indicated. The bone marrow residing components are the hematopoietic stem cell (HSC, light blue), multipotent progenitor (MPP, dark blue), lymphoid-primed multipotent progenitor (LMPP), common lymphoid progenitor (CLP, light green), common myeloid progenitor (CMP, dark green), granulocyte monocyte progenitor (GMP, light red), megakaryocyte erythrocyte progenitor (MEP, red), erythroblast (EB, light orange), megakaryocyte (MK, orange). The blood residing components are platelets (P), erythrocyte (E), neutrophil (N), eosinophil (Eo), monocyte (M) and lymphocyte (L). (B) Data analysis strategy. Reads were mapped to the transcriptome to quantify expression at the gene and transcript levels as well as the transcript proportion (defined as the fraction of gene expression level from a given transcript). Complementary to that, reads were mapped to the genome to identify novel splice junctions and sites where alternative splicing occurs. (C) Schematic highlighting the difference between assessing differential expression by looking at transcript expression or at transcript proportion.
We sequenced 25 poly-(A)+ RNA samples, yielding a total of 2.4 × 109 uniquely aligned reads, ranging from 36 to 150 × 106 reads per sample (Table S1). We employed a Bayesian framework implemented in MMSEQ (26) to quantify gene and transcript expression by aligning reads to the transcriptome (Fig. 1B). Transcript usage ratio, the proportion of a gene’s expression contributed by each of its transcripts, was also estimated (Fig. 1C). The latter provides an alternative to assessing differential transcript usage, which is less sensitive to depth of coverage and data normalization. To validate MMSEQ transcript expression estimates we purified 16 additional samples, representing 5 cell types, and performed quantitative RT-PCR (RT-qPCR) using the Fluidigm BioMark HD system for 36 transcript specific-assays. Linear regression between RNA-seq and RT-qPCR expression estimates indicated high reproducibility in biological replicates (R2 = 0.70) (fig. S2, Table S2).
We confirmed the identity of each cell population by assessing the expression patterns of a set of well-characterized TF genes that are known to be essential for lineage commitment (Figs. 2A, S3) (1, 27). For instance, EBF1 expression peaks in CLPs, as expected from its role in B-cell development (28). Moreover the estimated gene expression of GATA1 and GATA2 reflects their switch in the differentiation of MEPs to EBs and MKs (29).
Fig. 2.
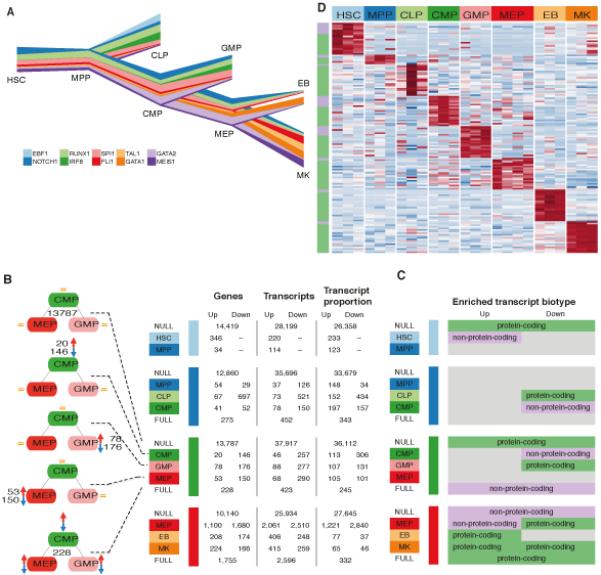
Transcriptional changes at lineage commitment events. (A) River plot representing gene expression levels across cell types for key transcription factors (TFs) required for lineage commitment. Line width represents expression level in log2(FPKM+1) normalized to the highest expression per gene across cell types. The relative changes in gene expression recapitulate the current understanding of the role of these TFs in hematopoietic differentiation. (B) Summary of the number of transcriptional classes - genes, transcripts and transcript proportions - changing at each lineage commitment point. Bayesian polytomous analysis was used to classify these 3 quantities into 5 possible models, from top to bottom: NULL model (no change); three single models (only one cell type different); and a FULL model (all three estimates differ). The number of events up or down regulated were tallied only when the change occurred in at least two samples at each branching event with an expression FPKM >1. (C) Cell-specific enrichment of protein-coding and non-protein coding biotypes in up and down regulated transcripts for the polytomous models at each branching event. (D) Heatmap of expression of lineage specific transcripts. Polytomous analysis was used to identify genes that were expressed significantly higher in a given cell type relative to all others. Top 20 highest scoring transcripts based on the posterior probability of the model are displayed. The colors along the left axis reflect whether the gene is protein coding (green) or otherwise (lilac).
Classification of differential expression patterns during lineage commitment
To assess differential expression during hematopoietic lineage commitment at each branching point, such that all possible patterns of expression changes are considered, we used MMDIFF (30) to perform Bayesian polytomous model selection between the five possible modes of expression change involving three cell types (see Materials and Methods for further details and significance thresholds, Fig. 2B). This methodology identifies, for example, transcripts that are downregulated during the transition from CMP to GMP but retain similar expression between CMP and MEP.
Across all fating events we detected 6,711 genes, 10,724 transcripts and 7,017 transcript usage ratios with significant differences at least at one of the branching points (Fig. 2B). In total, we detected transcriptional changes per cell type in 22-33% of the 20,459 genes expressed across our dataset (defined as expression level ≥ 1 fragment per kilobase of transcript per million fragments mapped [FPKM] in at least two samples). Changes at the transcript level did not imply measurable differences at the gene level. The overlap between sets of differentially expressed transcripts (at the transcript and usage ratio levels) and of the genes they belong to was low, ranging from 0% to 35% (fig. S4). The extent of overlap did not increase when the threshold applied to ascertain expression (i.e. FPKM ≥ 1) was relaxed. Our analysis strategy highlights the advantages of using RNA-seq for assessing the richness of changes in expression at gene and transcript levels compared to probe-based technologies.
Of the 54,386 transcripts, expressed at an FPKM≤1 in at least two samples, 28,563 (52.5%) were protein-coding. The second and third most abundant classes were transcripts with retained introns (8,661, 15.9%) and processed transcripts without open reading frames (8,140, 15.0%). Assessment of the transcript biotypes of differentially expressed transcripts revealed that some modes of expression, at specific branching points, were significantly enriched for non-protein coding isoforms (Fig. 2C, Table S3). For example, during the HSC to MPP transition, transcripts upregulated in HSCs were enriched for non-protein-coding biotypes, such as lincRNAs (FDR = 0.043), whereas transcripts with similar expression in both cell types were enriched for protein-coding biotypes (FDR = 0.014). In contrast, differentially expressed transcripts at the terminal differentiation stage (MEP to EB or MK branching point) were enriched for transcripts with protein-coding biotypes (FDR < 0.016). These results suggest that a proportion of the regulation of lineage commitment, in the early stages of hematopoiesis, involves non-protein-coding elements and that lincRNAs may counteract differentiation programs, as observed in ES cells (31).
Cell-type specific genes and transcripts
Having evaluated expression patterns for genes differentially expressed between cell types at a branching point, we next focused on those genes and transcripts that were more highly expressed in one given cell population while displaying similar levels of relatively low expression in all other seven cell types (cf. Materials and Methods).
These cell-type specific genes or transcripts are likely to be important in conferring cellular identity (Fig. 2D, Tables S4, S5). Using conservative thresholds on the posterior probability and the extent of differential expression (cf. Materials and Methods), we identified between 6 (for MPP) and 631 (for EB) genes that were cell-type specific. We tested whether our cell-type specific gene sets were able to discriminate between cell populations in two microarray atlases of gene expression in human hematopoiesis (14, 16), achieving high concordance (fig S5 A-D). The number of cell-type specific transcripts ranged from between 19 for MPPs (belonging to 18 genes) and 1,807 for EBs (belonging to 1,141 genes). The low number of cell-type specific transcripts in MPPs is consistent with the small number of transcripts we identified as upregulated in this cell type (Fig. 2B). Thus MPP not only displayed a lower number of upregulated genes compared to HSCs, CMPs and CLPs, but also an overall lower number of cell-specific transcripts, suggesting a less distinct transcriptional identity of MPPs compared to other progenitor cells.
Consistent with our findings at branching events, cell-type specific gene and transcript sets show different patterns of enrichment of biotypes. Non protein-coding biotypes were over-represented in HSC-specific transcripts (FDR = 1.48 × 10−3), while protein-coding transcripts were significantly enriched among transcripts specific in cells at terminally differentiated stages (EBs: FDR < 1.00 × 10−60; MKs: FDR = 1.96 × 10−55).
These cell-specific genes may play important roles in determining cell identity and proliferation of the different mature blood cells. We tested the hypothesis that these genes are directly implicated in mature cell differentiation and proliferation by overlapping these sets with genes harboring variants associated with RBC (32) and platelet (33) quantitative traits through genome wide association studies. Genes near loci associated with platelet count and volume were enriched in the MK-specific gene set (FDR=1.7×10−8). In contrast, genes in loci associated with RBC count, volume and hemoglobin concentration were not enriched in the EB-specific gene set (FDR=0.42) or in any other cell specific set. This suggests that the regulation of platelet production is primarily intrinsic to MKs, while RBC production is regulated by mechanisms extrinsic to the erythroid lineage.
Our data add to the repertoire of genes and transcripts associated with cell identity in early and late stages of cell development in hematopoiesis, informing downstream examination of the role of transcriptional isoforms expressed in each cell population and their changes at each lineage commitment event.
Discovery and characterization of unannotated splice junctions
Owing to their low abundance and anatomical compartmentalization in the bone marrow, blood progenitor cells are systematically under-represented in existing transcript sequence databases. We analyzed an average of 137 million aligned reads per sample obtained across the 25 samples (Table S1, Fig. 1B) to explore the magnitude of unannotated splice junctions in human hematopoietic progenitors. We intersected splice junction calls from three splice-aware aligners (fig. S6) and required the splice junction to be observed in at least two samples. A total of 159,495 unique splice junctions were detected, of which 29,736 were not annotated in Ensembl v70. We categorized these unannotated splice junctions into four classes depending on whether their donor and acceptor sites were annotated within Ensembl (Fig. 3A). For 8,382 (28.2%) junctions both donor and acceptor sites were known but not the splicing pattern. 8,112 (27.3%) and 8,321 (28.0%) splice sites included unannotated splice donors or acceptors. Finally, 4,921 (16.5%) splice events had both donor and acceptor sites unannotated. The frequency of the four different categories of splicing events did not differ between the eight cell populations (Fig. 3A).
Fig. 3.
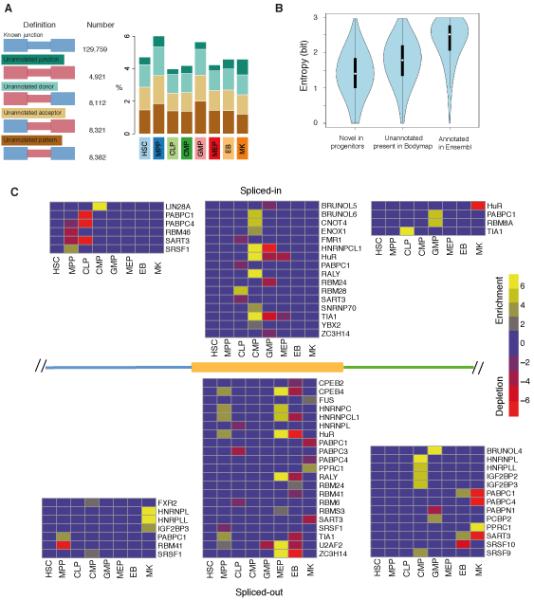
Cell-type specific splicing and RNA-binding motif enrichment in hematopoiesis. (A) Distribution of splice junction definition, absolute count and cell-type-specific fractions within unannotated splice junctions. Blue: annotated exons and junctions; Red: unannotated exons and junctions. (B) Cell-type specificity of known, unannotated and novel splice junctions measured with Shannon’s entropy (37). Lower entropy indicates that splice junctions are observed in a smaller number of cell types. (C) Region-specific patterns of RNA-binding protein motifs around spliced-in and spliced-out DSU cassette exons. The enrichment or depletion of motifs in three regions: the 300bp intronic region adjacent to the upstream of the 5′ splice site (blue); the exonic region of the cassette exon (orange); and the 300bp intronic region adjacent to the downstream of the 3′ splice site (green). The heatmaps present significant enrichment (yellow) or depletion (red) in -log10 P value, FDR< 0.05.
To characterize the 29,736 putative unannotated splice junctions, we investigated their splice site probability scores, degree of conservation and coding potential. Splice site probability scores (34) for unannotated splice sites were similar in known and unannotated donor sites (>0.90, fig. S7). We observed that conservation scores for exonic regions with unannotated splice sites were higher (mean 0.28) than for intronic regions in the same splice sites (mean 0.20; P<2.2×10−16; Wilcoxon rank sum test, fig. S8). Finally, protein-coding potential of the unannotated exons was assessed with the frequency of stop codons, in all three reading frames for both directions, within a 100 base pairs (bp) window around the splice sites. A similar distribution of coding potential was identified in unannotated and annotated exons, with both containing an average of 1.2 fewer stop codons than the equivalent intronic regions (fig. S9).
To identify novel splice junctions, we compared the unannotated splice junctions to splice junctions identified in the UCSC ESTs/mRNA dataset and in poly-(A)+ RNA-seq dataset from 16 human tissues in Illumina BodyMap 2.0 (35). In total, 73.5% of our unannotated events were detected in these external datasets, with 23.0% detected in UCSC ESTs/mRNA data and 72.0% in our re-analysis of the BodyMap 2.0 dataset (fig. S10). The remaining 7,881 (26.5%) splice junctions were specific to our dataset (hereafter called novel). Analysis of novel splice junctions revealed a higher proportion of the non-canonical splicing motif GC-AG (2.2% and 7.3% in unannotated and novel, respectively) compared to annotated sites (0.9%, fig. S11). While both the GT-AG and GC-AG splice sites are processed by the canonical U2-type spliceosome, GC-AG splice sites tend to be alternatively spliced (36). We calculated Shannon’s entropy (37) for the three classes of splice junctions: annotated, unannotated and novel (Fig 3B). A lower entropy distribution in the novel splice junction set indicates that these tend to be population-specific events, when compared to the unannotated splice junctions present in BodyMap 2.0 data, or all annotated junctions (P<2.2×10−16; Wilcoxon rank sum test, fig. S12). Enrichment analysis of genes containing novel splice junctions highlighted GO terms related to cell cycle, DNA metabolism and RNA processing (FDR < 5.0×10−17, Table S8), suggesting that these novel splice junctions may alter the function of genes involved in critical cellular processes.
Our results suggest that a number of the unannotated and novel splice junctions are indeed functional and used in a cell-type specific manner based on their splice probability, conservation score, coding potential and entropy.
Validation of novel splice junctions
We used PCR to independently validate 23 of the novel splice junctions. The PCR assays (Table S9) performed on 5 samples showed > 90% concordance rate (105/115 reactions, figs. S13-S16, Table S10). We performed additional sequencing of poly-(A)+ transcripts from sample MK_3 using the PacBio RS sequencing platform, which enables sequencing of full-length transcripts and overcomes the limitations of transcriptome assembly on the basis of short Illumina reads (38). PacBio sequencing yielded 67,110 reads identified as full-length molecules (originating from individual RNA transcripts) ranging between 322 and 13,170 bp in length (median 2,272 bp). These were further combined into 35,663 consensus sequence clusters – transcript structures (cf. Material and Methods). Two novel splice junctions validated by PCR in MKs were also observed within the PacBio dataset (fig. S15). Using these data we investigated the transcriptional context of the novel splice junctions in MKs. Visual inspection of the PacBio alignments indicated that a number novel splice junctions are part of full-length transcripts, including a previously unobserved intergenic locus on chromosome 12 and an antisense transcript within an annotated protein encoding region in the GNG12 locus (fig. S15).
Of the 94,423 splice junctions with 10 or more Illumina reads in MK_3, 54% were supported by PacBio data. In contrast, 7% (66/956) of novel and 11% (773/7,234) of unannotated splice junctions identified in MK_3 were recapitulated in the PacBio dataset. We used the annotated splice junctions to estimate the probability of detection by PacBio as a function of read depth and transcript length. The observed validation rates of unannotated and novel junctions, after accounting for read depth, would be consistent with the majority of these junctions originating from transcripts less than 300 bp in length (fig. S17 and (39)). Notwithstanding PacBio’s lower depth of sequencing and other unaccounted technical aspects, this analysis provides support to the idea that a large fraction of novel splicing events involve very short transcripts not captured by PacBio.
Differential usage of alternative splice junctions
To investigate the prevalence of cell-type specific alternative splicing, we identified 42,001 splice junction sets where two or more splice junctions shared either the donor or the acceptor sites. Of these, we focused on the 20,924 (49.8%) junctions that contained only two splicing alternatives and were detected in at least two biological replicates. To determine if an alternative splice site displays differential splicing usage (DSU), that is, that the relative contribution of splicing alternatives (usage proportion) differs between a given cell-type and the average proportion across all other cell types, we fitted a beta binomial model and established statistical significance using a likelihood ratio test. The beta binomial model accounts for the overdispersion - beyond the expected binomial variance - present in the data. It is an extension to the binomial model (i.e. logistic regression) and is akin to using a negative binomial distribution to model overdispersed counts data. This analysis identified 2,301 DSU sets (FDR<0.1). The number of DSU events ranged between 4 for HSC and 1,034 for CLP (Table S11, figs. S14, S18, S19). The DSU set was enriched with novel splice junctions compared to all junctions (P= 4.39×10−7, Chi-square test).
To better characterize the biological relevance of cell-type specific DSU, we classified splicing events according to their transcriptional consequences: 73.4% lead to exon skipping events, 8.3% of junctions have an alternative 3′ acceptor, and 6.2% have an alternative 5′ donor. 12.1% of events could not be annotated using the reference transcriptome (fig. S20). Although unannotated, the length distribution of this fraction suggests that the majority is composed of exon skipping events (fig. S20).
In the alternative spliced regions displaying DSU, 26.1% contain a premature stop codon and 38.5% contained at least one predicted protein structure or domain, therefore resulting in gain or loss of protein functions. No one type of domain was significantly overrepresented in the DSU set. Of the alternative acceptor sites displaying DSU, 39% (84/216) resulted from a 3 bp shift in the alternative acceptor sites (fig. S21, left panel) displaying a NAGNAG motif (40) (fig. S21, right panel). This motif maintains the translation frame and may introduce a single amino-acid (aa) insertion or a substitution (fig. S22). The 2,301 DSU events could be assigned to 1,704 genes. GO enrichment analysis of these genes indicates that these genes may be directly involved in the regulation of transcription and splicing (Table S12).
We validated 11 DSU events, four novel and seven known events, using PCR (figs. S14, S15, S23). Densitometry estimates of the percentage-spliced-in (PSI) of these PCRs correlates with the PSI estimated from the RNA-seq data (N=26, R2=0.78, fig. S24). In addition we validated a novel DSU in NFIB (see below and, fig. 4a) and a DSU event in GFI1B (39). The DSU event in GFI1B, in CMPs, EBs and MKs results in an alternatively spliced-out exon 4 that encodes for two Zn finger domains critical for megakaryopoiesis (39). Overall, the DSU analysis confirms alternative splicing as an additional key mechanism through which fundamental processes during hematopoiesis are regulated.
Fig. 4.

A novel isoform of the transcription factor NFIB regulates megakaryopoiesis. (A) A novel TSS and novel exon of NFIB was detected using RNA-seq (blue) and validated using 5′ race PCR (red) and PacBio sequencing (green). Ensembl annotated transcripts in black. (B) Cartoon representation of the short and long isoforms of NFIB (NFIB-S and NFIB-L) highlighting the functional domains. (C) Western blot (WB) for NFIB, NFIC and Tubulin in megakaryocytes (MK), erythroblasts (EB) and monocytes (M) confirms that the short form of NFIB (NFIB-S) is predominantly expressed in MKs (* is either the protein product of one of the shorter transcripts of NFIB observed in the 5′ race, or is unspecific). (D) Coimmunoprecipitation of overexpressed combinations of NFIC-HA together with TAP (Flag plus CBP) tagged NFIC, NFIB-L and NFIB-S. The upper panel was probed with anti-NFIC antibodies, showing both NFIC TAP tagged (upper band) and NFIC-HA tagged (lower band); note the absence of NFIC-HA in lane 4 showing lack of interaction between NFIC and NFIB-S. The lower panel was probed with anti-Flag antibody (part of the TAP tag), showing the immunoprecipitated NFIC (lane 2), NFIB-L (lane 3) and NFIB-S (lane 4) (see also Figs. S30 and S31), (E). Flow cytometry dot plots of CD41a and CD61 staining of megakaryocyte cultures at day 10 after infection with shRNA of control, NFIB and NFIC. The proportions of double positive, upper right (megakaryocytic), versus double negative, lower left (undifferentiated), cells decreased relative to control shRNA by silencing either NFIB or NFIC. (F) Overexpression of NFIC or NFIB-S lead to a higher proportion of megakaryocytic cells relative to NFIB-L or control. CD41a and CD42b double positive MKs in cultures at day 10 after infection. The y-axis is the probit proportion of double positive MKs after adjusting for batch effects.
RNA-binding motif enrichment in DSU
Alternative splicing is regulated by trans-acting splicing factors that recognize cis-acting sequences in exons or introns, to promote or suppress the assembly of the spliceosome at the adjacent splice site. We therefore investigated the molecular regulation of cell-specific alternative splicing by examining the sequences around alternatively spliced exons. We used 102 recently described RNA-binding motifs of 80 human RNA-binding proteins (41) to identify sets of motifs significantly enriched or depleted in the regions surrounding DSU junctions (Table S13). Of the 80 RNA-binding proteins with known binding motifs, 59 were expressed in our data with FPKM>1 and displayed variable cell-type specificity (fig. S25). RNA-binding motif enrichment analysis was performed on cassette exons and proximal intronic regions. The patterns of enrichment and depletion, in addition to the identity of the motifs, varied widely across cell types (Fig. 3C).
The proteins BRUNOL, SRSF, TIA1 and the HNRNP (heterogeneous ribonucleoprotein) family of proteins are known to regulate tissue-specific splicing (42). The patterns of enrichment and depletion in our dataset for these proteins suggest their role in regulating tissue-specific splicing also extends to hematopoietic cells (Fig. 3C). For example, we identified that the motifs of the HNRNP protein family, which typically bind to exonic splicing silencers (43), were enriched in exonic regions of MPPs and MEPs that are spliced out.
Novel isoform of NFIB regulates megakaryopoiesis
To investigate the impact of different transcript isoforms in a biological system we focused our attention on the role of two TFs in megakaryopoiesis: NFIB, described below, and GFI1B (39), as an example of how our analysis informs the interpretation of patient sequencing data. NFIB was identified at the MEP/EB/MK branching point (fig. S26), containing a novel MK-specific DSU event (FDR < 0.05). The role of NFIB has been extensively studied in lung maturation, the nervous system (44) and epithelial stem cell development (45). The NFI family of TFs, constituted by four members (A, B, C and X), has previously been implicated in regulating hematopoiesis: with Nfix identified as functional in murine HSCs and progenitors (46), and NFIA implicated in human erythropoiesis (47). NFIC has been observed as being differentially expressed between MKs of fetal and postnatal origin (48). In addition, NFIB has been identified as one of the TFs down-regulated in the HSC to MPP transition (49). However, its role in the later stages of hematopoiesis has remained unexplored.
By examining genomic alignments we identified a novel NFIB transcript (chr9:14,179,779-14,214,332bp) and annotated the position of the transcription start site (TSS) in the novel first exon. The isoform that results from this novel transcript was primarily expressed in HSCs and MKs, and was only present in white blood cells in the BodyMap 2.0 dataset, while the canonical isoform is widely expressed across other BodyMap 2.0 tissues. The novel TSS lies in a region of open chromatin in primary MKs (50) that is occupied by the TFs MEIS1 (this study), FLI1, GATA1, SCL/TAL1, but not GATA2 or RUNX1 (51). The TSS is also marked by the promoter mark H3K4me3 in MKs (fig. S27). We validated the novel TSS by 5′ race RT-PCR and observed multiple PacBio reads supporting it (Fig. 4A). Western blotting (WB) confirmed the presence of the protein encoded by the novel short isoform, NFIB-S, as the major isoform in MKs (Figs. 4B, S28) while the longer isoform encoded by the canonically spliced transcript could not be detected.
NFIB is known to bind DNA preferentially as a homodimer or a heterodimer in combination with other NFI family members(52). Since NFIB-S lacks the DNA binding and protein interaction domains (53), we investigated its ability to interact with NFIC in MKs, given its previously hypothesized role in definitive postnatal megakaryopoiesis (48). Co-transfection experiments followed by immunoprecipitation showed that the novel isoform, NFIB-S, lacked the ability to interact with NFIC (Fig. 4C). To determine the role of both NFIB and NFIC during megakaryopoiesis, we induced peripheral blood CD34+ cells to differentiate towards MKs and infected them with pools of shRNA lentiviruses targeting NFIB, NFIC, or a non-silencing control. Knockdown of either gene resulted in a marked reduction in differentiation towards MKs as assessed by flow cytometry (Fig. 4D) and confirmed by morphological analysis (fig. S29). This indicated that both NFIB-S and NFIC have an essential role in megakaryopoiesis despite the absence of a DNA binding domain in NFIB-S. Overexpression of both NFIB-S and NFIC in CD34+ cells increased cell maturation (Fig. 4E, P = 0.001 and P = 0.014, respectively), measured as double positivity for the MK maturation markers CD41a (ITGA2B) and CD42b (GP1BA) (54). In contrast, overexpression of the canonical isoform, NFIB-L, had no effect (Fig. 4E). These experiments indicate that both NFIC and the novel isoform of NFIB-S, identified in our analysis, play a critical interlinked role in the formation of MKs.
Discussion
Current knowledge of gene expression and function in hematopoiesis is mainly based on observations at gene level. However, it is clearly the transcript, rather than the gene, to which biological function should be ascribed, either as protein-coding or as non-protein-coding RNA.
Here, RNA-seq of HSCs and seven progenitor populations has enabled the identification, quantification and differential expression analysis of cell-type specific transcript isoforms, novel and unannotated splice junctions and alternative splicing events at a genome-wide level. Analysis of lineage commitment events revealed a wealth of previously undetectable transcript switching and of shifts altering isoform usage ratio, without appreciable changes at gene level, providing evidence of additional layers of regulation in cell fating.
Generating an atlas of splicing events allowed us to explore the diversity and mechanisms behind alternative splicing in human hematopoiesis as well as to contribute further to the human genome functional annotation by reporting 7,881 novel splice junctions, specific to these rare cell populations.
To demonstrate the importance of specific isoforms in driving lineage fating events we investigated the role of a transcription factor highlighted by the polytomous analysis. We envisage that integration of this Blueprint RNA-seq dataset with the deep catalogues generated by Blueprint and other epigenome consortia, will aid the annotation of the functional genomic landscape of the hematopoietic system. This is essential in the continued effort to interpret the functional consequences of mutations in patients with rare hematological disorders and support the next enhancements of personalized treatments for patients with hematological malignancies.
Materials and Methods
Progenitor cell purification
Cord blood (CB) was collected after informed consent (ethical approval REC 12/EE/0040) and the mononuclear cells extracted. CD34+ cells were isolated using the EasySep® progenitor cell enrichment kit with platelet depletion (STEMCELL Technologies, Vancouver, Canada), stained using a panel of antibodies and flow sorted to purify HSC, MPP, CLP, CMP, MEP and GMP cells that were lysed in TRIZOL reagent (Life Technologies, Carlsbad, CA, USA).
Cell culture and purification
EBs and MKs were cultured from CD34+ cells isolated from CB mononuclear cells with the human CD34 isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). For EBs, CD34+ cells were cultured with erythropoietin, SCF and IL3 for 14 days. For MKs CD34+ cells were cultured for 10 days in thrombopoietin and IL1β. Both populations were immuno-selected to > 95% purity before lysis.
RNA-seq library preparation and sequencing
RNA was extracted from TRIZOL preparations. 100 pg of RNA was used to generate poly-(A)+ RNA libraries with the SMARTer Ultra Low RNA and Advantage 2 PCR kits (Clontech, Mountain View, CA, USA). Samples were indexed with NEXTflex adapters (Bioo-scientific, Austin, TX) and 100 base-pair paired-end sequencing was performed on Illumina HiSeq 2000 instruments with TruSeq reagents (Illumina, San Diego, CA, USA).
Quality control, trimming, alignment and expression analysis
RNA-seq libraries were initially subjected to a quality control step, where outliers were identified and discarded from further analysis on the basis of the duplication rates and gene coverage. Paired-end reads of the 25 independent samples were trimmed for both PCR and sequencing adapters with Trim Galore (55). Trimmed reads were aligned to the Ensembl v70 human transcriptome using Bowtie (56). Quantification of gene and transcript expression was performed with MMSEQ (26)
Differential expression analysis through polytomous model classification
Presence of significant differential expression was determined with MMDIFF (30) at three different levels: gene, transcript and isoform usage (generically called features).
Polytomous classification was carried out by first performing two-model comparisons to calculate Bayes factors, B(0,m), between a common baseline model and models representing the expression patterns of interest for a given feature. Under the baseline model, 0, the feature’s mean expression level is the same in all cell types and under the alternative model, m, the mean expression level is allowed to differ according to the desired pattern (e.g. CMP=GMP≠MEP). Bayes’ theorem was used to compute the posterior probability that the true model γ is equal to m under the assumption that the alternative models are exhaustive: P(γ = m|x) = B(0,m)×P(γ = m) / ΣB(0,m′)×P(γ =m′), where x denotes the MMSEQ estimates for that feature.
For the transition from HSCs to MPPs, we used a two-model comparison, where we used a prior probability that the baseline model was true of 0.9. This can be interpreted as a prior belief that 10% of features are differentially expressed. Features with a posterior probability for the alternative model above 0.5 (equivalent to a Bayes factor threshold of 9, representing strong evidence for the alternative model) and an FPKM > 1 in at least two of the samples involved, were considered differentially expressed.
At each cell-fating point involving three cell types, we studied all patterns of expression amongst the progenitor cell and its immediate progeny. We classified feature expression patterns according to five models. The simplest model assumes that the mean expression level is the same across cell types. The most complex model assumes that the mean expression level is different for each cell type. The remaining three models assume that two of the three cell types have the same mean expression level. We specified a prior probability of 80% for the simplest model and distributed the remaining probability evenly across the four alternative models. The model with the highest posterior probability was selected.
Gene set enrichment analysis
Gene and transcript sets derived from the polytomous and the cell-type specific expression analyses of the RNA-seq data were tested for gene set enrichment with the goseq R/Bioconductor package version 1.14.0 (57), which accounts for the relationship between power of detection and transcript length. All P values were corrected for multiple testing using the Benjamini-Hochberg method (58).
Selection of Cell-type specific genes and transcripts
We selected cell-type specific genes and transcripts by performing a 9-model polytomous comparison. The simplest model assumes that the mean expression level is the same across cell types. Each of the remaining 8 models assumes that the expression is the same across all cell types except for one of the progenitors. We specified a prior probability of 0.5 for the simplest model and distributed the remaining probability evenly across the eight alternative models. Genes and transcripts were required to have a posterior probability greater than 0.5 and a fold change in expression greater than 1 in order to be declared cell-type specific. To compare cell-type specific gene expression estimates between our RNA-seq data and publicly available microarray datasets, we retrieved probe annotations for the Illumina (16) and Affymetrix (14) platforms from Ensembl v70.
Splice junction analysis
Identification of splice junctions for each sample was based on the alignment of the trimmed reads to the human genome (GRCh37) with three different aligners: GSNAP (59), STAR (60) and GEM (61). Splice junctions were considered for further analysis if supported by all three aligners and by at least 10 reads in at least two samples, where reads covered a minimum of 10-bp at both ends of the splice junction. We defined a splice junction as unannotated if not present in Ensembl v70. These were further compared to the EST/mRNA data from UCSC and the Illumina BodyMap 2.0 dataset (35) to identify novel splice junctions.
Splice site probability scores were extracted from the GSNAP output. PhastCons conservation scores were used to plot evolutionary conservation in the 100-bp surrounding each splice junction. Coding potential was estimated by summing the number of possible stop codons in exonic and intronic regions, in all reading frames and in 100-bp flanking the unannotated splice site. Shannon’s entropy (37) was calculated on the basis of the read coverage of the splice junctions.
DSU was identified with a beta binomial model. The characterization of the protein domains in cassette exons with DSU was performed with InterProscan 5 (62) to search for domains predicted by Pfam.
Validation of transcript isoform expression and splicing events
To validate the quantification of transcript levels determined by analysis of the RNA-seq data with MMSEQ, we performed RT-PCR assays with 40 transcript-specific assays and five positive control assays in multiple cell subsets. Quantitation of each transcript was performed in multiple progenitor cell subsets with the BioMark HD system (Fluidigm, San Francisco, CA). After requiring call quality scores > 0.9 (Fluidigm Real-Time PCR Analysis software, http://www.fluidigm.com/software.html), 36 transcripts were analyzed. For each probe, 55Cq values were calculated with the B2M transcript as control and the average 5Cq for MKs. Linear regression was then performed between 55Cq values and the corresponding MMSEQ estimates relative to mean MK.
To validate progenitor-specific novel splice junctions and exon-skipping events we designed PCR primers to amplify 30 junctions identified by RNA-seq. PCR was performed on pools of the RNA-seq libraries. PCR products were run on agarose gel, imaged and densitometry performed.
PacBio libraries were generated (Pacific Biosciences, Menlo Park, CA) from cDNA obtained by reverse transcription of 10 ng of MK_3 total RNA and sequenced in five SMRTcells on the PacBio RSII. SMRTpipe and ICE were used to filter reads to generate consensus sequence clusters that were mapped to the reference human genome (GRCh37) using GMAP.
Enrichment analysis of RNA-binding motifs around cassette exons
Motif enrichment analysis of 102 RNA binding motifs (41) was performed on DSU cassette exons (FDR < 0.05 and usage proportion change > 0.05) over three genomic regions: upstream intronic (300-bp), exonic and downstream intronic (300-bp). Enrichment and depletion of RNA binding motifs was determined with cumulative hypergeometric testing and P values were corrected for multiple testing.
Cloning, shRNA, lentivirus production and transduction
TRC shRNA lentivirus targeting NFIB and NFIC and a non-silencing control were purchased from Thermo Scientific Open Biosystems (Little Chalfont, UK). NFIB full length and NFIC cDNA were cloned into pWPI TAP tagged vector. Packaging was performed in 293T cells and viral stocks were titrated and quantified using qPCR and for pWPI using qPCR and GFP FACS. CD34+ cells were purified from NHS Blood and Transplant apheresis filters, as above (Miltenyi), and then infected with lentiviral particles in the presence of polybrene, in media supplemented with thrombopoietin and IL1b. On day 2, media was replaced and cells cultured to MKs. At day 10, MKs were counted and assessed by morphology and flow cytometry for maturation.
Transfections, immunoprecipitations and Western blots
To detect protein-protein interactions, NFI proteins were expressed by co-transfection in 293T cells and immunoprecipitated with an anti-flag antibody. Western blots were probed with NFIB, NFIC, β-Tubulin.
Data presentation
River plots were generated with the ggplot2 R package (version 0.9.3.1), which was obtained from Bioconductor (http://www.bioconductor.org/). Heatmaps were generated with both the gplots (version 2.13.0) and pheatmap (version 0.7.7) R packages, which were both obtained from CRAN (http://cran.r-project.org/). For sequence logos of the splice site motifs we used seqLogo R package (version 1.30.0) available from Bioconductor. The IGV genome browser (version 2.3.34) was used for visualization (http://www.broadinstitute.org/igv/).
Supplementary Material
Structured Abstract.
Introduction
Blood production in humans culminates in the daily release of around 1011 cells into the circulation, mainly platelets and red blood cells. All blood cells originate from a minute population of hematopoietic stem cells (HSCs) that expands and differentiates into progenitor cells with increasingly restricted lineage choice. Here, we explore the transcriptional diversity of these populations, generating a comprehensive repertoire of transcript isoform usage during lineage commitment of blood progenitors. Characterizing the transcriptional programs and alternative splicing events involved in hematopoiesis is critical for interpreting the effects of mutations leading to inherited disorders and blood cancers, and for the rational design of strategies to advance transplantation and regenerative medicine.
Methods
We sequenced RNA from six progenitor and two precursor populations representing the classical myeloid commitment stages of hematopoiesis and the main lymphoid stage. We aligned to the human reference transcriptome and genome to quantify known transcript isoforms and identify novel splicing events, respectively. We used Bayesian polytomous model selection to classify transcripts into distinct expression patterns across the three cell types that comprise each differentiation step. We employed enrichment analysis of RNA-binding motifs to elucidate the regulation of cell-type specific splicing events.
Results
We identified extensive transcriptional changes involving 6,711 genes and 10,724 transcripts and validated a number of these. Many of the changes at the transcript isoform level did not result in significant changes at the gene expression level. Moreover, we identified transcripts unique to each of the progenitor populations, observing enrichment in non-protein coding elements at the early stages of differentiation. We discovered 7,881 novel splice junctions and 2,301 differentially used alternative splicing events, enriched in genes involved in regulatory processes and often resulting in the gain or loss of functional domains. Of the alternative splice sites displaying differential usage, 73% resulted in exon skipping events involving at least one protein domain (38.5%) or introducing a premature stop codon (26%).
To demonstrate the importance of specific isoforms in driving lineage fating events we investigated the role of a transcription factor highlighted by our analyses. Our data show that NFIB is highly expressed in megakaryocytes and that it is transcribed from an unannotated transcription start site preceding a novel exon. The novel NFIB isoform lacks the DNA binding/dimerization domain and therefore is unable to interact with its binding partner, NFIC. We further show that NFIB and NFIC are important in megakaryocyte differentiation.
Discussion
We produced a quantitative catalogue of transcriptional changes and splicing events representing the early progenitors of human blood. Our analyses unveil a previously undetected layer of regulation affecting cell fating, which involves transcriptional isoforms switching without noticeable changes at the gene level and resulting in the gain or loss of protein functions.
Figure.
RNA-seq reads from human blood progenitors (opaque cells in A) were mapped to the transcriptome to quantify gene and transcript expression. Reads were also mapped to the genome to identify novel splice junctions and characterize alternative splicing events (B).
Acknowledgements
The authors wish to acknowledge the following funding bodies: The work described in this manuscript was primarily supported by the European Commission Seventh Framework Program through the BLUEPRINT grant with code HEALTH-F5-2011-282510 (DH, FB, GC, JHAM, KD, LC, MF, SC, SF and SPG). Research in the Ouwehand laboratory is further supported by program grants from the National Institute for Health Research (NIHR, http://www.nihr.ac.uk; to AA, MK, PP, SBGJ, SN, and WHO); and the British Heart Foundation under numbers RP-PG-0310-1002 and RG/09/12/28096 (http://www.bhf.org.uk; to AR and WJA). KF and MK were supported by Marie Curie funding from the NETSIM FP7 program funded by the European Commission. The laboratory receives funding from the NHS Blood and Transplant for facilities. The Cambridge BioResource (http://www.cambridgebioresource.org.uk), the Cell Phenotyping Hub, and the Cambridge Translational GenOmics laboratory (http://www.catgo.org.uk) are supported by an NIHR grant to the Cambridge NIHR Biomedical Research Centre (BRC). The BRIDGE-Bleeding and Platelet Disorders Consortium is supported by the NIHR BioResource - Rare Diseases (http://bioresource.nihr.ac.uk/; to ET, NF, and Whole Exome Sequencing effort). Research in the Soranzo laboratory (LV, NS and SW) is further supported by the Wellcome Trust (Grant Codes WT098051 and WT091310) and the EU FP7 EPIGENESYS initiative (Grant Code 257082). Research in the Cvejic laboratory (AC and CL) is funded by the Cancer Research UK under grant number C45041/A14953. SJS is funded by NIHR. MEF is supported by a British Heart Foundation Clinical Research Training Fellowship, number FS/12/27/29405. EBM is supported by a Wellcome Trust grant, number 084183/Z/07/Z. Research in the Laffan laboratory is supported by Imperial College BRC. FAC, CL and SW are supported by MRC Clinical Training Fellowships and TB by a British Society of Haematology/NHS Blood and Transplant grant. RJR is a Principal Research Fellow of the Wellcome Trust, grant No. 082961/Z/07/Z. Research in the Flicek laboratory is also supported by the Wellcome Trust (grant number 095908) and EMBL. Research in the Bertone laboratory is supported by EMBL. KF and CvG are supported by FWO-Vlaanderen through grant G.0B17.13N.
PF is a compensated member of the Omicia, Inc Scientific Advisory Board.
This study made use of data generated by the UK10K Consortium, derived from samples from the Cohorts arm of the project. A full list of the investigators who contributed to the generation of the data is available from www.UK10K.org
The authors wish to acknowledge the following facilities and people: Cambridge BRC Cell Phenotyping Hub, Cambridge BRC Translational Genomics Laboratory, Cambridge BRC Blood and Stem Cell Biobank, NHS Blood and Transplant donation clinic, Sanger Institute-EBI Single-Cell Genomics Centre, J. Garwood, D. Fletcher, L. Guglielmi and team of the Ouwehand lab, M. Bussemakers from the University of Nijmegen. The critical comments from Prof. J.-P. Allain and Dr. D. Rico were greatly appreciated.
Footnotes
For further details please refer to the supplementary material.
References and Notes
- 1.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoggatt J, Speth JM, Pelus LM. Sowing the seeds of a fruitful harvest: Hematopoietic stem cell mobilization. Stem cells (Dayton, Ohio) 2013 doi: 10.1002/stem.1574. doi: 10.1002/stem.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slukvin Hematopoietic specification from human pluripotent stem cells: current advances and challenges toward de novo generation of hematopoietic stem cells. Blood. 2013;122:4035–4046. doi: 10.1182/blood-2013-07-474825. doi: 10.1182/blood-2013-07-474825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lambert MP, Sullivan SK, Fuentes R, French DL, Poncz M. Challenges and promises for the development of donor-independent platelet transfusions. Blood. 2013;121:3319–3324. doi: 10.1182/blood-2012-09-455428. doi: 10.1182/blood-2012-09-455428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giebel B, Bruns I. Self-renewal versus differentiation in hematopoietic stem and progenitor cells: a focus on asymmetric cell divisions. Current stem cell research & therapy. 2008;3:9–16. doi: 10.2174/157488808783489444. doi. [DOI] [PubMed] [Google Scholar]
- 6.Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell stem cell. 2007;1:635–645. doi: 10.1016/j.stem.2007.10.001. doi: 10.1016/j.stem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baum CM, Weissman IL, Tsukamoto AS, Buckle AM, Peault B. Isolation of a candidate human hematopoietic stem-cell population. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:2804–2808. doi: 10.1073/pnas.89.7.2804. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orkin SH. Diversification of haematopoietic stem cells to specific lineages. Nature reviews. Genetics. 2000;1:57–64. doi: 10.1038/35049577. doi: 10.1038/35049577. [DOI] [PubMed] [Google Scholar]
- 9.Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell stem cell. 2012;10:120–136. doi: 10.1016/j.stem.2012.01.006. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Iwasaki H, et al. The order of expression of transcription factors directs hierarchical specification of hematopoietic lineages. Genes & development. 2006;20:3010–3021. doi: 10.1101/gad.1493506. doi: 10.1101/gad.1493506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. Journal of immunology (Baltimore, Md.: 1950) 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. doi. [DOI] [PubMed] [Google Scholar]
- 12.Tenen DG, Hromas R, Licht JD, Zhang DE. Transcription factors, normal myeloid development, and leukemia. Blood. 1997;90:489–519. doi. [PubMed] [Google Scholar]
- 13.Dickinson RE, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118:2656–2658. doi: 10.1182/blood-2011-06-360313. doi: 10.1182/blood-2011-06-360313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Novershtern N, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watkins NA, et al. A HaemAtlas: characterizing gene expression in differentiated human blood cells. Blood. 2009;113:e1–9. doi: 10.1182/blood-2008-06-162958. doi: 10.1182/blood-2008-06-162958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laurenti E, et al. The transcriptional architecture of early human hematopoiesis identifies multilevel control of lymphoid commitment. Nature immunology. 2013;14:756–763. doi: 10.1038/ni.2615. doi: 10.1038/ni.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang ET, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature genetics. 2008;40:1413–1415. doi: 10.1038/ng.259. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 20.Barbosa-Morais NL, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science (New York, N.Y.) 2012;338:1587–1593. doi: 10.1126/science.1230612. doi: 10.1126/science.1230612. [DOI] [PubMed] [Google Scholar]
- 21.Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science (New York, N.Y.) 2012;338:1593–1599. doi: 10.1126/science.1228186. doi: 10.1126/science.1228186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh RK, Cooper TA. Pre-mRNA splicing in disease and therapeutics. Trends in molecular medicine. 2012;18:472–482. doi: 10.1016/j.molmed.2012.06.006. doi: 10.1016/j.molmed.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 24.Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood. 2013;122:4021–4034. doi: 10.1182/blood-2013-09-381665. doi: 10.1182/blood-2013-09-381665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wan Y, Wu CJ. SF3B1 mutations in chronic lymphocytic leukemia. Blood. 2013;121:4627–4634. doi: 10.1182/blood-2013-02-427641. doi: 10.1182/blood-2013-02-427641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turro E, et al. Haplotype and isoform specific expression estimation using multi-mapping RNA-seq reads. Genome biology. 2011;12:R13. doi: 10.1186/gb-2011-12-2-r13. doi: 10.1186/gb-2011-12-2-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilkinson AC, Gottgens B. Transcriptional regulation of haematopoietic stem cells. Advances in experimental medicine and biology. 2013;786:187–212. doi: 10.1007/978-94-007-6621-1_11. doi: 10.1007/978-94-007-6621-1_11. [DOI] [PubMed] [Google Scholar]
- 28.Bryder D, Sigvardsson M. Shaping up a lineage--lessons from B lymphopoesis. Current opinion in immunology. 2010;22:148–153. doi: 10.1016/j.coi.2010.02.001. doi: 10.1016/j.coi.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Kaneko H, Shimizu R, Yamamoto M. GATA factor switching during erythroid differentiation. Current opinion in hematology. 2010;17:163–168. doi: 10.1097/MOH.0b013e32833800b8. doi: 10.1097/MOH.0b013e32833800b8. [DOI] [PubMed] [Google Scholar]
- 30.Turro E, Astle WJ, Tavare S. Flexible analysis of RNA-seq data using mixed effects models. Bioinformatics (Oxford, England) 2014;30:180–188. doi: 10.1093/bioinformatics/btt624. doi: 10.1093/bioinformatics/btt624. [DOI] [PubMed] [Google Scholar]
- 31.Guttman M, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295–300. doi: 10.1038/nature10398. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Harst P, et al. Seventy-five genetic loci influencing the human red blood cell. Nature. 2012;492:369–375. doi: 10.1038/nature11677. doi: 10.1038/nature11677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gieger C, et al. New gene functions in megakaryopoiesis and platelet formation. Nature. 2011;480:201–208. doi: 10.1038/nature10659. doi: 10.1038/nature10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. Journal of computational biology: a journal of computational molecular cell biology. 2004;11:377–394. doi: 10.1089/1066527041410418. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 35.Farrell CM, et al. Current status and new features of the Consensus Coding Sequence database. Nucleic acids research. 2014;42:D865–872. doi: 10.1093/nar/gkt1059. doi: 10.1093/nar/gkt1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thanaraj TA, Clark F. Human GC-AG alternative intron isoforms with weak donor sites show enhanced consensus at acceptor exon positions. Nucleic acids research. 2001;29:2581–2593. doi: 10.1093/nar/29.12.2581. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schug J, et al. Promoter features related to tissue specificity as measured by Shannon entropy. Genome biology. 2005;6:R33. doi: 10.1186/gb-2005-6-4-r33. doi: 10.1186/gb-2005-6-4-r33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steijger T, et al. Assessment of transcript reconstruction methods for RNA-seq. Nature methods. 2013;10:1177–1184. doi: 10.1038/nmeth.2714. doi: 10.1038/nmeth.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.See supplemental material.
- 40.Hiller M, et al. Widespread occurrence of alternative splicing at NAGNAG acceptors contributes to proteome plasticity. Nature genetics. 2004;36:1255–1257. doi: 10.1038/ng1469. doi: 10.1038/ng1469. [DOI] [PubMed] [Google Scholar]
- 41.Ray D, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barash Y, et al. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- 43.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annual review of biochemistry. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 44.Steele-Perkins G, et al. The transcription factor gene Nfib is essential for both lung maturation and brain development. Molecular and cellular biology. 2005;25:685–698. doi: 10.1128/MCB.25.2.685-698.2005. doi: 10.1128/mcb.25.2.685-698.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang CY, et al. NFIB is a governor of epithelial-melanocyte stem cell behaviour in a shared niche. Nature. 2013;495:98–102. doi: 10.1038/nature11847. doi: 10.1038/nature11847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holmfeldt P, et al. Nfix is a novel regulator of murine hematopoietic stem and progenitor cell survival. Blood. 2013;122:2987–2996. doi: 10.1182/blood-2013-04-493973. doi: 10.1182/blood-2013-04-493973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Starnes LM, et al. A transcriptome-wide approach reveals the key contribution of NFI-A in promoting erythroid differentiation of human CD34(+) progenitors and CML cells. Leukemia. 2010;24:1220–1223. doi: 10.1038/leu.2010.78. doi: 10.1038/leu.2010.78. [DOI] [PubMed] [Google Scholar]
- 48.Bluteau O, et al. Developmental changes in human megakaryopoiesis. Journal of thrombosis and haemostasis: JTH. 2013;11:1730–1741. doi: 10.1111/jth.12326. doi: 10.1111/jth.12326. [DOI] [PubMed] [Google Scholar]
- 49.Notta F, et al. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science (New York, N.Y.) 2011;333:218–221. doi: 10.1126/science.1201219. doi: 10.1126/science.1201219. [DOI] [PubMed] [Google Scholar]
- 50.Paul DS, et al. Maps of open chromatin highlight cell type-restricted patterns of regulatory sequence variation at hematological trait loci. Genome research. 2013;23:1130–1141. doi: 10.1101/gr.155127.113. doi: 10.1101/gr.155127.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tijssen MR, et al. Genome-wide analysis of simultaneous GATA1/2, RUNX1, FLI1, and SCL binding in megakaryocytes identifies hematopoietic regulators. Developmental cell. 2011;20:597–609. doi: 10.1016/j.devcel.2011.04.008. doi: 10.1016/j.devcel.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Apt D, Chong T, Liu Y, Bernard HU. Nuclear factor I and epithelial cell-specific transcription of human papillomavirus type 16. Journal of virology. 1993;67:4455–4463. doi: 10.1128/jvi.67.8.4455-4463.1993. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dekker J, van Oosterhout JA, van der Vliet PC. Two regions within the DNA binding domain of nuclear factor I interact with DNA and stimulate adenovirus DNA replication independently. Molecular and cellular biology. 1996;16:4073–4080. doi: 10.1128/mcb.16.8.4073. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poirault-Chassac S, et al. Notch/Delta4 signaling inhibits human megakaryocytic terminal differentiation. Blood. 2010;116:5670–5678. doi: 10.1182/blood-2010-05-285957. doi: 10.1182/blood-2010-05-285957. [DOI] [PubMed] [Google Scholar]
- 55. http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- 56.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome biology. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Statistics in medicine. 1990;9:811–818. doi: 10.1002/sim.4780090710. doi. [DOI] [PubMed] [Google Scholar]
- 59.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics (Oxford, England) 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marco-Sola S, Sammeth M, Guigo R, Ribeca P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nature methods. 2012;9:1185–1188. doi: 10.1038/nmeth.2221. doi: 10.1038/nmeth.2221. [DOI] [PubMed] [Google Scholar]
- 62.Jones P, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics (Oxford, England) 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karolchik D, et al. The UCSC Genome Browser database: 2014 update. Nucleic acids research. 2014;42:D764–770. doi: 10.1093/nar/gkt1168. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics (Oxford, England) 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 65.Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome research. 2005;15:1034–1050. doi: 10.1101/gr.3715005. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Picelli S, et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 67.Castle JC, et al. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nature genetics. 2008;40:1416–1425. doi: 10.1038/ng.264. doi: 10.1038/ng.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nurnberg ST, et al. A GWAS sequence variant for platelet volume marks an alternative DNM3 promoter in megakaryocytes near a MEIS1 binding site. Blood. 2012;120:4859–4868. doi: 10.1182/blood-2012-01-401893. doi: 10.1182/blood-2012-01-401893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Diaz A, Nellore A, Song JS. CHANCE: comprehensive software for quality control and validation of ChIP-seq data. Genome biology. 2012;13:R98. doi: 10.1186/gb-2012-13-10-r98. doi: 10.1186/gb-2012-13-10-r98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saleque S, Cameron S, Orkin SH. The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes & development. 2002;16:301–306. doi: 10.1101/gad.959102. doi: 10.1101/gad.959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vassen L, et al. Growth factor independent 1b (Gfi1b) and a new splice variant of Gfi1b are highly expressed in patients with acute and chronic leukemia. International journal of hematology. 2009;89:422–430. doi: 10.1007/s12185-009-0286-5. doi: 10.1007/s12185-009-0286-5. [DOI] [PubMed] [Google Scholar]
- 72.Osawa M, et al. Erythroid expansion mediated by the Gfi-1B zinc finger protein: role in normal hematopoiesis. Blood. 2002;100:2769–2777. doi: 10.1182/blood-2002-01-0182. doi: 10.1182/blood-2002-01-0182. [DOI] [PubMed] [Google Scholar]
- 73.Laurent B, et al. A short Gfi-1B isoform controls erythroid differentiation by recruiting the LSD1-CoREST complex through the dimethylation of its SNAG domain. Journal of cell science. 2012;125:993–1002. doi: 10.1242/jcs.095877. doi: 10.1242/jcs.095877. [DOI] [PubMed] [Google Scholar]
- 74.Monteferrario D, et al. A dominant-negative GFI1B mutation in the gray platelet syndrome. The New England journal of medicine. 2014;370:245–253. doi: 10.1056/NEJMoa1308130. doi: 10.1056/NEJMoa1308130. [DOI] [PubMed] [Google Scholar]
- 75.Stevenson WS, et al. GFI1B mutation causes a bleeding disorder with abnormal platelet function. Journal of thrombosis and haemostasis: JTH. 2013;11:2039–2047. doi: 10.1111/jth.12368. doi: 10.1111/jth.12368. [DOI] [PubMed] [Google Scholar]
- 76.Wolfe SA, Grant RA, Elrod-Erickson M, Pabo CO. Beyond the “recognition code”: structures of two Cys2His2 zinc finger/TATA box complexes. Structure (London, England: 1993) 2001;9:717–723. doi: 10.1016/s0969-2126(01)00632-3. doi. [DOI] [PubMed] [Google Scholar]
- 77.Kohler S, et al. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic acids research. 2014;42:D966–974. doi: 10.1093/nar/gkt1026. doi: 10.1093/nar/gkt1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Abecasis GR, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics. 2011;43:491–498. doi: 10.1038/ng.806. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jun G, et al. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. American journal of human genetics. 2012;91:839–848. doi: 10.1016/j.ajhg.2012.09.004. doi: 10.1016/j.ajhg.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Westerfield M. In: The zebrafish book: a guide for the laboratory use of zebrafish (Danio rerio) ed. 4 Westerfield M, editor. Eugene, OR: 2000. [Google Scholar]
- 83.Knight R, et al. PyCogent: a toolkit for making sense from sequence. Genome biology. 2007;8:R171. doi: 10.1186/gb-2007-8-8-r171. doi: 10.1186/gb-2007-8-8-r171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Loytynoja A, Goldman N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science (New York, N.Y.) 2008;320:1632–1635. doi: 10.1126/science.1158395. doi: 10.1126/science.1158395. [DOI] [PubMed] [Google Scholar]
- 85.Massingham T, Goldman N. Detecting amino acid sites under positive selection and purifying selection. Genetics. 2005;169:1753–1762. doi: 10.1534/genetics.104.032144. doi: 10.1534/genetics.104.032144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sipos B, Massingham T, Jordan GE, Goldman N. PhyloSim - Monte Carlo simulation of sequence evolution in the R statistical computing environment. BMC bioinformatics. 2011;12:104. doi: 10.1186/1471-2105-12-104. doi: 10.1186/1471-2105-12-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular biology and evolution. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 88.UniProt C. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic acids research. 2013;41:D43–47. doi: 10.1093/nar/gks1068. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.