

Abstract
Mitochondrial-mediated oxidative stress in response to high glucose is proposed as a primary cause of dorsal root ganglia (DRG) neuron injury in the pathogenesis of diabetic neuropathy. In the present study, we report a greater number of mitochondria in both myelinated and unmyelinated dorsal root axons in a well-established model of murine diabetic neuropathy. No similar changes were seen in younger diabetic animals without neuropathy or in the ventral motor roots of any diabetic animals. These findings led us to examine mitochondrial biogenesis and fission in response to hyperglycemia in the neurites of cultured DRG neurons. We demonstrate overall mitochondrial biogenesis via increases in mitochondrial transcription factors and increases in mitochondrial DNA in both DRG neurons and axons. However, this process occurs over a longer time period than a rapidly observed increase in the number of mitochondria in DRG neurites that appears to result, at least in part, from mitochondrial fission. We conclude that during acute hyperglycemia, mitochondrial fission is a prominent response, and excessive mitochondrial fission may result in dysregulation of energy production, activation of caspase 3, and subsequent DRG neuron injury. During more prolonged hyperglycemia, there is evidence of compensatory mitochondrial biogenesis in axons. Our data suggest that an imbalance between mitochondrial biogenesis and fission may play a role in the pathogenesis of diabetic neuropathy.
Introduction
Mitochondria are important mediators of cellular function through regulation of energy metabolism, generation of ATP, calcium handling, and the biosynthesis of key metabolites including α-ketoglutarate and glutamate [18]. The high metabolic activity of mitochondria is coupled with substantial generation of reactive oxygen species [20]. Therefore, the ability to repair or replace oxidatively damaged mitochondria as well as to increase mitochondrial mass is critical to maintenance of functional mitochondria [21]. When mitochondria cannot withstand stress or excessive oxidative injury, they initiate programmed cell death [25].
Mitochondria operate a number of dynamic mechanisms in order to maintain a functional network. To replace aged or damaged mitochondria, they undergo biogenesis [44]. Mitochondrial biogenesis involves replication of the mitochondrial genome followed by division into two daughter mitochondria, a process known as fission [9]. In addition, mitochondria divide and fuse independently of biogenesis in order to disperse a metabolic load or to partition damaged and nascent mitochondrial proteins and lipids [6]. The ability to participate in these mechanisms depends upon the maintenance of functional mitochondria, energy generation, and prevention of programmed cell death.
The energy requirement of neurites, far from the site of transcription of nuclear-encoded genes, presents a significant and unique challenge to the mechanism of mitochondrial biogenesis [1]. The capacity for mitochondria to undergo biogenesis in neurites is documented, but principally in brain neurons where the distance from the cell body is relatively short compared to peripheral sensory neurons [1]. Diabetes imposes significant stress on sensory neurons via excess metabolic substrates and a highly oxidative environment, increasing the turnover of mitochondria [19, 39]. Therefore, we hypothesized that we would observe significant increases in neurite mitochondrial oxidative stress in a model of hyperglycemia. This would be coupled with attempts to increase mitochondrial biogenesis and fission, to handle the metabolic load.
In this study, we confirmed that mitochondrial density is altered in both myelinated and unmyelinated dorsal root axons of 24 wk old type 2 diabetic mice, an age where these animals are known to have developed diabetic neuropathy [7, 32, 41]. In contrast, there is no change in the mitochondrial density of ventral root fibers in these same animals or in either dorsal or ventral root fibers in young diabetic animals prior to the development of neuropathy. Next, we examined mitochondrial biology in the neurites of a cell culture model of adult mouse DRG neurons in the presence of hyperglycemia. We found a loss of balance between mitochondrial fission and biogenesis that may underlie our in vivo data. We observed mitochondrial fission in neurites in response to short-term hyperglycemia with evidence of oxidative stress in mitochondria within neurites and subsequent activation of caspase 3, indicative of neurite injury. Mitochondrial biogenesis, while present in neurites after long-term glucose exposure, was insufficiently robust to counter the observed neurite damage. Our study suggests disruption of normal mitochondrial function in axons may underlie the pathogenesis of diabetic neuropathy and underscores the importance of targeting therapeutic strategies to preserve mitochondrial function in order to prevent peripheral nerve damage in diabetes.
Materials and Methods
Materials
Primary antibody clones and their sources were as follows. Actin: sc-1616 (Santa Cruz); Drp1: 3B5 (Abnova); neurofilament M: AB1987 (Millipore); Nrf-1: ab34682 (Abcam); PGC1α: 4C1.3 (Calbiochem); pyruvate dehydrogenase: 13G2 (Molecular Probes); TFAM: NB600-1462 (Novus Biologicals); VDAC: Ab-5 (185–197, Calbiochem).
Diabetic Mice
Diabetic (BKS.Cg-m+/+Leprdb, BKS-db/db) and control (BKS-db+) mice were purchased from Jackson Laboratories (Bar Harbor, Maine). Mice were housed in a pathogen-free environment, with continuous access to food (Purina 5053 chow) and water on a 12 h light–dark schedule and were cared for following the University of Michigan Committee on the Care and Use of Animals guidelines. Blood glucose levels were measured every 4 wks to document the onset and duration of diabetes. Following a 6 h fast, one drop of tail blood was analyzed using a standard glucometer (One Touch Profile, LIFESCAN, Inc. Milpitas, CA). At the end of the experimental period, glycated hemoglobin (GHb) was measured using the Helena Laboratories Test Kit, Glyco-Tek Affinity Column Method and plasma insulin was measured by ELISA at the University of Washington Mouse Metabolic Phenotyping Core. Analyses and procedures were performed in compliance with protocols established by the Animal Models of Diabetic Complications Consortium (AMDCC, http://www.amdcc.org).
Morphological Analysis
Semithin and ultrathin morphological analyses of ventral and dorsal roots of db/db and db+ littermates were performed as described [26]. The mice were euthanized by sodium pentobarbital overdose. Age-matched BKS-db/db and db+ mice were euthanized at 12 or 24 wk of age as described above and perfused with 4% paraformaldehyde in phosphate buffered saline (0.1 M, pH 7.2; PBS). Tissues from five each of db/db and db+ mice at each time-point were evaluated. Tissues were post-fixed in 0.12 M phosphate buffer containing 2% glutaraldehyde followed by osmium tetroxide and embedded in Epon (Fluka, Buchs SG, Switzerland). Sections were obtained from the spinal roots. Semithin sections (0.5–1 μm thick) were stained with toluidine blue and examined by light microscopy (BX51; Olympus, Tokyo, Japan). Ultrathin sections (100–120 nm thick) were stained with uranile acetate and lead citrate and examined by electron microscopy (magnification x 3000, Leo 912, Omega, Brattleboro, VT). For quantitative ultrastructural analyses, blocks of tissue were selected for electron microscopy after light microscopy examination of semithin sections. All ultrastructural analyses were performed in a blinded and nonbiased manner from photomicrographs captured using the electron microscope. For all mice, nonoverlapping photographs were taken from the spinal roots. An average of 20 microscopic fields per mouse were analyzed to cover the entire cross section of the nerve. An average of 400 unmyelinated and 150 myelinated axons per mouse was analyzed. The axonal areas were determined in 286 axons per group. The number of mitochondria present in both single fibers and in total axon area was counted at a final magnification of x 8000. The number of mitochondria present in the cytoplasm of each fiber were counted and mitochondrial diameters were measured in the transverse sections [7]. Samples were compared using a Chi-square test [7].
Adult Mouse DRG Cultures
DRG neurons were cultured as previously described [35, 36, 41]. Briefly, adult (> 6 wk old) C57Bl6/J mice and dissociated in 0.2 % collagenase for 30 min followed by 1 % trypsin for 15 min. Cells were seeded on collagen-coated plates in DMEM:F-10, 50:50, containing 1 x B27 additives, 40 μM FUDR, 7 μM aphidicolin and 1,000 U/ml penicillin/streptomycin/neomycin. Cells were re-fed on day 2 and experiments performed on day 3. In this media the basal glucose concentration is 5.7 mM. For high glucose experiments, glucose is added to the media to a final concentration of 25.7 mM. In all experiments, additional controls were assessed by adding the same concentration of o-methyl-glucopyranose (OMG) instead of glucose to the media. This serves as an osmotic control per our previous studies [39, 40]. Unlike high glucose, an equimolar concentration of OMG produced results similar to an untreated control. These data are not shown in the figures.
MitoSOX
MitoSOX (Molecular Probes, Eugene, OR) is a cell-permeable probe that accumulates in mitochondria and fluoresces following oxidation by superoxide. MitoSOX is dissolved in DMSO, then diluted to 3 μM final concentration, as previously described [35, 41, 42]. A 50 X solution of MitoSOX is added to the culture media 30 min prior to the end of the experimental period. After 30 min, the cells are rinsed in PBS, then mean red fluorescence per well is read immediately at 485 nm excitation and 590 nm emission (Fluoroskan Ascent II plate reader, LabSystems, Helsinki). Alternatively, cells are fixed in 2% paraformaldehyde, mounted in Prolong Gold, and examined by confocal microscopy within 16 h as described [13, 27].
CaspaTag
After 0, 1, 3, or 5 h exposure to 20 mM excess glucose, coverslips were treated with fluorescent CaspaTag reagent per the manufacturer’s protocol (Chemicon, San Francisco, CA), fixed in 2% paraformaldehyde in PBS and examined by confocal microscopy as described [13, 27]. Quantification of CaspaTag labeling and fixed oxidized MitoSOX (described in the previous paragraph) was performed using Scion Image software. Five images were randomly selected from each coverslip, with 3 coverslips per condition, and converted to grayscale. The integrated pixel intensity in an area 500μm x 500 μm was measured in all 15 images. The mean pixel intensity over the cell bodies was measured in at least 200 neurons per condition, randomly selected in the 15 images. The mean pixel intensity along a line drawn in the neurites was assessed over 50 μm length segments, with at least 120 segments per condition, giving a final mean pixel intensity over at least 6 mm of neurite per condition. The mean and standard error of the measurements were plotted. The differences between time-points and between db+ and db/db samples were compared using the unpaired t-test.
Western blotting
Western blotting was performed per previous studies [38, 40, 41]. 40 μg protein/lane, separated on a 12.5% polyacrylamide gel, was transferred to nitrocellulose and probed with 5 μg/ml primary antibody in 5% non-fat milk overnight at 4°C, HRP-conjugated secondary antibody (Santa Cruz, CA), 1:3000) for 1 h at room temperature, then visualized with ECL (Amersham, Arlington Heights, IL), and exposed to film (Hyperfilm-ECI, Amersham). Blots were stripped in 2 ml 10% SDS, 0.15 g dithiothreitol, 2 ml 0.5 M Tris pH 6.8 in 6 ml H2O at 70°C for 20 min. Membranes were then re-blocked and probed for actin as a loading control.
RNA Isolation and Real-Time RT-PCR
Assessment of mRNA for specific mitochondrial proteins and transcription factors was performed as previously described [7] and data is presented in Supplementary Fig. 1. Briefly, total RNA was extracted using an RNeasy Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Reverse transcription was performed using iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA). Real-time PCR reactions were carried out in a 96-well 0.2 ml PCR plates sealed with iCycler Optical Sealing tapes (Bio-Rad, Hercules, CA, USA). The PCR amplification profile was: 95°C for 5 min, 40 cycles of denaturation at 95°C for 30 s, annealing at 58°C for 1 min, and extension at 72°C for 30 s with a final phase of 72°C for 5 min. The fluorescence threshold CT value was calculated by the iCycler iQ system software and the levels were first normalized to the endogenous reference GAPDH (ΔCT), then relative to the control group (ΔΔCT) and expressed as 2−ΔΔCT. Levels of PCR products were demonstrated as mean ± SEM and a two-sample equal variance t-test was performed.
Mitochondrial DNA Quantitation
DNA was extracted using the Genelute Mammalian Genomic DNA Kit (Sigma, St. Louis, MO) according to the manufacturer’s instructions. Real-time PCR amplification and SYBR Green fluorescence detection were performed using iCycler iQ Real-time Detection System (Bio-Rad, Hercules, CA). Mitochondrial DNA was quantitated by normalizing the mitochondrial gene (cytochrome b) to the nuclear gene (actin), then relative to the control group (ΔΔCT). The 2−ΔΔCT method was used. A total of 10ng genomic DNA was used for mitochondrial DNA and nuclear DNA markers, in a 25ul reaction containing 1X SYBR Green iCycler iQ mixture and 0.2 μM each of forward and reverse gene-specific primers as previously described [7].
Immunohistochemical staining
DRG neurons were fixed in 4% paraformaldehyde in 0.1 M phosphate buffered saline solution (PBS) for 5 min. Primary antibodies were applied in 0.1 M PBS containing 1% Triton X100 and 5% serum for 16 h at 4°C. Cells were washed for 3x 10 min in PBS, then species specific secondary antibody conjugated to AlexaFluor 594, (Molecular Probes, Eugene, OR) diluted 1:1000 in 0.1 M PBS containing 1% Triton X100 and 5% serum was applied. After a further 1 h at room temperature, cells were washed 3 x 10 min in PBS. Fluorescence was quantitated in the fluorescent plate reader at 485 ex, 590 em. Immunofluorescence was examined on an Olympus Fluoview inverted microscope.
EdU labeling
This technique was modified in our laboratory [14]. Cultured DRG neurons were exposed to 10 μM EdU for the duration of the experimental treatment. Control untreated cultures were labeled in parallel with EdU only. After fixing, neurons were permeabilized using 0.1% triton-X-100 solution then endogenous peroxidase was quenched with 0.3% H2O2. Incorporated EdU was then detected using the reagents in a Click-it EdU Imaging Kit (Invitrogen) according to the manufacturer’s instructions. DRG neurons were then mounted in Prolong Gold and examined by confocal microscopy. Two coverslips from two separate experiments were examined per condition. All of the cells on a microscope field were examined; approx. 3–6 neuron cell bodies/field, and 3 fields per coverslip. All of the neurofilament positive neurites on or crossing the field were measured. Thus, the mean and std error for n = 18–25 cell bodies and for > 30 μm of neurites from > 15 separate neurites were calculated.
Statistical Analysis
Data analyses were performed using Prism, version 3 (GraphPad Software, Inc.). Assumptions about the Gaussian distribution of data and rules for transformation of non-normative data were made as previously described [28]. In mouse studies, there were 5 mice per group. Experimental groups were compared using the Chi-Squared test. For all cell culture studies, n is at least three separate replicate experiments and is defined under each separate method. Comparison of dependent variables was performed using unpaired t-test with significance assigned when P<0.05. All measurements were made by an observer blinded to the experimental condition. Bar graphs illustrate the mean ± standard error of the mean (SEM).
Results
Altered Numbers of Mitochondria in Axons
To determine whether axonal mitochondria are affected in diabetes, we used electron microscopy to compare mitochondria in dorsal and ventral root axons in diabetic (12 wk and 24 wk old db/db) and non-diabetic age-matched (db+) mice. The db/db mice had significantly elevated body weights, blood glucose, insulin, and glycated hemoglobin (GHb) compared to age-matched db+ littermates (Table 1).
Table 1.
Glycemia Status of 24 wk Old db+ and db/db Mice
| Weight (g) | Blood Glucose (mg/dL) | Insulin (ng/mL) | GHb (%) | |
|---|---|---|---|---|
| db+ | 29.8±0.6 | 131.7±3.1 | 1.78±0.3 | 6.23±.07 |
| db/db | 53.6±1.8* | 562.7±18.8* | 5.01±1.2* | 12.53±.40* |
p<0.01 when comparing db+ and db/db
At 24 wk of age, there was an increase in the number of mitochondria per axon in dorsal root myelinated fibers of db/db mice compared to db+ littermates (Fig. 1b versus 1a). There was no difference in the number of mitochondria in ventral root myelinated fibers of db/db (Fig. 1d) and db+ (Fig. 1c) animals. Quantitative analysis of the dorsal roots revealed a significantly increased number of mitochondria in myelinated fibers at 24 wk in db/db mice compared to db+ (Fig. 1g, P = 0.0356). There was no significant difference in the number of mitochondria in the myelinated fibers of ventral roots (P = 0.24, data not shown).
Fig. 1. Mitochondrial Density and Morphology in Dorsal and Ventral Root Axons of Diabetic Mice.

Dorsal (a–b) and ventral (c–d) nerve roots of 24 wk old db+ and db/db littermates were prepared for ultrathin cross sections for TEM. Arrowhead indicates accumulation of mitochondria (b). High magnification images of axons from dorsal roots of db+ animals (e) show normal mitochondrial number while axons from dorsal roots of db/db animals (f) show increased numbers of mitochondria in axons (arrowhead) and increased neurofilaments and microtubules. In (e) and (f), each inset contains a higher magnification of the mitochondria in (e; normal mitochondria) and (f; mitochondria with slightly dilated cristae) respectively. Scale bars = 2 μm in (a–d), 1 μm in (e–f), and 10 μm in inset; a= axon. Quantitative analysis of mitochondrial density in myelinated axons from dorsal roots is presented in the distribution plots (g, h). There were increased numbers of mitochondria in the dorsal root myelinated fibers of the diabetic (db/db) compared to control (db+) mice at 24 wk (P<0.05, g). There were no statistical differences in mitochondrial density in dorsal root myelinated fibers between dbdb and db+ animals 12 wk of age. In (i), the areas of myelinated axons were measured in the 24 wk samples and presented as an area frequency plot. There is a trend to decrease axonal area in the db/db mice, confirming that in increase in mitochondrial number per axon represents an increase in total mitochondria and in mitochondrial density.
In parallel, there was an increase in the number of mitochondria in the dorsal root unmyelinated fibers of 24 wk old db/db mice (Fig. 2c–d) when compared to db+ littermates (Fig. 2a–b). Quantitative analysis of the mitochondrial density in these unmyelinated fibers revealed a highly significant difference between the two groups (Fig. 2e, P < 0.0001).
Fig. 2. Mitochondrial Density in Unmyelinated Dorsal Root Axons.
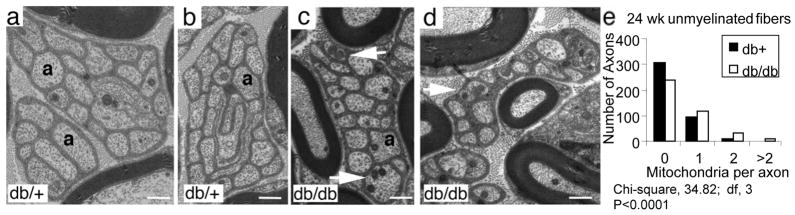
Transverse sections of dorsal root unmyelinated fibers from db/+ (a–b) and db/db mice (c–d) indicate increased numbers of mitochondria in unmyelinated fibers of db/db mice (arrows in c and d). Scale bars = 1 μm in (a–d). Quantitative analysis of mitochondrial density in unmyelinated axons from dorsal roots is presented in the distribution plot (e). There were increased numbers of mitochondria in the dorsal root unmyelinated fibers of the diabetic (db/db) compared to control (db+) mice at 24 wk (P<0.0001).
We have previously reported a decrease in the diameter of mitochondria in DRG neurons from 24 wk old db/db animals when compared to mitochondria from nondiabetic db+ age matched animals [7]. In contrast to these data, there was no quantitative differences in diameters when comparing mitochondria in the ventral roots of 24 wk old db/db (237.5 nm ± 2.3 SEM, n= 526) to db+ (233.1 ± 2.6, SEM, n = 629) animals. Dilated intracristal spaces were commonly seen in the mitochondria in the dorsal roots of the 24 wk old db/db samples (Fig. 1f inset), while similar morphological changes were not present in the mitochondria from the db+ animals (Fig. 1e inset). These changes in cristae morphology may explain why the mitochondria between the two groups of animals have similar diameters.
At 12 wk of age, there were no observable differences in the number of mitochondria when comparing the dorsal root myelinated fibers of db+ to db/db animals. The quantitative analysis of the numbers of mitochondria in myelinated dorsal root fibers is presented in Fig. 1h and confirms no difference between the diabetic and control animals (P = 0.99, Fig. 1h). There was also no difference in the number of mitochondria in unmyelinated fibers from the same animals (P = 0.24, data not shown).
To confirm that increases in the number of mitochondria per axon represent an increase in mitochondrial density, we measured the areas of the axons. A frequency distribution plot for axonal areas of myelinated fibers demonstrates a modest shift to smaller axonal areas in 24 wk db/db compared to db+ dorsal roots (Fig. 1i). There was no difference in axonal area in unmyelinated fibers in dorsal or ventral roots at 12 or 24 wk, nor was there a difference in myelinated ventral roots or in any 12 wk samples (not shown).
Hyperglycemia Leads to Neurite Oxidative Stress
The findings in vivo in type 2 diabetic mice led us to examine mitochondria changes in vitro in response to hyperglycemia. Our previous studies demonstrate that exposure to an increase in extracellular glucose rapidly leads to DRG neuron oxidative stress, activation of the caspase cascade and neuronal injury [13, 28, 38, 41]. These responses are specific for glucose and not a related osmotic control glucose derivative, o-methyl-glucopyranose (OMG) [39, 40]. Using live cell stains and confocal microscopy we can now determine the effects of high glucose specifically in the neurites of cultured DRG neurons. We prepared cultures of DRG neurons from both db+ and db/db mice at 24 wk of age. After 3 d in culture, DRG neurons were exposed to control or high glucose media for 1, 3, or 5 h, then probed for mitochondrial oxidative stress using MitoSOX (Fig. 3). We observed that the neurites of DRG neurons from db/db mice under basal conditions tend to be shorter, more swollen, and more tortuous than db+ (Fig. 3a). Within 1 h of high glucose treatment, MitoSOX oxidation increased significantly in both db+ and db/db neurons (Fig. 3b). In the cell bodies, this increase was more pronounced in db/db than db+ neurons (Fig. 3c). MitoSOX oxidation was significantly higher in control glucose in db/db cultures compared to db+ in both the cell body and neurites (Fig. 3c–d), and this difference was reversed in high glucose, particularly in the neurites. Following the peak of MitoSOX oxidation at 1 h, the degree of mitochondrial oxidative stress declined, but the decrease was slower in db/db than db+ neurons (Fig. 3b).
Fig. 3. Mitochondrial Superoxide Generation in Response to Hyperglycemia in Cultured DRG Neurons from Diabetic Mice.

DRG neurons were cultured from 24 wk old diabetic db/db mice and non-diabetic db+ littermates. After 3 d in basal (5.7 mM) glucose, neurons were exposed to 25.7 mM glucose for 0, 1, 3, or 5 h and loaded with MitoSOX for the final 30 min, then fixed and examined by confocal microscopy. A representative image from each condition (total n=15 images collected per condition) is shown in (a). In (b–d), the pixel intensities of all images per condition were analyzed using Scion Image software. In (b), the integrated pixel density in a 500 x 500 μm square containing at least 3 neuron cell bodies with associated neurites was measured. In (c), the mean pixel intensity of at least 200 neuron cell bodies per condition was calculated. In (d), the mean pixel intensity of at least 120 distinct 50 μm length segments of neurites was calculated. Bars represent the mean and standard error of the mean for all images. *Indicates a significant change in pixel intensity between time-points, p<0.01. +Indicates a significant change in pixel intensity between db+ and db/db samples at a single time-point, p<0.05
Responses of Diabetic and Non-Diabetic DRG Neurons to Hyperglycemia
Sister cultures from the db+ and db/db mice described above, exposed to control or high glucose media for 1, 3, or 5 h, were then probed for activation of caspase 3 (Fig. 4). Using fluorimetry, we previously demonstrated the activation of caspase 3 in DRG neurons cultured in high glucose but not the osmotic control OMG [35, 37–39]. This increase in caspase 3 is detectable after 1 h high glucose, significant after 3 h, and peaks at 5 h. We now demonstrate that at the peak time point, the activation of caspase 3 is significant in the neurites and not in the cell body (Fig. 4). In DRG neurons from db+ mice, punctate active caspase 3 labeling was observed associated with neurites after 1 h high glucose, continuing to increase through 5 h (Fig. 4a). Quantitation of this signal demonstrates that this increase is statistically significant at 1 h, but does not further significantly increase from 1 h to 5 h. The DRG neurons from db/db mice had markedly more caspase 3 activation even in basal glucose compared with db+ mice (Fig. 4a–b). Initially, after 1 h glucose, caspase 3 activation was markedly increased in the cell bodies of db/db DRG neurons (Fig. 4a and 4c). Caspase 3 activation increased significantly in both the cell bodies and neurites of db/db neurons over 3–5 h with the biggest increase occurring in the neurites between 3–5 h (Fig. 4d).
Fig. 4. Hyperglycemia Activates Caspase-3 in Cultured DRG Neurons from Diabetic Mice.

DRG neurons were cultured from 24 wk old diabetic db/db mice and non-diabetic db+ littermates. After 3 d in basal (5.7 mM) glucose, neurons were exposed to 25.7 mM glucose for 0, 1, 3, or 5 h and loaded with CaspaTag for the final 1h, then fixed and examined by confocal microscopy. A representative image from each condition (total n=3 replicate experiments with 15 images collected per condition) is shown in (a). In (b–d), the pixel intensities of all images per condition were analyzed using Scion Image software. In (b), the integrated pixel density in a 500 x 500 μm square containing at least 3 neuron cell bodies with associated neurites was measured. In (c), the mean pixel intensity of at least 200 neuron cell bodies per condition was calculated. In (d), the mean pixel intensity in at least 6 mm length of neurites was calculated (50 μm length segments of neurite, with at least 120 segments per condition). Bars represent the mean and standard error for all images. *Indicates a significant change in pixel intensity between time-points, p<0.01. +Indicates a significant change in pixel intensity between db+ and db/db samples at a single time-point, p<0.05
High Glucose Increases Mitochondrial DNA Synthesis in Neurites
Following previous studies suggesting that hyperglycemia-induced oxidative stress is principally generated by the mitochondria, we examined mitochondria in the neurites of the cultured DRG neurons from non-diabetic adult C57/Bl6 mice. DRG neurons were concurrently exposed to EdU and high glucose media, then fixed after 3, 6, or 24 h. Subsequently, DRG neurons were labeled for a mitochondrial marker pyruvate dehydrogenase (PDH) and for an axonal marker, neurofilament (NF). We observed an increase in EdU (green) labeling with time in high glucose media (Fig. 5). This was more common in non-neuronal cells, identified by the absence of NF (blue) staining (* Fig. 5b). Nonetheless, EdU incorporation was clearly observed in neuronal cell bodies and neurites in high glucose (Fig. 5b–d). Higher magnification images of the neurites confirm that there are many more mitochondria in the neurites in high glucose conditions than in control cultures, and that a proportion of these are incorporating EdU (green and red overlay, producing yellow signal), suggesting that they are undergoing mitochondrial DNA synthesis.
Fig. 5. Hyperglycemia Increases Mitochondrial DNA Synthesis in Cultured DRG Neurons.

DRG neurons from C57/Bl6 mice were exposed to 25.7 mM glucose for 0, 3, 6, or 24 h in the presence of EdU, then fixed and stained for EdU incorporation (green), mitochondria (PDH, red), and neurons (neurofilament, blue). (a) No mitochondria or EdU labeling was observed in neurites under control conditions. (b) EdU labeling markedly increased in non-neuronal cells (*) after 3 h hyperglycemia (immunonegative for PDH); EdU incorporation also increased in neurites (arrows). In (c–d), both mitochondria labeling and EdU incorporation continued to increase in neurites with prolonged hyperglycemia. In (a′-d′), higher magnification images of neurites permit counting of the numbers of discrete mitochondria and the proportion of these incorporating EdU. Quantification of a′-d′ is shown in (e–f). 2 separate experiments, each with 3 replicates and 3 images per condition, giving n=18 images were analyzed. The number of yellow (PDH and EdU positive) and red (PDH only) mitochondria in neurites were counted. The number of mitochondria was divided by the neurite length to give a measure of mitochondria/mm and the percent yellow mitochondria was calculated. *Mean number of mitochondria/mm and the percent incorporating EdU both increased significantly by 3 h compared to control glucose and remained elevated up to 24 h, p<0.01.
By counting the numbers of red, green, and yellow dots in the blue-labeled neurites, we were able to quantitate mitochondria in the neurites and the degree of mitochondrial DNA synthesis (Fig. 5e–f). The total number of discrete mitochondria/mm along the neurites significantly increased from approximately 50 to 200 mitochondria/mm after 3 h high glucose exposure (Fig. 5e). The number of mitochondria/mm remained at approximately 200 over 24 h. The proportion of these mitochondria in the neurite that were incorporating EdU increased from approximately 4% in control media to 18% by 3 h of hyperglycemia treatment and continued to increase up to 23% by 24 h (Fig. 5f).
Alterations in Mitochondrial Transcription Factors in High Glucose
To further understand mitochondrial biogenesis in this system, we examined the expression of mitochondrial transcription factors (Supplementary Fig. 1) as well as the subcellular localization of the principal mitochondrial transcription regulator PCG1 α (Supplementary Fig. 2).
Mitochondria in Neurites Undergo Fission
To assess mitochondrial fission, the DRG neurons cultured from C57/Bl6 mice were immunolabeled for VDAC (red) and Drp1 (green) and confocal images were taken of the cells (Fig. 6). In control DRG neurons, modest VDAC (mitochondria) and Drp1 were observed in the neurites and cell bodies. There was no co-localization of the two markers in the neurites, although there was limited co-localization in the cell body. After 6 h of high glucose treatment, VDAC markedly increased in the neurites and Drp1 increased in many, but not all, neurites. Co-localization of Drp1 in the mitochondria increased in the neurites as well as the cell body. After 24 h high glucose, there was considerable Drp1 mitochondrial localization in both the cell body and neurites. These co-localizations were notably greater at sites of neurite beading.
Fig. 6. Hyperglycemia Promotes Mitochondrial Fission in DRG Neurons.

DRG neurons from C57/Bl6 mice were exposed to 25.7 mM glucose for 0, 6, or 24 h, then fixed and immunolabeled for Drp1 (green) and mitochondria (VDAC, red). Representative cell bodies appear above associated representative neurites. Co-localization of the labels, which demonstrates re-localization of Drp1 to mitochondria, suggesting mitochondrial fission, appears in yellow.
Discussion
Defects in mitochondrial function and trafficking are significant factors in a number of neurodegenerative disorders including Charcot-Marie Tooth disease [5, 48], amyotrophic lateral sclerosis [2], and Alzheimer’s disease [15, 43]. Mitochondrial oxidative stress and injury is also recognized as a unifying mechanism of cellular injury in the complications of diabetes [3, 12, 17, 23, 39, 41]. We have noted that common features exist between patients with mitofusion mutations and patients with diabetic neuropathy, including slowing peripheral nerve conduction velocities and chronic progressive distal axon degeneration, with diminished ability to regenerate [22]. In this study, we demonstrate changes in mitochondrial density in myelinated and unmyelinated dorsal root fibers in diabetic mice at an age when these mice are known to have neuropathy [7], then explore these changes in our in vitro model of adult DRG neuron cultures exposed to hyperglycemia. We suggest that mitochondrial dysfunction in sensory axons may underlie the development of diabetic neuropathy. Our studies form the foundation for mechanistic evaluation of mitochondrial injury in diabetes and will be a valuable tool for the assessment of novel treatment strategies targeted at mitochondria biogenesis, fission, and axonal injury.
In our well-characterized model of type 2 diabetes with diabetic neuropathy, the 24 wk old BKLS db/db mouse [32, 33], the increase in the number of axonal mitochondria agrees with our recent report of increased mitochondrial density in the cell bodies of the DRG neurons [7]. Mitochondrial density was never different between samples in younger 12 wk db/db animals, an age at which neuropathy has not yet developed [32, 41]. There was also no difference in mitochondrial density in ventral roots at either 12 or 24 wk of age between db/db and db+ animals. Our results are consistent with studies in diabetic rats showing greater effects on dorsal than ventral roots, and that selective effects of oxidative stress are present in the DRG cell body and dorsal roots [30].
The finding that mitochondrial diameters were not decreased in the dorsal roots was unanticipated, as we have previously reported that there is a decrease in diameter in the mitochondria located in the DRG cell body of these mice [7]. Closer inspection of the axonal mitochondria from the db/db mice revealed these organelles were misshapen with evidence of swollen cristae. This pathologic swelling of the cristae is likely the reason we did not observe the anticipated difference in mitochondrial size between the diabetic and non-diabetic animals. Similar morphological changes in mitochondrial cristae structure are reported in DRG cell bodies and nerve roots in streptozotocin rat models of diabetic neuropathy [28, 31]. Our data support these published results, and suggest that there are observable changes in the morphology of axonal mitochondria in animals with diabetic neuropathy.
The increase in the number of mitochondria in the dorsal root fibers of db/db animals with diabetic neuropathy supports our hypothesis that attempts to disperse metabolic overload in diabetes activate excessive mitochondrial fission leading to mitochondria damage and cellular injury. Similar mechanisms have been identified in the cardiovascular system [46] and in pancreatic beta cells [16]. Fission is an important mitochondrial mechanism believed to facilitate the segregation of damaged mitochondrial structures for autophagy, the dispersal of excessive ion gradients, and is the final step in the process of mitochondrial biogenesis [34].
Our MitoSOX measurements of mitochondrial oxidative stress demonstrated a significantly greater MitoSOX fluorescence in basal glucose in db/db compared to db+ neurons and neurites in vitro. Glucose-induced mitochondrial oxidative stress was greater and more prolonged in db/db than db+ cultures. A similar recent study also demonstrated neurite oxidative stress in hyperglycemia in adult rat DRG neurons, but only from diabetic, not non-diabetic control neurons [49]. This study used a type 1 diabetes model of streptozotocin-induced insulin deficiency, so the primary differences in the results are likely related to the insulin used in the culture system, as well as differences in the duration of hyperglycemia. In the cited study, the duration of hyperglycemia was at least 24 h, where the present study examines short time-points up to 24 h as a model of acute hyperglycemia.
Activation of caspase 3 followed mitochondrial oxidative stress in a similar pattern. In particular, caspase 3 activation also increased more in the neurites in db/db than db+ DRG neurons. These data indicate the increased susceptibility of DRG axons to hyperglycemia in the type 2 diabetic mice. Activation of caspase 3 occurs subsequent to mitochondria permeability transition and release of pro-apoptotic factors [8]. Caspase 3 activation is observed less robustly in vivo than in vitro, probably because of greater neuronal protection by accessory cells and other factors in vivo and because of early removal of injured neurons by phagocytosis. However, this remains a reliable marker of early neuronal injury in vitro [28, 41]. These data underscore the distal dying back of neurites in diabetic neuropathy, since we demonstrate hyperglycemia-induced mitochondria injury and activation of caspase 3 in the neurites.
We postulate that the significantly greater caspase 3 activation at baseline in db/db compared to db+ mice is related to long-term hyperlipidemia leading to an altered the lipid profile of the DRG neurons [36]. A different lipid profile will confer altered ability to withstand metabolic and oxidative insults [4]. Thus, hyperglycemia and hyperlipidemia in db/db mice will produce an additive injury in susceptible DRG neurons and axons similar to C57/Bl6 mice on a high fat diet [36]. This cumulative mechanism of glucose and lipid-induced injury is observed in pancreatic beta cells [24].
To explore the mechanism of mitochondria accumulation in neurites in hyperglycemia, we assessed mitochondria biogenesis, via mitochondrial EdU incorporation. Hyperglycemia produced a rapid increase in the number of mitochondria in the neurites as previously shown [13], as well as a greater increase in mitochondrial EdU incorporation in neuronal cell bodies and neurites. The ability for mitochondria to undergo biogenesis in neurites is likely important for supply of energy to the distal regions of the neurite and growth cones [1].
Further confirmation that hyperglycemia leads to an attempt to increase mitochondria biogenesis came from modest increases in the transcription factors TFAM, Nrf1, and PGC1α and the fission protein Fis1 without significant increases overall in mitochondrial protein (Supplementary Fig. 1). These data suggest that mitochondrial biogenesis is a slow response to hyperglycemic insult and is unlikely to be able to provide rapid increases in mitochondrial mass to accommodate acute changes in metabolic load in diabetes.
Using confocal microscopy, we made two observations regarding the localization of PCG1α (Supplementary Fig. 2). First, after 3–6 h of hyperglycemia, PGC1α increased in the neurites. This indicates that PGC1α may have been operating as a modulator of protein function rather than binding to DNA, as recently reported [10]. Second, there was a large amount of PGC1α-associated signal in the perinuclear region of DRG neurons after 24 h of hyperglycemia. This suggests that this transcription factor was being synthesized at this time, which supports the idea of a relatively slow response to increase mitochondrial mass. Since glucose-induced oxidative stress occurring within 1–2 h is sufficient to injure DRG neurons [38], this inability to rapidly generate more mitochondrial mass may underlie the acute sensitivity of mitochondria to hyperglycemia. The data suggest that fission may be part of an important self-defense mechanism. Over time, however, excessive fission leading to loss of balance between fission/fusion and biogenesis is detrimental to mitochondrial function [45].
To better explore this idea, we assessed Drp1 localization as a measure of neurite mitochondrial fission. In particular, exposure to hyperglycemia led to a marked increase in Drp1 labeling in neurites that was co-localized with mitochondria in punctate sites. We conclude that there is significant Drp1-dependent mitochondrial fission in response to hyperglycemia. This finding suggests that our initial discovery of increased numbers of mitochondria in axons of diabetic mice may in part be the result of glucose-induced increases in mitochondrial fission. We recently demonstrated hyperglycemia-induced mitochondrial fission in DRG neuronal cell bodies both in vivo in db/db mice and in vitro in embryonic rat DRG neuron cultures [7]. The significance of Drp1-mediated fission was confirmed by a decrease in hyperglycemia-induced injury when Drp1 expression was decreased with miRNA [7]. The current study extends the findings to the axons where there is increased susceptibility to hyperglycemia-induced injury [29, 49] and neurites of adult mouse DRG cultures.
The idea that dysregulation of mitochondrial fission/fusion dynamics underlies neurodegenerative diseases is gaining support [7, 11, 13, 34, 47]. We now provide evidence that mitochondrial fission occurs in cell bodies and neurites during hyperglycemic injury in DRG neurons and axons. Since we find that fission proceeds on a faster time-course than new mitochondria biogenesis, a balance between fission and biogenesis is lost and may account for the rapid loss of energy generation, ultimately leading to DRG neuron and axon injury in hyperglycemia. Our data support the idea that dysfunction of axonal mitochondria may play an important role in the pathogenesis of diabetic neuropathy. We contend the regulation of mitochondrial fission and/or biogenesis in axons may be a new therapeutic target for diabetic neuropathy.
Supplementary Material
Acknowledgments
This work is supported by the Chemistry Core of the MDRTC for mouse plasma insulin and GHb measurements. This work was supported by the Juvenile Diabetes Research Foundation (AMV, ELF), the American Diabetes Association (AMV), the Animal Models of Diabetes Complications Consortium (AMDCC; NIH UO1 DK076160, ELF), the Program for Neurology Research and Discovery (JLE), the A. Alfred Taubman Medical Institute (AMV, JLE, ELF), the Instituto Superiore di Sanita (AQ), and FIRB (AQ).
References
- 1.Amiri M, Hollenbeck PJ. Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev Neurobiol. 2008;68:1348–1361. doi: 10.1002/dneu.20668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bacman SR, Bradley WG, Moraes CT. Mitochondrial involvement in amyotrophic lateral sclerosis: trigger or target? Mol Neurobiol. 2006;33:113–131. doi: 10.1385/MN:33:2:113. [DOI] [PubMed] [Google Scholar]
- 3.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 4.Cameron NE, Cotter MA. Pro-inflammatory mechanisms in diabetic neuropathy: focus on the nuclear factor kappa B pathway. Current drug targets. 2008;9:60–67. doi: 10.2174/138945008783431718. [DOI] [PubMed] [Google Scholar]
- 5.Cartoni R, Martinou JC. Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp Neurol. 2009;218:268–273. doi: 10.1016/j.expneurol.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 7.Edwards JL, Quattrini A, Lentz SI, et al. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia. 2010;53:160–169. doi: 10.1007/s00125-009-1553-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. N Engl J Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 9.Garnier A, Fortin D, Zoll J, et al. Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. FASEB J. 2005;19:43–52. doi: 10.1096/fj.04-2173com. [DOI] [PubMed] [Google Scholar]
- 10.Handschin C. The biology of PGC-1alpha and its therapeutic potential. Trends in pharmacological sciences. 2009;30:322–329. doi: 10.1016/j.tips.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Knott AB, Perkins G, Schwarzenbacher R, et al. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowluru RA, Atasi L, Ho YS. Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2006;47:1594–1599. doi: 10.1167/iovs.05-1276. [DOI] [PubMed] [Google Scholar]
- 13.Leinninger GM, Backus C, Sastry AM, et al. Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiology of Disease. 2006;23:11–22. doi: 10.1016/j.nbd.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 14.Lentz SI, Edwards JL, Backus C, et al. Mitochondrial DNA (mtDNA) Biogenesis: Visualization and Duel Incorporation of BrdU and EdU Into Newly Synthesized mtDNA In Vitro. J Histochem Cytochem. 2009 doi: 10.1369/jhc.2009.954701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leuner K, Hauptmann S, Abdel-Kader R, et al. Mitochondrial dysfunction: the first domino in brain aging and Alzheimer’s disease? Antioxidants & redox signaling. 2007;9:1659–1675. doi: 10.1089/ars.2007.1763. [DOI] [PubMed] [Google Scholar]
- 16.Men X, Wang H, Li M, et al. Dynamin-related protein 1 mediates high glucose induced pancreatic beta cell apoptosis. The international journal of biochemistry & cell biology. 2009;41:879–890. doi: 10.1016/j.biocel.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 17.Midaoui AE, Elimadi A, Wu L, et al. Lipoic acid prevents hypertension, hyperglycemia, and the increase in heart mitochondrial superoxide production. Am J Hypertens. 2003;16:173–179. doi: 10.1016/s0895-7061(02)03253-3. [DOI] [PubMed] [Google Scholar]
- 18.Mironov SL. Complexity of mitochondrial dynamics in neurons and its control by ADP produced during synaptic activity. The international journal of biochemistry & cell biology. 2009;41:2005–2014. doi: 10.1016/j.biocel.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Moreira PI, Santos MS, Seica R, et al. Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. J Neurol Sci. 2007;257:206–214. doi: 10.1016/j.jns.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 20.Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. American journal of physiology. 2007;292:C670–686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 21.Nisoli E, Clementi E, Moncada S, et al. Mitochondrial biogenesis as a cellular signaling framework. Biochemical Pharmacology. 2004;67:1–15. doi: 10.1016/j.bcp.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 22.Ouvrier R, Grew S. Mechanisms of disease and clinical features of mutations of the gene for mitofusin 2: an important cause of hereditary peripheral neuropathy with striking clinical variability in children and adults. Developmental medicine and child neurology. doi: 10.1111/j.1469-8749.2010.03613.x. [DOI] [PubMed] [Google Scholar]
- 23.Pennathur S, Heinecke JW. Oxidative stress and endothelial dysfunction in vascular disease. Curr Diab Rep. 2007;7:257–264. doi: 10.1007/s11892-007-0041-3. [DOI] [PubMed] [Google Scholar]
- 24.Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocrine reviews. 2008;29:351–366. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem. 2004;90:1281–1289. doi: 10.1111/j.1471-4159.2004.02572.x. [DOI] [PubMed] [Google Scholar]
- 26.Quattrini A, Previtali S, Feltri ML, et al. β4 Integrin and other Schwann cell markers in axonal neuropathy. Glia. 1996;17:294–306. doi: 10.1002/(SICI)1098-1136(199608)17:4<294::AID-GLIA4>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 27.Russell JW, Golovoy D, Vincent AM, et al. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. Faseb J. 2002;16:1738–1748. doi: 10.1096/fj.01-1027com. [DOI] [PubMed] [Google Scholar]
- 28.Russell JW, Sullivan KA, Windebank AJ, et al. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol Dis. 1999;6:347–363. doi: 10.1006/nbdi.1999.0254. [DOI] [PubMed] [Google Scholar]
- 29.Said G. Diabetic neuropathy--a review. Nat Clin Pract Neurol. 2007;3:331–340. doi: 10.1038/ncpneuro0504. [DOI] [PubMed] [Google Scholar]
- 30.Sasaki H, Schmelzer JD, Zollman PJ, et al. Neuropathology and blood flow of nerve, spinal roots and dorsal root ganglia in longstanding diabetic rats. Acta Neuropathologica. 1997;93:118–128. doi: 10.1007/s004010050592. [DOI] [PubMed] [Google Scholar]
- 31.Schmeichel AM, Schmelzer JD, Low PA. Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes. 2003;52:165–171. doi: 10.2337/diabetes.52.1.165. [DOI] [PubMed] [Google Scholar]
- 32.Sullivan KA, Hayes JM, Wiggin TD, et al. Mouse models of diabetic neuropathy. Neurobiol Dis. 2007;28:276–285. doi: 10.1016/j.nbd.2007.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sullivan KA, Lentz SI, Roberts JL, Jr, et al. Criteria for creating and assessing mouse models of diabetic neuropathy. Current drug targets. 2008;9:3–13. doi: 10.2174/138945008783431763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Laar VS, Berman SB. Mitochondrial dynamics in Parkinson’s disease. Exp Neurol. 2009;218:247–256. doi: 10.1016/j.expneurol.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vincent AM, Feldman EL. Can drug screening lead to candidate therapies for testing in diabetic neuropathy? Antioxidants & redox signaling. 2008;10:387–393. doi: 10.1089/ars.2007.1815. [DOI] [PubMed] [Google Scholar]
- 36.Vincent AM, Hayes JM, McLean LL, et al. Dyslipidemia-induced neuropathy in mice: the role of oxLDL/LOX-1. Diabetes. 2009;58:2376–2385. doi: 10.2337/db09-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vincent AM, Kato K, McLean LL, et al. Sensory neurons and schwann cells respond to oxidative stress by increasing antioxidant defense mechanisms. Antioxidants & redox signaling. 2009;11:425–438. doi: 10.1089/ars.2008.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vincent AM, McLean LL, Backus C, et al. Short-term hyperglycemia produces oxidative damage and apoptosis in neurons. FASEB J. 2005;19:638–640. doi: 10.1096/fj.04-2513fje. [DOI] [PubMed] [Google Scholar]
- 39.Vincent AM, Olzmann JA, Brownlee M, et al. Uncoupling proteins prevent glucose-induced neuronal oxidative stress and programmed cell death. Diabetes. 2004;53:726–734. doi: 10.2337/diabetes.53.3.726. [DOI] [PubMed] [Google Scholar]
- 40.Vincent AM, Perrone L, Sullivan KA, et al. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology. 2007;148:548–558. doi: 10.1210/en.2006-0073. [DOI] [PubMed] [Google Scholar]
- 41.Vincent AM, Russell JW, Sullivan KA, et al. SOD2 protects neurons from injury in cell culture and animal models of diabetic neuropathy. Exp Neurol. 2007;208:216–227. doi: 10.1016/j.expneurol.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vincent AM, Stevens MJ, Backus C, et al. Cell culture modeling to test therapies against hyperglycemia-mediated oxidative stress and injury. Antioxidants & redox signaling. 2005;7:1494–1506. doi: 10.1089/ars.2005.7.1494. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Su B, Lee HG, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yin W, Signore AP, Iwai M, et al. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke. 2008;39:3057–3063. doi: 10.1161/STROKEAHA.108.520114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6:657–663. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- 46.Yu T, Sheu SS, Robotham JL, et al. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovascular research. 2008;79:341–351. doi: 10.1093/cvr/cvn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan H, Gerencser AA, Liot G, et al. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ. 2007;14:462–471. doi: 10.1038/sj.cdd.4402046. [DOI] [PubMed] [Google Scholar]
- 48.Zhao C, Takita J, Tanaka Y, et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587–597. doi: 10.1016/s0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]
- 49.Zherebitskaya E, Akude E, Smith DR, et al. Development of selective axonopathy in adult sensory neurons isolated from diabetic rats: role of glucose-induced oxidative stress. Diabetes. 2009;58:1356–1364. doi: 10.2337/db09-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.