

Abstract
5-HT4 receptor agonists facilitate synaptic transmission in the enteric nervous system and these drugs are used to treat constipation. In the present study, we investigated the effects of the 5-HT4 receptor agonist, renzapride, on rundown and recovery of fEPSPs during and after trains of stimulation and on transmitter release from individual myenteric neuronal varicosities. Intracellular electrophysiological methods were used to record fEPSPs from neurons in longitudinal muscle myenteric plexus preparations of guinea pig ileum in vitro. During trains of supramaximal electrical stimulation (10 Hz, 2s), fEPSP amplitude declined (time constant = 0.6 ± 0.1 s) from 17 ± 2 mV to 0.7 ± 0.3 mV. Renzapride (0.1 μM) did not change the time constant for fEPSP rundown but it decreased the time constant for recovery of fEPSP amplitude after the stimulus train from 7 ± 2 s to 1.6 ± 0.2 s (P < 0.05). 5-HT (0.1 μM) also increased fEPSPs and facilitated recovery from rundown. The adenylate cyclase activator, forskolin (1 μM) mimicked the actions of renzapride and 5-HT while H-89, a protein kinase (PKA) inhibitor, blocked the effects of renzapride. We used nicotinic acetylcholine receptor containing outside-out patches obtained from myenteric neurons maintained in primary culture to detect acetylcholine release from single varicosities. Renzapride (0.1 μM) increased release probability by 2-fold. We conclude that 5-HT4 receptors activate the adenylyl cyclase-PKA pathway to increase acetylcholine release from single varicosities and to accelerate recovery from synaptic rundown. These responses may contribute to the prokinetic actions of 5-HT4 receptor agonists.
Keywords: fast synaptic transmission, enteric nervous system, electrophysiology, renzapride, single nicotinic acetylcholine receptor channels
Gastrointestinal (GI) function is controlled in part by the enteric nervous system (14). Two types of enteric neurons can be distinguished based on electrophysiological criteria (3,6,22,30). In acutely isolated longitudinal muscle myenteric plexus preparations (LMMP), single electrical stimuli applied to interganglionic connectives elicit fast excitatory postsynaptic potentials (fEPSP) in S neurons. Acetylcholine (ACh) acting at nicotinic acetylcholine receptors (nAChRs) is the principal mediator of fEPSPs in S neurons (17,20). The second type of neuron is the AH neuron and slow excitatory postsynaptic potentials are the primary mechanism of synaptic excitation in these neurons (17). S neurons are interneurons and motorneurons while AH neurons function as intrinsic primary afferent neurons (IPANs)(14).
IPANs are excited by mechanical and chemical stimulation of the mucosa and these stimuli can initiate reflex motor responses throughout the gut (15). In small segments of guinea pig ileum maintained in vitro, mechanical distortion of mucosal villi which mimics physiological stimulation in vivo evokes bursts of fEPSPs and action potentials in S neurons (4). These observations indicate that AH neurons responding to the mechanical stimulus release the mediators of the fEPSPs onto S neurons to produce bursts of fEPSPs. The observation that physiological stimuli can elicit bursts of fEPSPs is important as it has been shown that fEPSP amplitude declines during short trains of electrical stimulation and this synaptic rundown is likely due to depletion of a releasable pool of ACh (32). It was also shown that synapses in the myenteric plexus recover relatively slowly from rundown (32). Controlling the rate of fEPSP rundown and recovery may be a mechanism by which propulsive motility patterns are regulated so that content is propelled through the intestine in a controlled and timely manner. Alternatively, pathophysiological processes might accelerate rundown or delay recovery from rundown leading to impaired motility and delayed intestinal propulsion. Therefore, the mechanisms responsible for fEPSP rundown and recovery might be targets for drugs which can be used to treat motility disorders.
5-HT4 receptor agonists are know to increase ACh release from myenteric neurons (8,25,26) and fast synaptic transmission can be modulated by presynaptic 5-HT4 receptors as 5-HT4 receptor agonists facilitate fEPSPs (30,31). 5-HT4 receptors are G-protein coupled receptors that link to the stimulatory G-protein Gs and activation of the adenylate cyclase-protein kinase A (PKA) signaling pathway (1). Additional studies have shown that forskolin mimics 5-HT4 receptor-mediated facilitation of fEPSPs and PKA selective inhibitors block synaptic facilitation (18). 5-HT acting at 5-HT4 receptors also reduces the threshold for initiation of the peristaltic reflex (11,21). This effect provides the basis for the therapeutic effects of 5-HT4 receptor agonists like renzapride which can be used to treat constipation (7). However, the underlying molecular and cellular mechanisms by which 5-HT4 receptor agonists act on myenteric neurons have not been fully characterized. In addition, as physiological stimulation elicits bursts of fEPSPs, it would be informative to study 5-HT4-mediated effects on fEPSPs during trains of stimulation.
The present study tested the hypothesis that 5-HT4 receptor agonists delay rundown of fEPSPs and facilitate recovery from synaptic rundown during and after trains of stimulation, respectively. Furthermore, we tested the hypothesis that 5-HT4 receptor agonists would act on individual myenteric neuronal varicosities to increase transmitter release probability.
MATERIALS AND METHODS
Intracellular electrophysiological recordings from neurons in the longitudinal muscle myenteric plexus (LMMP) preparation
Animal use procedures were approved by the Institutional Animal Care and Use Committee at Michigan State University. Male guinea pigs (250–350 g) were anesthetized via short-term halothane inhalation, stunned and exsanguinated by severing the major neck blood vessels. A segment of ileum was harvested 15–20 cm proximal to the ileocecal junction and placed in oxygenated (95% O2 and 5% CO2) Krebs’ solution of the following composition (mM): NaCl, 117; KCl, 4.7; CaCl2, 2.5; MgCl2, 1.2; NaHCO3, 25; NaH2PO4, 1.2; glucose, 11. The Krebs’ solution contained nifedipine (1 μM) for blocking L-type calcium channels and scopolamine (1 μM) for blocking muscarinic cholinergic receptors to inhibit longitudinal muscle contractions. Details of the LMMP preparation used in these studies have been published previously (18,31,32).
Individual myenteric ganglia were visualized at 200x magnification using an inverted microscope (Olympus CK-2) with differential interface contrast optics. Intracellular recordings were obtained from single neurons using glass microelectrodes filled with 2 M KCl (tip resistance = ~ 100 MΩ). An amplifier (Axoclamp 2A, Molecular Devices, Sunnyvale, CA) was used to record membrane potential. When recording fEPSPs, the membrane potential was adjusted to −70 mV using constant DC current to avoid generating action potentials.
A Krebs’ solution filled glass pipette (tip diameter ~100 μm) was used as a focal stimulating electrode. A Ag+/AgCl wire was inserted into the pipette and a second Ag+/AgCl wire positioned in the recording chamber was used as the circuit ground. The stimulating wires were connected to a Grass Technologies (West Warwick, RI) S88 stimulator via a stimulus isolation unit (SIU5, Grass Technologies). The stimulating electrode was positioned on an interganglionic connective supplying the ganglia containing impaled neurons. Trains of electrical stimulation (10 Hz, for 2s, 0.5 ms pulse duration, 40–60 V) were used to study rundown of fEPSP amplitude. Slow excitatory postsynaptic potentials were not elicited in most S neurons studied. Neurons in which a stimulus train did evoke a sEPSP were not studied further. When studying drug effects on fEPSP rundown, trains of stimulation were applied at 3 minute intervals during drug application. We also studied drug effects on recovery from synaptic rundown. In these studies, a 10 Hz, 2s train of stimulation was used to cause synaptic rundown. Then a single stimulus was applied at 1 s intervals for up to 10 s until fEPSP amplitude had recovered to the amplitude of the first fEPSP in the preceding train. This protocol allowed measurements of the time constant for recovery in the absence and presence of drug treatment. Drugs were applied for 10 minutes before assessing drug effect on recovery.
Tissue culture
Tissues for establishing primary cultures of intestinal myenteric neurons were obtained from newborn guinea pigs (n = 5; ~36 h postnatal; ~70 g). Newborn guinea pigs were sacrificed by severing the major neck blood vessels and spinal cord after deep halothane anesthesia. The protocol for primary culture of guinea pig small intestinal myenteric neurons has been published previously (40,41,42).
Patch-clamp recording
Whole-cell and outside-out patch-clamp recordings were carried out at room temperature using patch pipettes with tip resistances of 5–10 MΩ. Recordings were obtained from myenteric neurons that had been maintained in primary culture for a minimum of 1 week. This allowed enough time for the neurons to develop axonal projections and to make synaptic contacts with other neurons. Whole cell and outside-out patch seal resistances were >5 GΩ. The tips of pipettes used for single-channel recordings in outside-out patches were coated with Sylgard (Dow Corning, Midland, MI) to reduce pipette capacitance. The composition of the pipette solution (millimolar) was: CsCl, 160; MgCl2, 2; EGTA, 1; HEPES, 10; ATP, 1 and GTP, 0.25. The pH and osmolarity were adjusted to 7.4 (with CsOH) and 315 mosmol/kg (with CsCl), respectively.
Recordings were obtained using an upright microscope (Nikon Diaphot, Mager Scientific, Dexter, MI) equipped with Hoffman modulation contrast optics. The recording pipette was positioned using a 3-dimensional mechanical micromanipulator (MX-2 Narishige Instruments, USA East Meadow, NY). After establishing the whole-cell recording configuration from an individual neuron, ACh (1 mM) was applied from a flow tube positioned near the neuron to verify that the neuron expressed nAChRs. Then the pipette was slowly withdrawn from the cell to establish the outside-out patch configuration. ACh was applied to the patch to verify that the patch contained nAChRs. The pipette containing the patch was then positioned within 2 μm of an individual myenteric neuronal varicosity under visual control (400 X magnification). The axon containing the targeted varicosity was stimulated with single stimuli (0.5 ms duration, 40 V, 0.5 Hz) using a bipolar stimulating electrode. The stimulating electrode was prepared using double-barreled glass capillary tubing. The individual barrels were drawn out to a final tip diameter of ~100 μm using a microelectrode puller. The focal stimulating electrode reduced the stimulus artifact when recording single channel currents caused by transmitter released from the targeted varicosity. Single channel activity was recorded during successive 200 ms long episodes triggered by the stimulus. Release was evoked using single stimuli applied at low frequency (0.1 Hz) as preliminary studies revealed that trains of stimuli applied at frequencies ≥2 Hz disrupted the seal between the patch membrane and the pipette. Recordings were obtained using an Axopatch 200B amplifier (Molecular Devices). Data were acquired using pClamp 9.0 software (Molecular Devices). Whole cell currents were sampled at 2 kHz and were filtered at 1 kHz (4-pole Bessel filter, Warner Instruments, Hamden, CT). Single channel currents were sampled at 5 kHz and filtered at 2 kHz. Data were stored on a computer hard drive for offline analysis. Single channel activity in outside-out patches was studied at a patch potential of −90 mV (inside negative). At this patch potential, the single channel current amplitude carried by nACRs is 1.9 pA (40,41). Therefore, single channel currents evoked by agonist or nerve stimulation were defined as those events crossing a 50% threshold (0.95 pA) from the mean baseline current level prior to ACh application. Channel activity during each 200 ms sweep was quantitated by measuring total charge transfer as the area under the single channel current curve.
Drug application
In the intracellular recordings made in acutely isolated preparations, receptor agonists, antagonists and enzyme activators and inhibitors were added into superfusing Krebs’ solution. Recordings were made 10–15 minutes after drug application. In patch-clamp recordings made in the cultured neurons, agonists were applied through gravity-driven flow tubes where flow was gated by computer controlled solenoids (General Valve Co., Fairfield, NJ). The tip of tube was positioned ~200 μm away from the target neuron or from the target patch.
Drugs
Renzapride was a gift from SmithKline Beecham Pharmaceuticals. All other drugs and chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
Statistics
All data are expressed as mean ± S.E.M. Two way analysis of variance was used to analyze drug effects on the extent of fEPSP rundown during trains of stimulation. Drug effects on the time course of rundown and recovery from synaptic rundown were determined by fitting rundown and recovery curves obtained in individual neurons to the following single exponential function:
where y0 was set to 1.0 (initial fEPSP amplitude during rundown or full recovery of fEPSP amplitude after rundown), A is fEPSP amplitude at 2 s (the end of the stimulus train) for rundown studies and 0 for recovery and t is the time constant for rundown or recovery of fEPSP amplitude. Differences in the average time constant for rundown or recovery were assessed using Student’s t-test for paired data; “n” indicates the number of neurons. Fischer’s Exact test was used to test for differences in the proportion of success vs. failures in evoking single channel activity following nerve stimulation in the outside-out patch measurements of ACh release. For all statistical analysis, P <0.05 was the level set for significant differences.
Results
Renzapride increased fEPSP amplitude during trains of stimulation
Previous studies showed that a train of nerve stimulation at 10 Hz evoked fEPSPs that declined in amplitude (rundown) to near 0 mV in ~ 1 s (32). Therefore, we used a 10 Hz stimulus train for 2 s to evoke fEPSPs in order to study synaptic rundown (Fig. 1A). The amplitude of the first fEPSP in the train was 17 ± 2 mV and it declined to 0.7 ± 0.3 mV (n = 9) at the 20th stimulus (Fig. 1B). The time constant for fEPSP rundown was 0.6 ± 0.1 s. In the presence of renzapride, the amplitude of the first fEPSP was 28 ± 3 mV and it declined to 6 ± 2 mV (n = 9) at the 20th stimulation (Fig. 1B); analysis of variance revealed that renzapride increased the amplitude of fEPSPs throughout the stimulus train (P < 0.05). Although renzapride increased fEPSP amplitude, it did not change the time constant of synaptic rundown (P > 0.05, Fig. 1C).
Fig. 1.

Renzapride increased fEPSPs elicited by trains of stimulation (10 Hz, 2s). A. Recordings of fEPSPs before and during renzapride. Fast EPSPs declined to 0 mV by the 12th stimulus (arrow) under control conditions. In the presence of renzapride, fEPSPs were larger and they did not decline to 0 mV by the last stimulus (arrow). B. Pooled data showing that renzapride increased fEPSPs throughout the stimulus train. Data are mean ± s.e.m. (n = 9). C. Renzapride did not alter the time constant (τ) of synaptic rundown.
Stimuli paired closely in time elicit paired pulse facilitation or depression where fEPSPs caused by the second stimulus are either larger (facilitation) or smaller (depression) than fEPSPs caused by the first stimulus. Paired pulse depression is a property of synapses in the myenteric plexus (32). This result was confirmed here where we found 17% depression in control solutions and 29% depression with renzapride (0.1 μM) present.
Renzapride accelerates recovery from synaptic rundown
Recovery from synaptic rundown was tested by applying 20 stimuli at 10 Hz and then waiting intervals of 1 to 12 s before a single test stimulus was used to elicit a fEPSP (fEPSPtest); the amplitude of fEPSPtest was expressed as a fraction of the amplitude of the first fEPSP in the stimulus train (fEPSPfirst). Recovery from rundown followed a single exponential time constant and renzapride (0.1 μM)(Fig. 2A) decreased the time constant for full recovery of fEPSP amplitude (Fig. 2B).
Fig. 2.

Renzapride accelerates recovery of fEPSP amplitude after rundown. A. Time course of recovery of fEPSP amplitude after rundown. Data points are mean ± s.e.m. (n = 10). Recovery is expressed as the ratio of fEPSPtest to fEPSPfirst where fEPSPfirst is the first fEPSP in the stimulus train and fEPSPtest is fEPSP amplitude at various times after the end of the stimulus train. Solid lines are monoexponential fits to the data points. B. Renzapride shortened the time constant (τ) for full recovery of fEPSP amplitude. Data are the mean ± s.e.m. of the recovery time constant determined from monoexponential fits of the recovery time course before and during renzapride (0.1 μM)(n = 10) application. *Indicates different from control (P < 0.05)
5-HT potentiates fEPSPs and accelerates recovery from rundown
5-HT1A receptors are localized to some myenteric nerve terminals (31) and activation of these receptors inhibits fEPSPs. We used NAN-190 (1 μM) to block presynaptic 5-HT1A receptors which allowed 5-HT to act at 5-HT4 receptors to facilitate fEPSPs; NAN-190 itself does not alter fEPSPs (31). Under these conditions, 5-HT (0.1 μM) increased fEPSP amplitude throughout the stimulus train but it did not alter the time constant of synaptic rundown (Fig. 3A)(control = 0.42 ± 0.04 s; 5-HT = 0.40 ± 0.03 s, n = 5). Paired pulse depression also occurred in the presence of 5-HT. Before 5-HT application, the amplitude of the first and second fEPSPs in the train were 17 ± 2 and 11 ± 1 mV respectively (35% depression); in the presence of 5-HT these values were 22 ± 1 and 15 ± 2 mV (32% depression). The time constant for complete recovery from synaptic rundown was reduced by 5-HT (control = 4.3 ± 0.5 s; 5-HT = 1.8 ± 0.3 s, n = 5, P < 0.05)(Fig. 3B).
Fig. 3.
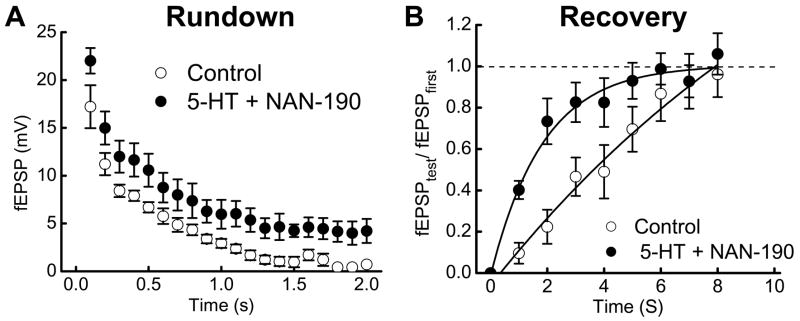
5-HT (1 μM) reduces synaptic rundown and accelerates recovery of fEPSP amplitude after rundown. A. 5-HT (with 1 μM NAN-190) increased fEPSP amplitude but it did not change the time constant of synaptic rundown. Data are mean ± s.e.m. (n = 5). B. The time course of recovery from synaptic rundown was shortened by 5-HT (with 1 μM NAN-190). Recovery is expressed as the ratio of fEPSPtest to fEPSPfirst where fEPSPfirst is the first fEPSP in the stimulus train and fEPSPtest is the amplitude of the fEPSP evoked at various times after the end of the stimulus train. Solid lines are monoexponential fits to the data points in each group.
Signaling pathway linked to acceleration of synaptic recovery
5-HT4 receptors couple to Gs type G-protein and activation of the adenylate cyclase-cyclic AMP (cAMP)-protein kinase A (PKA) pathway. To investigate if renzapride changes the time constants for rundown or full recovery from rundown by stimulating adenylate cyclase, forskolin (1 μM), an adenylate cyclase activator was added to the superfusing Krebs’ solution. Forskolin increased fEPSPs during a 10 Hz stimulus train (control = 17 ± 3 mV; forskolin = 25 ± 3 mV, n = 8, P < 0.01)(Fig. 4A). fEPSP amplitude declined with a similar time constant before and after forskolin treatment (Fig. 4A). Forskolin did not change paired-pulse depression (control: 1st = 17 ± 3, 2nd = 13 ± 2 mV, 24% depression; forskolin: 1st = 25 ± 3 mV 2nd = 20 ± 3 mV, 20% depression). Forskolin also reduced the recovery time constant (control = 3.7 ± 0.5 s; forskolin = 1.8 ± 0.2 s, n = 8, P < 0.01) (Fig. 4B).
Fig. 4.

Forskolin reduces synaptic rundown and accelerates rundown recovery after a stimulus train. A. Forskolin (1 μM) increased fEPSP amplitude but it did not change the time constant of rundown. Data are mean ± s.e.m. (n = 8). B. Forskolin decreased the time constant for recovery of fEPSP amplitude after synaptic rundown. Recovery is expressed as the ratio of fEPSPtest to fEPSPfirst where fEPSPfirst is the first fEPSP in the stimulus train and fEPSPtest is the amplitude of the fEPSP evoked at various times after the end of the stimulus train. Solid lines are monoexponential fits to the data points in each group. Data points are mean ± s.e.m. (n = 8).
H-89 (10 μM), a PKA blocker, was used to determine if activation of PKA is needed for renzapride-induced changes in the time constant for recovery of fEPSP amplitude. Previous work has shown that H-89 does not alter control fEPSP amplitude (18). The time constant for rundown of fEPSP amplitude during the stimulus train was not changed by renzapride or renzapride plus H-89. However, renzapride decreased the time constant for recovery from rundown and this effect was blocked by H-89 (Fig. 5B). The control time constant was 3.2 ± 0.05 s. In the presence of renzapride or renzapride and H-89 this value was 1.6 ± 0.2 (P < 0.05) and 3.4 ± 0.9 s respectively.
Fig. 5.

The PKA inhibitor, H-89, blocks the effects of renzapride on fEPSPs during and after trains of stimulation. A. Renzapride (0.1 μM) fEPSPs throughout the stimulus train (10 Hz) and H-89 (10 μM) blocked this effect. Renzapride or renzapride with H-89 did not alter the time constant of synaptic rundown. Data are mean ± s.e.m. (n = 3) B. Renzapride accelerated recovery after fEPSP rundown. Recovery at each time point was expressed as the ratio of fEPSPtest to fEPSPfirst where fEPSPfirst is the first fEPSP in the stimulus train and fEPSPtest is the amplitude of the fEPSP evoked at various times after the end of the stimulus train. The renzapride induced decrease in the recovery time constant was blocked by H-89. Solid lines are monoexponential fits to the data points.
Renzapride increases probability of ACh release from single varicosities
The above studies show that renzapride facilitated synaptic transmission by activating PKA. A mechanism underlying this response could be that renzapride increases ACh release probability from single varicosities. However, this hypothesis is difficult to test using the acutely isolated LMMP preparation where electrical stimulation of interganglionic connectives activates multiple axons and synaptic inputs to individual neurons. Therefore, it is not possible to make direct assessments of drug action on individual varicosities. To more directly study the effects of renzapride on ACh release from individual varicosities, we used primary cultures of myenteric neurons and outside-out patches containing nAChRs as ACh detectors. Outside-out patches were pulled from donor neurons and were tested for the presence of nAChRs by applying a saturating concentration of ACh (1 mM) onto the patch. At a patch potential of −90 mV, ACh evoked a peak current of 10–20 pA. Preliminary studies indicated that recordings of single channel activity were stable for up to 40 minutes following regular ACh applications (1 per minute) although typical experiments lasted ≤ 20 minutes. At −90 mV, the single channel current carried by nAChRs in myenteric neurons is 1.9 pA (40,41) indicating that outside-out patches contained 5 – 10 nAChR channels.
The patch was positioned within 2 μm of a varicosity and 10 stimuli (0.5 Hz) were used to generate an ensemble average of single channel currents caused by ACh release (Fig. 6A,B). Varicosities were identified as swellings that occurred periodically along axons (Fig. 6A′). When the detector patch was moved a few μm away from the target varicosity, it was no longer possible to detect single channel currents (Fig. 6A,B) suggesting that we were measuring transmitter release from a single varicosity and not from overflow from nearby varicosities. The single channel currents were due to nerve released ACh acting at nAChRs as these currents were blocked completely and reversibly by the nAChR antagonist hexamethonium (100 μM)(Fig. 6B). We further verified that the single channel currents were due to nAChR activation by measuring their reversal potential. This was accomplished by adjusting the patch potential to potentials between −90 and 50 mV and measuring the amplitude of the currents evoked by nerve stimulation when the patch was positioned near a varicosity. The current-voltage relationship was linear with a reversal potential near 0 mV (not shown). These data are similar to those reported previously for single channel currents carried by nAChRs in myenteric neurons (41,42).
Fig. 6.

Detection of ACh released from individual varicosities along the axons of myenteric neurons maintained in primary culture. A. Schematic diagram illustrating that outside out patches detect ACh release from a single varicosity (A′ shows a photomicrograph of a single axon and the arrows indicate two typical varicosities from which recordings like those shown in B and C were obtained, scale = 3 μm). “1” indicates position of an outside-out patch attached to a recording pipette near a single varicosity. A focal stimulating electrode was used to evoke action potentials in the axon containing the target varicosity. Single channel currents evoked by 10 stimuli were used to construct the ensemble average labeled “1” in panel B. Arrow in B indicates the stimulus artifact. When the patch-containing pipette was moved to the position labeled “2” it was not longer possible to evoke single channel activity in the outside-out patch as shown in the trace labeled “2” in B. C. Single channel activity evoked by axonal stimulation (arrows) is due to ACh release. Hexamethonium (100 μM) blocked single channel activity evoked by nerve stimulation. Traces are ensemble averages of response caused by axonal stimulation (arrows) before, during and after hexamethonium.
The data above demonstrate that the outside-out patches containing nAChRs could be used to evaluate the actions of renzapride on ACh release from individual varicosities. In these studies 20 stimuli were used to evoke ACh release in five experiments using 5 different culture preparations and single channel activity was assessed in each sweep. Under control conditions, 27 of 100 stimuli elicited detectable ACh release while renzapride (0.1 μM) increased the success rate to 52% (Fig. 7, Table 1). Renzapride also increased total channel activity measured as charge transfer or area under the single channel current-time plots (Table 1). These data show that renzapride did not increase the amount of ACh released per stimulus (Table 1).
Fig. 7.

Representative recordings of single channel activity in an outside-out patch obtained in the absence and presence of renzapride (0.1 μM). Traces on the left are 6 examples of individual responses to axonal stimulation (at arrow). Under control conditions axonal stimulation elicited single channel activity after 2 of 6 stimuli. Traces on the right show recordings from the same patch during renzapride application. Single channel activity was detected after 4 of 6 stimulations. It is important to note that the traces shown in this figure are individual sweeps recorded after single stimuli. They are qualitatively different from the ensemble averages of multiple sweeps shown in Fig. 6.
Table 1.
Analysis of detector patch data obtained from outside-out patches obtained from neurons in 5 different culture preparations. A patch was positioned within 2 μm of a varicosity and each patch was tested against 20 stimuli applied to the axon containing the test varicosity before and after renzapride (0.1 μM) application. Failures were stimuli that did not evoke single channel activity in the detector patch; successes evoked single channel activity. Renzapride increased the proportion of successful stimuli (#P < 0.001 vs. control, Fischer’s Exact test). Charge transfer was measured as the area under the single channel current time curve (200 ms). Renzapride increased total charge transfer because it increased the number of successful stimuli (* P < 0.05 vs control). Charge transfer per success was not changed by renzapride.
| Failures | Successes | Total charge transfer (pA·ms) | Charge transfer per success (pA·ms) | |
|---|---|---|---|---|
| Control | 73 | 27 | 64 ± 9 | 283 ± 47 |
| Renzapride | 48 | 52# | 216 ± 47* | 372 ± 52 |
Discussion
Renzapride increases ACh release probability
Fast EPSP amplitude declines during short trains of electrical stimulation. We showed previously that synaptic rundown is not due to presynaptic action potential failure or nAChR desensitization and that synaptic rundown may be due to depletion of a readily releasable pool of ACh-containing synaptic vesicles (32). In the present study, we tested the hypothesis that renzapride, would prevent synaptic rundown. Under control conditions, fEPSP amplitude declined to almost 0 mV by the end of the stimulus train. Although renzapride did not change the time constant of rundown, fEPSP amplitude declined to a steady state level above 0 mV suggesting that renzapride can help to maintain synaptic excitation during burst of action potentials. Paired pulse depression is a property of synapses with high release probability (43). In the myenteric plexus, release probability is high as paired-pulse depression occurs in response to two closely spaced stimuli as shown here and in previous work (32). Paired pulse depression persisted in the presence of renzapride suggesting that renzapride increases release probability perhaps by increasing the size of a pool of ACh-containing synaptic vesicles. Renzapride could increase the size of this pool by increasing the number of readily releasable vesicles in individual varicosities or by increasing the number of varicosities that release transmitter in response to a stimulus.
Transmitter release from autonomic nerve endings does not occur after every action potential (2,5,34) so renzapride could facilitate fEPSPs in the myenteric plexus by increasing the amount of ACh released from a single varicosity or by increasing release probability from silent varicosities. Conventional approaches for studying synaptic plasticity are based on quantal analysis (12) and determination of the parameters “m”, “n” and “p” where m is the number of vesicles (quanta) contributing to a fEPSP, n is the size of the postsynaptic response (miniature EPSP) caused by neurotransmitter released from a single vesicle and p is release probability. This analysis can not be applied to studies of synaptic transmission in the LMMP preparation due to technical limitations. Chief among these limitations is that miniature EPSPs have not been recorded from neurons in the myenteric plexus and therefore measurements of “n” required for quantal analysis are not possible.
In light of the limitations described above, we assessed release probability from individual varicosities using primary cultures of myenteric neurons and a detector patch technique (23). Studies using exogenously applied ACh, hexamethonium, and reversal potential measurements verified that the patches contained functional nAChRs. Previous work in myenteric neurons maintained in primary culture showed that most fEPSPs recorded from these neurons are mediated by ACh acting at nAChRs (41). Our data using the detector patch confirmed these previous findings. We also verified that release from a single autonomic varicosities is intermittent as release events were detected after only 27% of stimuli but renzapride doubled this success rate. These data suggest that, in the myenteric plexus, renzapride (and possibly other 5-HT4 receptor agonists) increases fEPSP amplitude by increasing the likelihood that varicosities will release ACh when an action potential invades those varicosities. Release probability at an individual release site (a varicosity in our studies) is determined by the number of vesicles available for release and the probability that each vesicle will be released in response to an action potential and calcium entry into the varicosity (27). Although the number of release events increased significantly in the presence of renzapride the average size of each event was not altered by renzapride. This conclusion is based on our observation that renzapride did not change mean charge transfer occurring during each release event. This result suggests that within each varicosity there is a fixed pool of readily releasable ACh-containing vesicles. Renzapride does not change the size of this pool but it does increase the likelihood that action potential-induced calcium entry will release some of this pool of ACh. Previous work at the crayfish neuromuscular junction showed that 5-HT enhances transmitter release via a mechanism downstream from calcium entry possibly by recruiting previously silent synapses (38). This scheme would account for the increased fEPSP amplitude occurring after a single stimulus in our studies. As more varicosities are releasing transmitter in the intact myenteric plexus, fEPSPs will be larger.
Renzapride and 5-HT accelerate recovery from synaptic rundown in the myenteric plexus
Short trains of stimulation evoke fEPSPs that rundown in amplitude and fEPSP amplitude recovers with a time constant of about 7 s. A novel effect reported here is that recovery from synaptic rundown is markedly accelerated by 5-HT (in the presence of a 5-HT1A receptor antagonist) and renzapride. Synaptic rundown caused by high frequency stimulation occurs throughout the nervous system and recovery from rundown is required for long term maintenance of synaptic transmission. Renzapride and 5-HT may shorten the time constant for recovery from synaptic rundown by accelerating replenishment of nerve terminal stores of transmitter
Renzapride activates the adenylate cyclase-cAMP-PKA signaling pathway
Our data indicate that renzapride and 5-HT increase ACh release probability and accelerate recovery of the availability of a readily releasable pool of ACh after depletion. 5-HT4 receptors are G-protein coupled receptors that link to activation of the adenylate cyclase-cAMP-PKA signaling pathway. Previous work showed that this pathway mediates the increase in fEPSP amplitude caused by 5-HT4 receptor agonists (18). We show here that this signaling pathway is also responsible for renzapride-induced acceleration of recovery of fEPSP amplitude after synaptic rundown as forskolin mimics and H-89 blocked the effects renzapride.
Our data suggest that there are at least two points where the adenylate cyclase-cAMP-PKA signaling pathway interacts with ACh release in myenteric nerve terminals. Firstly, release probability following a single action potential is increased as discussed above. As 5-HT-induced increases in release are likely to occur downstream of calcium entry (38), renzapride must act through a pathway that directly affects the release machinery (36). For example, PKA could increase coupling of the calcium sensing protein, synaptotagmin to the proteins involved in vesicle fusion with the plasma membrane (27). Secondly, the adenylate cyclase-cAMP-PKA signaling pathway accelerates recovery from synaptic rundown, so it might increase vesicle refilling, recycling and perhaps mobilization from a reserve pool. PKA phosphorylates several proteins involved in neurotransmitter release and synaptic vesicle trafficking (9,29,34,35,39). For example, synapsin is a vesicle phosphoprotein that anchors synaptic vesicles to the actin cytoskeleton (10,33). Phosphorylation of synapsin mobilizes synaptic vesicles from a reserve pool which may help to accelerate recovery from synaptic rundown (24,37).
Summary and conclusion
We found that 5-HT and the 5-HT4 receptor agonist renzapride facilitate fast synaptic excitation by increasing ACh release probability. 5-HT and renzapride also shorten the time to recovery of fEPSP amplitude after depletion of ACh stores by a train of high frequency stimulation. These effects are mediated by activation of the adenylate cyclase-cAMP-PKA pathway. Both effects will strengthen synaptic transmission in myenteric neuronal pathways controlling propulsive motility and these effects would contribute to the prokinetic actions of renzapride and possibly other 5-HT4 receptor agonists. Acceleration of recovery from synaptic rundown after a burst of synaptic activity would be particularly important as this would allow the neural circuits to maintain the high levels of synaptic activity required for prolonged periods of propulsive motility. It is possible that the pathophysiology responsible for impaired motility, as can occur in chronic constipation, might be due partly to disruption of the mechanisms responsible for release and replenishment of ACh-containing vesicles in nerve terminals in the myenteric plexus.
Acknowledgments
The authors would like to thank Smith Kline and Beecham Pharmaceuticals for the generous gift of renzapride.
Grants: This work was supported by NIH DK57039
References
- 1.Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083–1152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- 2.Bennett MR. Autonomic neuromuscular transmission at a varicosity. Prog Neurobiol. 1996;50:505–532. doi: 10.1016/s0301-0082(96)00039-1. [DOI] [PubMed] [Google Scholar]
- 3.Bornstein JC, Furness JB, Kunze WAA. Electrophysiological characterization of myenteric neurons: how do classification schemes relate? J Auton Nerv Syst. 1994;48:1–15. doi: 10.1016/0165-1838(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 4.Bornstein JC, Furness JB, Smith TK, Trussell DC. Synaptic responses evoked by mechanical stimulation of the mucosa in morphologically characterized myenteric neurons of the guinea-pig ileum. J Neurosci. 1991;11:505–518. doi: 10.1523/JNEUROSCI.11-02-00505.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brain KL, Jackson VM, Trout SJ, Cunnane TC. Intermittent ATP release from nerve terminals elicits focal smooth muscle Ca2+ transients in mouse vas deferens. J Physiol. 2002;541:849–862. doi: 10.1113/jphysiol.2002.019612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brookes SJ. Classes of enteric nerve cells in the guinea-pig small intestine. Anat Rec. 2001;262:58–70. doi: 10.1002/1097-0185(20010101)262:1<58::AID-AR1011>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 7.Camilleri M, McKinzie S, Fox J, Foxx-Orenstein A, Burton D, Thomforde G, Baxter K, Zinsmeister AR. Effect of renzapride on transit in constipation-predominant irritable bowel syndrome. Clin Gastroenterol Hepatol. 2004;2:895–904. doi: 10.1016/s1542-3565(04)00391-x. [DOI] [PubMed] [Google Scholar]
- 8.Cellek S, John AK, Thangiah R, Dass NB, Bassil AK, Jarvie EM, Lalude O, Vivekanandan S, Sanger GJ. 5-HT4 receptor agonists enhance both cholinergic and nitrergic activities in human isolated colon circular muscle. Neurogastroenterol Motil. 2006;18:853–861. doi: 10.1111/j.1365-2982.2006.00810.x. [DOI] [PubMed] [Google Scholar]
- 9.Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/s0896-6273(03)00151-x. [DOI] [PubMed] [Google Scholar]
- 11.Craig DA, Clarke DE. Peristalsis evoked by 5-HT and renzapride: evidence for putative 5-HT4 receptor activation. Br J Pharmacol. 1991;102:563–564. doi: 10.1111/j.1476-5381.1991.tb12211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.del Castillo J, Katz B. Quantal components of the end-plate potential. J Physiol. 1954;124:560–73. doi: 10.1113/jphysiol.1954.sp005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang X, Liu S, Wang XY, Gao N, Hu HZ, Wang GD, Cook CH, Needleman BJ, Mikami DJ, Xia Y, Fei GJ, Hicks GA, Wood JD. Neurogastroenterology of tegaserod (HTF 919) in the submucosal division of the guinea-pig and human enteric nervous system. Neurogastroenterol Motil. 2008;20:80–93. doi: 10.1111/j.1365-2982.2007.00983.x. [DOI] [PubMed] [Google Scholar]
- 14.Furness JB. The enteric nervous system. Wiley-Blackwell Publishing; 2006. [Google Scholar]
- 15.Furness JB, Jones C, Nurgali K, Clerc N. Intrinsic primary afferent neurons and nerve circuits within the intestine. Prog Neurobiol. 2004;72:143–164. doi: 10.1016/j.pneurobio.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 16.Galli T, Haucke V. Cycling of synaptic vesicles: how far? How fast! Sci STKE. 2004;2004(264):re19. doi: 10.1126/stke.2642004re19. [DOI] [PubMed] [Google Scholar]
- 17.Galligan JJ. Pharmacology of synaptic transmission in the enteric nervous system. Curr Opin Pharmacol. 2002;2:623–629. doi: 10.1016/s1471-4892(02)00212-6. [DOI] [PubMed] [Google Scholar]
- 18.Galligan JJ, Pan H, Messori E. Signalling mechanism coupled to 5-hydroxytryptamine4 receptor-mediated facilitation of fast synaptic transmission in the guinea-pig ileum myenteric plexus. Neurogastroenterol Motil. 2003;15:523–529. doi: 10.1046/j.1365-2982.2003.00428.x. [DOI] [PubMed] [Google Scholar]
- 19.Galligan JJ, Vanner S. Basic and clinical pharmacology of new motility promoting agents. Neurogastroenterol Motil. 2005;17:643–653. doi: 10.1111/j.1365-2982.2005.00675.x. [DOI] [PubMed] [Google Scholar]
- 20.Galligan JJ, LePard KJ, Schneider DA, Zhou X. Multiple mechanisms of fast excitatory synaptic transmission in the enteric nervous system. J Auton Nerv Syst. 2001;81:97–103. doi: 10.1016/s0165-1838(00)00130-2. [DOI] [PubMed] [Google Scholar]
- 21.Grider JR, Foxx-Orenstein AE, Jin JG. 5-Hydroxytryptamine4 receptor agonists initiate the peristaltic reflex in human, rat, and guinea pig intestine. Gastroenterology. 1998;115:370–380. doi: 10.1016/s0016-5085(98)70203-3. [DOI] [PubMed] [Google Scholar]
- 22.Hirst GDS, Holman ME, Spence I. Two types of neurons in the myenteric plexus of duodenum in the guinea pig. J Physiol. 1974;236:303–326. doi: 10.1113/jphysiol.1974.sp010436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hume RL, Role LW, Fischbach GD. Acetylcholine release from growth cones detected with patches of acetylcholine receptor-rich membranes. Nature. 1983;305:632–634. doi: 10.1038/305632a0. [DOI] [PubMed] [Google Scholar]
- 24.Hosaka M, Hammer RE, Sudhof TC. A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron. 1999;24:377–387. doi: 10.1016/s0896-6273(00)80851-x. [DOI] [PubMed] [Google Scholar]
- 25.Kilbinger H, Gebauer A, Haas J, Ladinsky H, Rizzi CA. Benzimidazolones and renzapride facilitate acetylcholine release from guinea-pig myenteric plexus via 5-HT4 receptors. Naunyn Schmiedebergs Arch Pharmacol. 1995;351:229–236. doi: 10.1007/BF00233241. [DOI] [PubMed] [Google Scholar]
- 26.Kilbinger H, Wolf D. Effects of 5HT4 receptor stimulation on basal and elecrically evoked release of acetylchoine from guinea pig myenteric plexus. Naunyn-Schmiedeberg’s Arch Pharmacol. 1992;345:270–275. doi: 10.1007/BF00168686. [DOI] [PubMed] [Google Scholar]
- 27.Leenders AG, Sheng ZH. Modulation of neurotransmitter release by the second messenger-activated protein kinases: implications for presynaptic plasticity. Pharmacol Ther. 2005;105:69–84. doi: 10.1016/j.pharmthera.2004.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu M, Geddis MS, Wen Y, Setlik W, Gershon MD. Expression and function of 5-HT4 receptors in the mouse enteric nervous system. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1148–1163. doi: 10.1152/ajpgi.00245.2005. [DOI] [PubMed] [Google Scholar]
- 29.Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, Sorensen JB. Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron. 2004;41:311–313. doi: 10.1016/s0896-6273(04)00038-8. [DOI] [PubMed] [Google Scholar]
- 30.Nishi S, North RA. Intracellular recording from the myenteric plexus of the guinea pig ileum. J Physiol. 1973;231:471–491. doi: 10.1113/jphysiol.1973.sp010244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pan H, Galligan JJ. 5-HT1A and 5-HT4 receptors mediate inhibition and facilitation of fast synaptic transmission in enteric neurons. Am J Physiol Gastrointest Liver Physiol. 1994;266:230–8. doi: 10.1152/ajpgi.1994.266.2.G230. [DOI] [PubMed] [Google Scholar]
- 32.Ren J, Galligan JJ. Dynamics of fast synaptic excitation during trains of stimulation in myenteric neurons of guinea-pig ileum. Auton Neurosci. 2005;117:67–78. doi: 10.1016/j.autneu.2004.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sudhof TC, Czernik AJ, Kao HT, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS, De Camilli P. Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science. 1989;245:1474–1480. doi: 10.1126/science.2506642. [DOI] [PubMed] [Google Scholar]
- 34.Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 35.Thakur P, Stevens DR, Sheng ZH, Rettig J. Effects of PKA-mediated phosphorylation of Snapin on synaptic transmission in cultured hippocampal neurons. J Neurosci. 2004;24:6476–6481. doi: 10.1523/JNEUROSCI.0590-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trudeau LE, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- 37.Turner KM, Burgoyne RD, Morgan A. Protein phosphorylation and the regulation of synaptic membrane traffic. Trends Neurosci. 1999;22:459–464. doi: 10.1016/s0166-2236(99)01436-8. [DOI] [PubMed] [Google Scholar]
- 38.Wang C, Zucker RS. Regulation of synaptic vesicle recycling by calcium and serotonin. Neuron. 1998;21:155–167. doi: 10.1016/s0896-6273(00)80523-1. [DOI] [PubMed] [Google Scholar]
- 39.Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- 40.Zhou X, Galligan JJ. Non-additive interaction between nicotinic cholinergic and P2X purine receptors in guinea-pig enteric neurons in culture. J Physiol. 1998;513:685–697. doi: 10.1111/j.1469-7793.1998.685ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou X, Galligan JJ. P2X purinoceptors in cultured myenteric neurons of guinea-pig small intestine. J Physiol. 1996;496:719–729. doi: 10.1113/jphysiol.1996.sp021722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou X, Ren J, Brown E, Schneider D, Caraballo-Lopez Y, Galligan JJ. Pharmacological properties of nicotinic acetylcholine receptors expressed by guinea pig small intestinal myenteric neurons. J Pharmacol Exp Ther. 2002;302:889–97. doi: 10.1124/jpet.102.033548. [DOI] [PubMed] [Google Scholar]
- 43.Zucker RS, Regehr WG. Short-term synaptic plasticity. Ann Rev Physiol. 64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]