

Abstract
Thirty years after the discovery of HIV-1, the early transmission, dissemination, and establishment of the virus in human populations remain unclear. Using statistical approaches applied to HIV-1 sequence data from central Africa, we show that from the 1920s Kinshasa (in what is now the Democratic Republic of Congo) was the focus of early transmission and the source of pre-1960 pandemic viruses elsewhere. Location and dating estimates were validated using the earliest HIV-1 archival sample, also from Kinshasa. The epidemic histories of HIV-1 group M and nonpandemic group O were similar until ~1960, after which group M underwent an epidemiological transition and outpaced regional population growth. Our results reconstruct the early dynamics of HIV-1 and emphasize the role of social changes and transport networks in the establishment of this virus in human populations.
AIDS is one of the most devastating infectious diseases in human history, and its cause, HIV, has been responsible for nearly 75 million infections (1). Shortly after the first reports of AIDS in the United States in 1981 (2) and the isolation of HIV-1 2 years later (3, 4), the disease was discovered to be established in heterosexual populations of central and east Africa (5, 6), suggesting a much older—and, to that point, hidden—history of the pandemic in Africa.
Surveys of African apes identified chimpanzee [Pan troglodytes troglodytes (Ptt)] populations in southern Cameroon harboring simian immunodeficiency viruses (SIVs) most closely related to the pandemic lineage of HIV-1, group M (7, 8). HIV-1 group M comprises numerous genetically distinct virus subtypes (A, B, C, etc.) and recombinant forms. Although only group M viruses established pandemic spread, other separate cross-species transmissions of SIV to humans in the Congo River basin led to nonpandemic transmission of HIV-1 groups O, N, and P, which are still largely confined to Cameroon and its surrounding countries (9–11).
By the end of 1980s, the genetic diversity of HIV-1 group M in the Democratic Republic of Congo (DRC), then known as Zaire, was greater and more complex than that in the rest of the world (12, 13). HIV-1 strains collected in central Africa form phylogenetic outgroups to the subtypes of group M (14), suggesting that the latter are the products of incomplete sampling and exportation events (15). Two HIV-1 sequences substantially predate the discovery of AIDS and were retrospectively recovered from blood and tissue samples (16, 17) collected in Kinshasa, capital of the DRC, in 1959–1960. Other countries in the Congo River basin—notably the Republic of Congo (RC) (18, 19), as well as Cameroon and Gabon (20, 21)—also harbor very high diversities of HIV-1 comparable to that observed in the DRC. Nevertheless, hypotheses concerning the geographic source of the pandemic and its early dissemination in humans remain controversial and have yet to be formally tested.
Although critical to our understanding of the establishment and evolution of human pathogens, a substantial period of HIV pandemic history is unclear. Despite our increased understanding of the cross-species transmissions of SIV to humans, we know very little about the early dissemination routes of HIV-1 and how group M became established as a continental epidemic in the decades immediately following its spillover from chimpanzees. Further, the genesis of major HIV-1 lineages, such as subtypes B and C, remains obscure. The lack of direct evidence about the early transmission of HIV-1 group M has led to several competing hypotheses for the emergence of AIDS (22). The two most widely accepted hypotheses for the establishment of the group M pandemic argue that urbanization and/or viral genetic factors, such as adaptation of the HIV-1 vpu gene (23), were decisive in the epidemiological success of group M compared with other SIV cross-species transmissions, such as group O, that did not cause pandemics.
By probing information contained in sampled viral sequences, evolutionary analyses can reveal the epidemic history of fast-evolving pathogens (24). Molecular clocks agree that a common ancestor of HIV-1 group M existed in the first half of the 20th century (16, 25–27), and models that link viral phylogenies to past transmission rates have been used to infer the epidemic history of group M (16, 27). However, several aspects of the evolutionary models used remain vulnerable to criticism (28), and the impact of recombination [a driver of HIV-1 genetic diversity (29)] on estimates of the time scale of group M spread has not been fully addressed. Using alternative methods of evolutionary analysis applied to a compilation of HIV-1 sequences from central Africa, we have uncovered the dynamics of the establishment of HIV-1 in humans, which explain how just one of many cross-species transmission events gave rise to the global pandemic we see today.
The spatiotemporal origins of pandemic HIV-1
A preliminary analysis of all available env C2V3 HIV-1 sequence data (30) from countries in the Congo River basin, as well as the range of Ptt chimpanzees, indicated that group M spread from the DRC to other countries (figs. S1 and S3); hence, we focused on this area in subsequent analyses. A very high genetic diversity of HIV-1 has been reported, not only in Kinshasa and the north and south of the DRC (12, 13, 31, 32), but also in Brazzaville in the RC and, to a lesser extent, in the Mayombe area of RC near Pointe-Noire, all of which have been suggested as potential source locations of the pandemic (22, 33, 34). We therefore performed phylogeographic analyses of viruses collected in both the DRC and RC (table S1) and compared sequence sampling locations with phylogenetic history to formally test hypotheses concerning the location of ancestral viral lineages (30). Our analyses robustly place the spatial origin of the HIV-1 group M pandemic in Kinshasa [posterior probability (PP) = 0.99] (Figs. 1 and 2). In line with previous approaches, we estimated the time of the most recent common ancestor (TMRCA) of group M to be around 1920 [95% Bayesian credible interval (BCI): 1909–1930] (Figs. 1 and 3A). Although we focus on estimates under the best-fitting demographic model for data set A, which reduces by 39% the BCIs of previous estimates (16, 25–27), the epidemic time scale we infer is robust to the evolutionary models chosen (fig. S8) and the data sets analyzed (fig. S9). Because sequence fragments for the earliest HIV-1 sample [ZR59, sampled in 1959 in Kinshasa (17)] partly overlap with the C2V3 region analyzed here, we included ZR59 as an internal control and estimated both the age and location of this strain. The estimate of the age of ZR59 is centered on 1958 (95% BCI: 1946–1970) (Fig. 3A), with little variation across data sets (fig. S9). In Fig. 3A, the posterior probability distribution of this age estimate is stratified according to the estimated location of ZR59; crucially, Kinshasa receives the highest support as the estimated location (PP = 0.81). The decisive support for Kinshasa as the epicenter of pandemic group M is robust to differences in spatial model specification and sampling heterogeneity (30) (tables S2 to S5 and fig. S4). To further test robustness, we deliberately excluded Kinshasa sequences sampled at the earliest time point (1985, representing 51% of strains for this location), which resulted in a root location at Brazzaville (PP = 0.97), located just 6 km from Kinshasa across the Congo River.
Fig. 1. Time-scaled phylogeographic history of pandemic HIV-1.
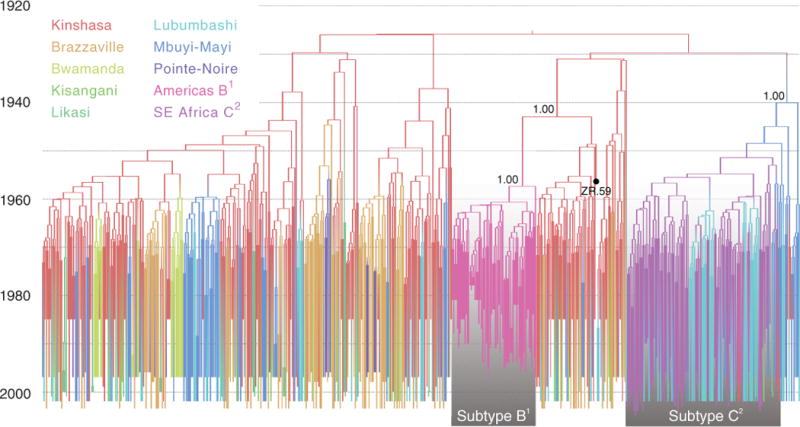
Branch colors represent the most probable location of the parental node of each branch. The respective colors for each location are shown in the upper left. U.S./Haiti/Trinidad subtype B and southeast African subtype C lineages are highlighted by boxes with a gradient shading, along with the posterior probabilities for their ancestral nodes. The tip for the ZR59 sequence is highlighted with a black circle.
Fig. 2. Spatial dynamics of HIV-1 group M spread.
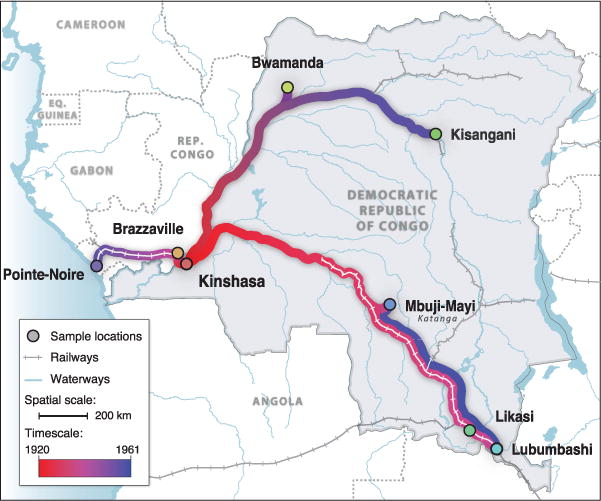
Circles represent sampled locations and are colored according to the estimated time of introduction of HIV-1 group M from Kinshasa. Strongly supported rates of virus spatial movement (table S6) are projected along the transportation network for the DRC (railways and waterways), which was fully operational until 1960 (38). Gradient colors depict the time scale of spatial movements (bottom left).
Fig. 3. HIV-1 group M establishment, human mobility, and urbanization.
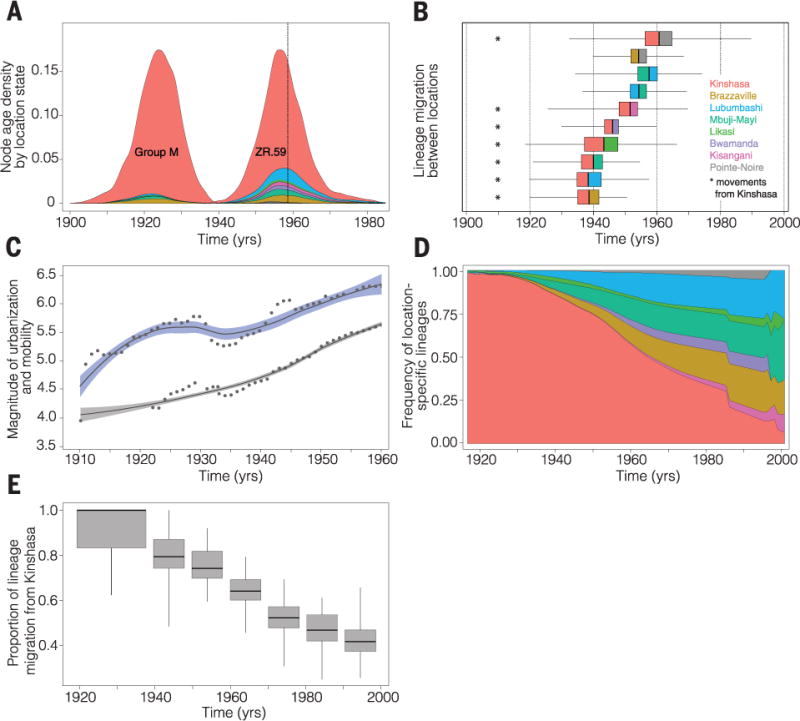
(A) Posterior probability densities for the estimated age of the most recent common ancestor of HIV-1 group M and of the archival ZR59 sequence. Distributions are stratified according to the estimated locations of both nodes (location-specific colors correspond to those in Fig. 1). A vertical dotted line shows the known sampling date for ZR59 (1959). (B) Earliest dates of lineage migration for significant routes of group M dispersal in the DRC and RC (table S6). Each box-and-whisker plot represents movement between a pair of locations. The vertical bar in each box represents the earliest date of movement, and colors to the left and right of this bar represent the seeding and receiving locations, respectively. The width of the boxes and the whiskers represents the 25-to-75% and 2.5-to-97.5% percentiles, respectively, of the estimated date of earliest movement. (C) Locally weighted regression curves for the official total number of passengers (log10) transported along railways (95% of journeys) and waterways (5%) in the DRC (38) (blue) and for the human population size (log10) of Kinshasa (gray) between 1900 and 1960 (43), after which reliable transportation data are unavailable. Dots show regression data points. (D) Estimated frequency of group M lineages at each location in the DRC and RC through time. (E) Estimated proportion of all migration events that began in Kinshasa until 1940 and, per decade, between 1940 and 2000. [Box-and-whisker widths are defined in (B).] This percentage drops to 43.5% between 1990 and 2000 (fig. S6 shows the estimated proportion of movement events originating from each location).
Our estimated location of pandemic origin explains the observation that Kinshasa exhibits more contemporary HIV-1 genetic diversity than anywhere else (12, 13). It clarifies why the oldest known HIV-1 sequences were sourced from this city (16, 17) and why several early cases indicative of AIDS are linked to Kinshasa (35). The cross-species transmission of SIV to humans predates the group M common ancestor (36) and probably occurred in southeast Cameroon, where the chimpanzees with SIVcpz strains most similar to group M have been identified (7, 8). After localized transmission, presumably resulting from the hunting of primates, the virus probably traveled via ferry along the Sangha River system to Kinshasa (37). During the period of German colonization of Cameroon (1884–1916), fluvial connections between southern Cameroon and Kinshasa were frequent due to the exploitation of rubber and ivory (36).
Early spatial expansion from Kinshasa
With the geographic origins of pandemic group M clear, we next sought to investigate its spread from Kinshasa to the rest of Africa. To identify statistically significant epidemiological links among locations and quantify virus exchange, we estimated rates of viral lineage migration using an established “robust counting” approach (30). In addition to identifying Kinshasa as the location of the group M common ancestor, our analyses showed a dynamic pattern of HIV-1 movement in the DRC and RC, dominated initially by viral dispersal away from Kinshasa and toward other population centers (Fig. 2). Overall, 57% (95% BCI: 48 to 65%) of all viral lineage movements originated from Kinshasa. Of these, one-third were directed to the neighboring city of Brazzaville (fig. S5), explaining the high genetic diversity of group M reported there (18, 19). Further, our results revealed that the earliest introductions of HIV-1 to Brazzaville occurred by 1937 (95% BCI: 1920–1953) (Fig. 3B). We note that these estimates pertain to viral lineages that survived to be sampled in each location; thus, HIV-1 may have been introduced earlier (e.g., to Brazzaville) but without successful onward transmission. Historical transportation data from the DRC during 1900–1960 (38) (Fig. 3C) suggests that viral lineages in migrant populations living in or around Kinshasa would have had many opportunities for introduction to DRC regions connected to other population centers in central Africa (39).
Our genetic analyses indicated that the virus reached the southern DRC locations Lubumbashi and Mbuji-Mayi by ~1937 (95% BCI: 1919–1957) and ~1939 (95% BCI: 1922–1954), respectively (Fig. 3B). These two locations received ~41% of viral lineage export from Kinshasa (fig. S5). Even if we consider our most conservative dating estimates, our results indicate that group M viruses were circulating in Brazzaville and southern DRC before the date of the earliest known HIV-1 samples (1959–1960), and therefore, similar samples may exist in historical collections in locations outside Kinshasa. However, it took another decade for pandemic HIV-1 strains to seed central and northern DRC locations, reaching Bwamanda by 1946 (95% BCI: 1929–1959) (Fig. 3C) and Kisangani by 1953 (95% BCI: 1926–1970). The comparatively late arrival of pandemic HIV-1 in northeastern DRC is consistent with historical records indicating that only 5% of human journeys within the DRC occurred on the fluvial network connecting Kinshasa and Kisangani (38).
Group M arrived first at the three largest population centers—Brazzaville, Lubumbashi, and Mbuji-Mayi (40, 41)—that were better connected to Kinshasa (38), indicating a critical role for mobility networks in the early spread and establishment of HIV-1 from its epicenter (42). Within the DRC, the majority of journeys took place along the railway network, which was used by >300,000 passengers per year in 1922, peaking at >1 million annual passengers in 1948 (Fig. 3C). Mbuji-Mayi, the world’s second largest producer of industrial diamonds, and Lubumbashi, also a mining city and the second largest of the DRC, were connected via the most active section of the DRC railway network (38). Although most viral movement consisted of lineage dissemination away from Kinshasa (Fig. 3D), we also identified one instance of significant bidirectional virus exchange, between Mbuji-Mayi and Lubumbashi (table S6), with the two earliest migrations between them dating back to 1957 (95% BCI: 1934–1974) and 1954 (95% BCI: 1936–1968), respectively (Fig. 3B). To further quantify changes in HIV-1 dissemination through time, we estimated, for each decade, the relative proportion of viral lineage movements that began in Kinshasa. We found a significant decline (8% per year) in this measure (Fig. 3, D and E). By the mid 1980s, approximately half of all dispersal events were seeded from secondary locations (Fig. 3E), thereby establishing the geographically heterogeneous distribution of HIV subtypes observed across eastern and southern Africa (39).
Divergent epidemic dynamics of HIV-1 groups M and O
Whereas our data show that HIV-1 group M was already established in several DRC locations before 1960, group O remained nonpandemic and largely confined to west-central Africa. To investigate how the spatial expansion of HIV-1 in the DRC relates to its epidemic history, we estimated past growth rates for HIV-1 groups M and O in central Africa using methods based on coalescent theory (30), a population genetic model that links phylogenetic tree shape to the demographic history of the sampled population (24).
Our analyses provide an estimate of the effective number of HIV-1 infections through time for HIV-1 groups M and O. Between 1920 and 1960, group M underwent an early phase of relatively slow exponential growth (Fig. 4). Using a two-phase exponential-logistic model of population growth (30), we estimate the exponential growth rate of group M during the early phase to be 0.1 year−1 (95% BCI: 0.064 to 0.15 year−1), close to the population growth rate of Kinshasa (0.081 year−1, SD: 0.00077) (Fig. 4) (43). HIV-1 was largely restricted to Kinshasa for most of this period (Figs. 1 and 3D). For group O, we estimate slightly slower exponential growth rates of 0.071 year−1 (95% BCI: 0.046 to 0.099 year−1), which may reflect lower infectivity caused by the greater susceptibility of group O to the antiviral host protein tetherin (23). This suggests that genetic factors specific to the SIV ancestors of HIV-1 that infected chimpanzees and gorillas may have been most important in the period immediately after cross-species transmission.
Fig. 4. Population dynamics of HIV-1 groups M and O.
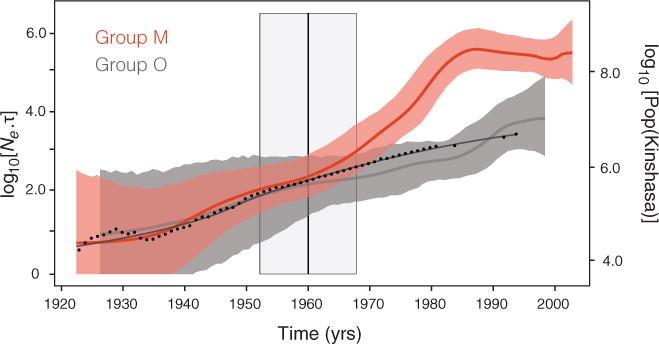
Bayesian skygrid estimates of past population dynamics for group M (red) and group O (gray) (30). The left y axis represents the effective number of infections (Ne) multiplied by the mean viral generation time (τ). Group O dynamics were obtained using the same best-fitting demographic model as for group M (table S7), applied to an alignment of 50 concatenated gag, pol, and env sequences sampled between 1987 and 1999 from west-central African patients (62). The superimposed black curve represents a locally weighted regression of human population growth in Kinshasa between 1920 and 1994 (43, 64). Dots show regression data points. The vertical line at 1960 corresponds to the estimated time at which group M transitioned from slow to faster exponential growth. The 95% BCI for this estimate is highlighted in gray.
However, around 1960 (95% BCI: 1952–1968) group M transitioned to a second, faster phase of exponential growth (Fig. 4; see also fig. S8, which demonstrates robustness of the estimated growth parameters to the molecular clock model used). During this second period, group M growth rates more than doubled to 0.27 year−1 (95% BCI: 0.20 to 0.33 year−1), substantially outpacing the concurrent rate of Kinshasa population growth. Our results thus call into question the role of human population expansion in HIV-1 emergence (33). Crucially, the estimated time of this transition also marks the time at which the epidemic histories of groups M and O diverge. Although the TMRCA of group O (1926; 95% BCI: 1903–1948) is similar to that of group M and both grew at similar rates until ~1960, group O exhibits no subsequent increase in growth rate and remains largely confined to Cameroon and surrounding countries (10). Whereas virus-specific factors may explain the differences in early-phase growth rates, invoking this hypothesis to explain the group M transition between 1952 and 1968 would require the implausible proposition that viral accessory genes evolved concurrently and convergently in multiple lineages already present in different central African locations (Figs. 1 and 3B and fig. S3). Lastly, Fig. 4 indicates a stabilization in epidemic growth over the past two decades. Although the methods used here may sometimes underestimate growth rates near the present (44), this slowdown agrees with reports of relatively stable HIV prevalence in the DRC from 1976 to 1997 (45, 46).
The observation that HIV-1 group M growth rates nearly tripled around 1960 is a consequence of a relative increase (at that time) in the rate at which sampled viral lineages join together, or coalesce, as time proceeds backward toward the phylogeny root. Theory suggests three non–mutually exclusive explanations for this change in the coalescence rate. (i) Group M viruses expanded geographically and established new subpopulations around 1960 (47), resulting from the dispersal of sampled lineages from Kinshasa (Fig. 3). (ii) Group M transmission rates increased, in Kinshasa or elsewhere, such that the number of infections was substantially lower before ~1960. (iii) Onward transmission per capita was more homogeneous and, on average, less frequent after the estimated transition (95% BCI: 1952–1968). This counterintuitive result arises because the lineage coalescence rate will be faster when only a small fraction of infections generate the majority of new cases and when the viral generation time is reduced (48). To discriminate among these hypotheses, we first reconstructed the epidemic history of lineages that maintained ancestry within Kinshasa (i.e., 84 taxa in Fig. 1 that have exclusively red branches in their ancestry that were sampled between 1985 and 2002; PP cut-off > 0.80). Because this procedure recovers a similar epidemic profile (fig. S7) to that in Fig. 4, it seems unlikely that geographic expansion directly drove the change in coalescence rate, despite it being a necessary condition for the international establishment of the pandemic.
Explanations (ii) and (iii) are compatible with an early establishment of group M in high-risk groups of small size—for example, commercial sex workers with higher rates of partner exchange and/or exposure to contaminated injections—before later spreading to the larger, general DRC population from the 1950s onward. Specifically, the transition to faster exponential growth (Fig. 4) agrees with available public health data (34) and the hypothesis that transmission rates of group M increased as a result of the administration of unsterilized injections at sexually transmitted disease clinics in the 1950s and/or subsequent changes in the nature of commercial sex work in Kinshasa from the early 1960s, which led to increased client numbers (34). The idea that early HIV spread included an iatrogenic component is supported by data from other blood-borne viruses. A study of hepatitis C virus (HCV) in the DRC showed that it exhibits an age cohort effect (49), and an epidemic of hepatitis [presumably hepatitis B virus (HBV)] was reported in Kinshasa in 1951–1952 (50). Both events indicate an important role for past iatrogenic transmission. Additional genetic data may allow the past dynamics of HCV, HBV, and other viruses in the DRC to also be reconstructed.
It seems less likely that genital ulcer disease (GUD) or circumcision practices played a role in the group M transition. In 1920, GUD incidence was ~10% for primary and secondary syphilis and chancroid but dropped by a magnitude of 1.5 to 2.5 until 1960 (51). It is conceivable that post-independence changes in sexual behavior could have increased GUD incidence, but unfortunately this is difficult to assess, as postcolonial medical records are scarce or nonexistent. Further, a lack of circumcision was unlikely to have played a role, as nearly all males in Kinshasa were circumcised by 1960 (51).
The emergence of HIV-1 subtypes
Unlike HIV-1 strains from outside Africa, group M viruses from the DRC are not structured into clearly distinct subtypes (14). The former are exemplified by subtype B, which forms a distinct monophyletic cluster within the group M phylogeny (Fig. 1). It is thought that subtype B originated as a viral lineage exported from Africa to Haiti (52) and then to the United States, from where it spread internationally to become the most geographically dispersed subtype worldwide (53). Our analyses indicate that the lineage ancestral to subtype B originated in Kinshasa (PP = 0.99) (Fig. 1). This lineage was already present in Kinshasa by 1944 (95% BCI: 1935–1951) and, in agreement with previous findings (52), arrived in Haiti around 1964 (95% BCI: 1960–1967). It has been suggested this occurred with the return of Haitian professionals who worked in the newly independent Congo in the 1960s (54, 55). Our results strengthen this hypothesis, as a large proportion of these professionals were based in Kinshasa (55, 56).
In contrast to subtype B, subtype C spread successfully within Africa and currently accounts for ~50% of HIV-1 infections worldwide (53). Our phylogeographic reconstruction suggests Mbuji-Mayi as the most likely ancestral location of subtype C (PP = 0.56) (Fig. 1). Moreover, south and east African subtype C sequences are phylogenetically interspersed with sequences from Lubumbashi, capital of the southern Katanga province. Therefore genetic and historical data indicate independently that the DRC transportation network provided the key connection between the Kinshasa region and other human population centers in sub-Saharan Africa (Figs. 2 and 3 and table S6), and additionally provided a link between southern DRC and neighboring Zambia and Angola (38). This indicates subtype C as a lineage that developed in the DRC mining regions, from where it spread south and east, probably through migrant labor. The impact of migrant labor on the spread of HIV-1 is well established in southern Africa (57), where subtype C dominates with high prevalences (53).
Impact of recombination and evolutionary rate heterogeneity
The cocirculation of divergent HIV-1 subtypes has facilitated the identification of recombinant HIV lineages (29). Recombination may confound phylogenetic reconstructions and may adversely affect molecular clock estimates (58, 59). Although we perform evolutionary reconstructions on relatively short sequence fragments (averaging 391 nucleotides) with limited opportunity to contain recombination breakpoints (60), we also conducted extensive simulations to assess the potential effect of recombination on the estimation of divergence times, evolutionary rates, and viral growth rates (30). These analyses confirm that recombination does not significantly affect our TMRCA estimation. Even for rates of 3 × 10−4 recombinations per site per year [about one order of magnitude higher than rates reported for group M (60, 61)], the variance of the TMRCA of group M increased only by 5.3% (tables S8 and S9). Thus, even for levels of recombination that are much higher than expected, the potential bias on key parameters inferred here is limited (62).
Evolutionary rates may also vary among subtypes, and it has been suggested that relaxed molecular clock models may have difficulties accommodating this rate heterogeneity (63). We addressed this by investigating the robustness of our estimates with respect to the inclusion of subtype B and C sequences. Although evolutionary rate estimates for data sets comprising only subtype B or C result in slightly slower rates (2.90 × 10−3 and 2.47 × 10−3 substitutions per site per year, respectively) than those estimated for the complete group M data set (3.26 × 10−3 substitutions per site per year), analyses of group M divergence times without subtypes B and C produce a similar TMRCA estimate (1926; 95% BCI: 1918–1934), indicating that evolutionary rate variation is accommodated satisfactorily here by a relaxed clock model (fig. S9).
Conclusions
We show that the HIV-1 group M pandemic ignited in Kinshasa around the early 1920s and that its spatial expansion in central Africa was contingent upon an active transportation network that connected the country’s main population centers to other regions of sub-Saharan Africa. Further, the increase in the exponential growth rate of group M around 1960 stands in contrast to that of the spatially confined, non-pandemic group O. Our results are consistent with hypotheses that iatrogenic interventions in Kinshasa and its surroundings and/or post-independence changes in sexual behavior were critical for the emergence of group M (22). We suggest that a distinct combination of circumstances during a particular spatial and socio-historical window permitted the establishment, spatial dissemination, and epidemic growth of the HIV-1 group M pandemic. Similar arguments may underlie the emergence of other blood-borne pathogens, particularly that of HCV.
Supplementary Material
Acknowledgments
We thank the researchers whose publicly available data made this work possible and A. M. Vandamme, M. L. Kalish, M. Worobey, and G. Leonard for helpful discussions. The research leading to these results has received funding from the European Union Seventh Framework Programme for research, technological development, and demonstration under grant agreement no. 278433-PREDEMICS and European Research Council grant agreement no. 260864. P.L. was partly supported by the “Onderzoeksfonds KU Leuven/Research Fund KU Leuven.” M.A.S. is partly supported by NSF grant DMS 1264153 and NIH grant R01 HG006139. Collaboration between M.A.S., A.R., and P.L. was supported by the National Evolutionary Synthesis Center (NESCent) and NSF grant EF-0423641. This work was supported by the Wellcome Trust (grant 092807) to A.R. T.B. was supported by the Royal Society. J.D.S. is partly supported by the Fonds voor Wetenschappelijk Onderzoek Flanders grant G.0692.14. The data reported in this paper are deposited in the Dryad Repository (http://dx.doi.org/10.5061/dryad.nn952). Author contributions: P.L., O.G.P., A.R., M.A.S., M.P., and N.R.F. conceived the experiments and designed the study. N.R.F. and P.L. conducted the phylodynamic analyses. D.P. performed the recombination analysis and G.B. the model selection analysis. M.A.S. and T.B. contributed methodology. T.B., M.J.W., and N.A. assisted the sequence analysis. J.P., A.J.T., and J.D.S. contributed historical and spatial data. M.P. provided sequence data. N.R.F., P.L., O.G.P., and A.R. wrote the paper. All authors discussed the results and approved the final manuscript.
Footnotes
We declare no competing financial interests.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/346/6205/56/suppl/DC1
Materials and Methods
References (65–98)
REFERENCES AND NOTES
- 1.UNAIDS. Global Reports – UNAIDS report on the global AIDS epidemic 2013. UNAIDS; Geneva: 2013. [Google Scholar]
- 2.Gottlieb MS, Schanker MD, Fan PT, Saxon MD, Weisman JD, Centers for Disease Control (CDC) MMWR Morb Mortal Wkly Rep. 1981;30:250–252. [Google Scholar]
- 3.Barré-Sinoussi F, et al. Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 4.Gallo RC, et al. Science. 1983;220:865–867. doi: 10.1126/science.6601823. [DOI] [PubMed] [Google Scholar]
- 5.Piot P, et al. Lancet. 1984;2:65–69. doi: 10.1016/s0140-6736(84)90241-1. [DOI] [PubMed] [Google Scholar]
- 6.Van de Perre P, et al. Lancet. 1984;2:62–65. doi: 10.1016/s0140-6736(84)90240-x. [DOI] [PubMed] [Google Scholar]
- 7.Keele BF, et al. Science. 2006;313:523–526. doi: 10.1126/science.1126531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Heuverswyn F, et al. Virology. 2007;368:155–171. doi: 10.1016/j.virol.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 9.Ayouba A, et al. AIDS. 2000;14:2623–2625. doi: 10.1097/00002030-200011100-00033. [DOI] [PubMed] [Google Scholar]
- 10.Peeters M, et al. AIDS. 1997;11:493–498. doi: 10.1097/00002030-199704000-00013. [DOI] [PubMed] [Google Scholar]
- 11.Vallari A, et al. J Virol. 2011;85:1403–1407. doi: 10.1128/JVI.02005-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalish ML, et al. Emerg Infect Dis. 2004;10:1227–1234. doi: 10.3201/eid1007.030904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vidal N, et al. J Virol. 2000;74:10498–10507. doi: 10.1128/jvi.74.22.10498-10507.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rambaut A, Robertson DL, Pybus OG, Peeters M, Holmes EC. Nature. 2001;410:1047–1048. doi: 10.1038/35074179. [DOI] [PubMed] [Google Scholar]
- 15.Rambaut A, Posada D, Crandall KA, Holmes EC. Nat Rev Genet. 2004;5:52–61. doi: 10.1038/nrg1246. [DOI] [PubMed] [Google Scholar]
- 16.Worobey M, et al. Nature. 2008;455:661–664. doi: 10.1038/nature07390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu T, et al. Nature. 1998;391:594–597. doi: 10.1038/35400. [DOI] [PubMed] [Google Scholar]
- 18.Bikandou B, et al. AIDS Res Hum Retroviruses. 2004;20:1005–1009. doi: 10.1089/aid.2004.20.1005. [DOI] [PubMed] [Google Scholar]
- 19.Niama FR, et al. Infect Genet Evol. 2006;6:337–343. doi: 10.1016/j.meegid.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 20.Pandrea I, et al. AIDS Res Hum Retroviruses. 2002;18:1103–1116. doi: 10.1089/088922202320567842. [DOI] [PubMed] [Google Scholar]
- 21.Carr JK, et al. Retrovirology. 2010;7:39. doi: 10.1186/1742-4690-7-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pepin J. The Origins of AIDS. Cambridge Univ. Press; Cambridge: 2011. [Google Scholar]
- 23.Sauter D, et al. Cell Host Microbe. 2009;6:409–421. doi: 10.1016/j.chom.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pybus OG, Rambaut A. Nat Rev Genet. 2009;10:540–550. doi: 10.1038/nrg2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korber B, et al. Science. 2000;288:1789–1796. doi: 10.1126/science.288.5472.1789. [DOI] [PubMed] [Google Scholar]
- 26.Salemi M, et al. FASEB J. 2001;15:276–278. doi: 10.1096/fj.00-0449fje. [DOI] [PubMed] [Google Scholar]
- 27.Yusim K, et al. Philos Trans R Soc London Ser B. 2001;356:855–866. doi: 10.1098/rstb.2001.0859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baele G, et al. Mol Biol Evol. 2012;29:2157–2167. doi: 10.1093/molbev/mss084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robertson DL, Sharp PM, McCutchan FE, Hahn BH. Nature. 1995;374:124–126. doi: 10.1038/374124b0. [DOI] [PubMed] [Google Scholar]
- 30.Materials and methods are available as supplementary materials on Science Online.
- 31.Kita K, et al. AIDS Res Hum Retroviruses. 2004;20:1352–1357. doi: 10.1089/aid.2004.20.1352. [DOI] [PubMed] [Google Scholar]
- 32.Vidal N, et al. J Acquir Immune Defic Syndr. 2005;40:456–462. doi: 10.1097/01.qai.0000159670.18326.94. [DOI] [PubMed] [Google Scholar]
- 33.Chitnis A, Rawls D, Moore J. AIDS Res Hum Retroviruses. 2000;16:5–8. doi: 10.1089/088922200309548. [DOI] [PubMed] [Google Scholar]
- 34.Pépin J. Sex Transm Infect. 2012;88:307–312. doi: 10.1136/sextrans-2011-050277. [DOI] [PubMed] [Google Scholar]
- 35.Vangroenweghe D. Philos Trans R Soc London Ser B. 2001;356:923–925. doi: 10.1098/rstb.2001.0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Sousa JD, Alvarez C, Vandamme AM, Müller V. Viruses. 2012;4:1950–1983. doi: 10.3390/v4101950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharp PM, Hahn BH. Nature. 2008;455:605–606. doi: 10.1038/455605a. [DOI] [PubMed] [Google Scholar]
- 38.Huybrechts A. Transports et Structures de Development au Congo: Etude du Progres Economique de 1900–1970. Mouton; Paris: 1970. [Google Scholar]
- 39.Gray RR, et al. AIDS. 2009;23:F9–F17. doi: 10.1097/QAD.0b013e32832faf61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flouriot J. Introduction a la Geographique Physique et Humaine du Zaire. Lyon, France: 1994. mimeographed. [Google Scholar]
- 41.Hance WA. Population, Migration, and Urbanization in Africa. Columbia Univ Press; New York: 1970. [Google Scholar]
- 42.Quinn TC. Proc Natl Acad Sci USA. 1994;91:2407–2414. doi: 10.1073/pnas.91.7.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ngimbi M. Kinshasa, 1881–1981:100 Ans Après Stanley: Problèmes et Avenir d’une Ville. Centre de Recherches Pédagogiques; Kinshasa: 1982. [Google Scholar]
- 44.Lemey P, Rambaut A, Pybus OG. AIDS Rev. 2006;8:125–140. [PubMed] [Google Scholar]
- 45.Mulanga-Kabeya C, et al. AIDS. 1998;12:905–910. doi: 10.1097/00002030-199808000-00013. [DOI] [PubMed] [Google Scholar]
- 46.Nzilambi N, et al. N Engl J Med. 1988;318:276–279. doi: 10.1056/NEJM198802043180503. [DOI] [PubMed] [Google Scholar]
- 47.Duke-Sylvester SM, Biek R, Real LA. Philos Trans R Soc London Ser B. 2013;368:20120194. doi: 10.1098/rstb.2012.0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Magiorkinis G, et al. PLOS Comput Biol. 2013;9:e1002876. doi: 10.1371/journal.pcbi.1002876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iles JC, et al. Infect Genet Evol. 2013;19:386–394. doi: 10.1016/j.meegid.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 50.Beheyt P. Ann Soc Belg Med Trop. 1953;33:297–340. [PubMed] [Google Scholar]
- 51.de Sousa JD, Müller V, Lemey P, Vandamme AM. PLOS ONE. 2010;5:e9936. doi: 10.1371/journal.pone.0009936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilbert MT, et al. Proc Natl Acad Sci USA. 2007;104:18566–18570. doi: 10.1073/pnas.0705329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hemelaar J, Gouws E, Ghys PD, Osmanov S, WHO-UNAIDS Network for HIV Isolation and Characterisation AIDS. 2011;25:679–689. [Google Scholar]
- 54.Kuyu C. Les Haitiens au Congo. L’Harmattan; Paris: 2006. [Google Scholar]
- 55.Institut National de la Statistique. Étude Socio-Demographique de Kinshasa, 1967: Rapport General. Institut National de la Statistique; Kinshasa: 1969. [Google Scholar]
- 56.Bonacci G. Cahiers d’Etudes Africaines. Vol. 192. L’Harmattan; Paris: 2008. Kuyu, Camille – Les Haïtiens au Congo; p. 895. [Google Scholar]
- 57.Jochelson K, Mothibeli M, Leger JP. Int J Health Serv. 1991;21:157–173. doi: 10.2190/11UE-L88J-46HN-HR0K. [DOI] [PubMed] [Google Scholar]
- 58.Schierup MH, Forsberg R. Proceedings of the Conference: Origins of HIV and Emerging Persistent Viruses, 28 to 29 September 2001. Vol. 187. Accademia Nazionale dei Lincei; Rome: 2003. pp. 231–245. [Google Scholar]
- 59.Schierup MH, Hein J. Genetics. 2000;156:879–891. doi: 10.1093/genetics/156.2.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neher RA, Leitner T. PLOS Comput Biol. 2010;6:e1000660. doi: 10.1371/journal.pcbi.1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ward MJ, Lycett SJ, Kalish ML, Rambaut A, Leigh Brown AJ. J Virol. 2013;87:1967–1973. doi: 10.1128/JVI.02478-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lemey P, et al. Genetics. 2004;167:1059–1068. doi: 10.1534/genetics.104.026666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wertheim JO, Fourment M, Kosakovsky Pond SL. Mol Biol Evol. 2012;29:451–456. doi: 10.1093/molbev/msr266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Saint-Moulin L. Villes et Organisation de l’Espace en République Démocratique du Congo. L’Harmattan; Paris: 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.