

Evolving the Definition of Dystonia
The term dystonia originated in 1911 with Oppenheim's description of 4 individuals who were floppy at rest, yet developed stiffness when they tried to move.1 He believed the core problem was a defect in muscle tone, so he coined the term dys‐tonia, which literally means abnormal muscle tone. Over the next decades, additional clinical manifestations of dystonia were recognized, and various authorities began to alter Oppenheim's definition according to their own views. It was not until 1984 that the Dystonia Medical Research Foundation (DMRF) created a committee to develop a more unified definition: “Dystonia is a syndrome of sustained involuntary muscle contractions, frequently causing twisting or repetitive movements, or abnormal postures.”
In the decades that followed this definition, more was learned about the manifestations of dystonia, and expert opinions again led to varying adjustments to the definition. As a result, a new consensus committee revised the definition.2 This committee was again supported by the DMRF, but also by Dystonia Europe, the International Parkinson and Movement Disorder Society (formerly the Movement Disorders Society), and an international panel with broad expertise in the clinical and biological basis of the dystonias: “Dystonia is a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive movements, postures, or both. Dystonic movements are typically patterned, twisting, and may be tremulous. Dystonia is often initiated or worsened by voluntary action and associated with overflow muscle activation.”
The essential change was the addition of the word intermittent and two other sentences. The word intermittent was added to address the frequent misconception that dystonic movements are always sustained, slow, or twisting.3, 4 Examples of rapid, intermittent dystonic movements include blinking in blepharospasm, voice breaks in spasmodic dysphonia, tremor‐dominant dystonias, and myoclonic dystonia. One sentence was added to emphasize that the varied clinical manifestations may be tremulous or twisting, but they tend to be patterned. The second sentence was added to emphasize characteristic worsening with voluntary action, a feature noted in Oppenheim's original concept.
Evolving the Classification of the Dystonias
An increasing appreciation of the varied clinical manifestations and causes for dystonia over the years also led to varying proposals for how to classify them (Table 1). The committee that recently revised the definition agreed that a more widely acceptable system would be useful, and it began by delineating the problems that were motivating many of the recently proposed changes for classification (Table 2).
Table 1.
Evolving classifications for the dystonias
| Fahn and Eldridge (1976) | Fahn (1988) | Fahn (1998) | Bressman (2004) | Albanese (2011) |
|---|---|---|---|---|
| Primary | Idiopathic | Primary | Primary | Primary |
| Hereditary | Hereditary | Hereditary | Pure | |
| Sporadic | Sporadic | Sporadic | Dystonia plus | |
| Paroxysmal | ||||
| Secondary | Symptomatic | Dystonia Plus | Secondary | Heredodegenerative |
| Heredodegenerative | Dystonia plus | |||
| Environmental | Degenerative | |||
| Complex | ||||
| Acquired | ||||
| Psychological | Heredodegenerative | Secondary | ||
| Acquired |
This table shows a representative sample of some of the most commonly used classifications for dystonia.
Table 2.
Weaknesses of historical terminology
| Basis for Classification | Weaknesses Identified |
|---|---|
| Primary vs. secondary |
Inconsistent use of terms across proposals Inappropriate cultural connotations following translation Concept not logically based on etiology |
| Dystonia‐plus |
Inconsistent inclusion of different dystonias across proposals Arbitrary meaning of “plus” Concept not logically based on etiology |
| Heredodegenerative |
Some disorders are hereditary, but not degenerative. Some disorders are degenerative, but not hereditary. Waste‐basket category with little value for organizing knowledge |
The changes recommended by the committee are summarized in Table 3. The most significant change involved dividing the classification system into two separate axes, one for clinical features and the other for etiologies. The reason was that the design of any classification system depends, in large part, on its goals, and for the dystonias, there are two main goals. A major goal for the clinician is to organize the many dystonias into clinically meaningful groups to facilitate diagnosis, treatment, and clinical research. On the other hand, an etiology‐based classification is directly related to treatment algorithms and for organizing biological knowledge to guide the development of novel concepts regarding pathogenesis and the design of new therapies.5, 6
Table 3.
Classification of the dystonias
| Axis | Dimension for Classification | Subgroups |
|---|---|---|
| Axis I: clinical features | Age at onset | Infancy (birth to 2 years) |
| Childhood (3–12 years) | ||
| Adolescence (13–20 years) | ||
| Early adulthood (21–40 years) | ||
| Late adulthood (40 years and older) | ||
| Body distribution | Focal (one isolated body region) | |
| Segmental (two or more contiguous regions) | ||
| Multifocal (two or more noncontiguous regions) | ||
| Hemidystonia (half the body) | ||
| Generalized (trunk plus two other sites) | ||
| Temporal pattern | Disease course (static vs. progressive) | |
| Short‐term variation (e.g., persistent, action specific, diurnal, or paroxysmal) | ||
| Associated features | Isolated (with or without tremor) | |
| Combined (with other neurological or systemic features) | ||
| Axis II: etiology | Nervous system pathology | Degenerative |
| Structural (e.g., focal static lesions) | ||
| No degenerative or structural pathology | ||
| Heritability | Inherited (e.g., sex linked or autosomal, dominant or recessive, or mitochondrial) | |
| Acquired (e.g., brain injury, drugs/toxins, vascular, or neoplastic) | ||
| Idiopathic | Sporadic | |
| Familial |
These two goals are only partly overlapping, so the importance of both was addressed by dividing the classification of the dystonias into two main axes.2 The first axis focuses on clinical manifestations with four dimensions that address the character of dystonia, including body region affected, age at onset, temporal aspects, and any associated clinical manifestations. The second axis focuses on etiology with two dimensions for inheritance or underlying neuropathology. The two different axes were not envisioned as being independent. Instead, the goal of delineating the clinical phenomenology (Axis I) is to build a comprehensive picture of a syndromic pattern (Fig. 1). This pattern can then be used to narrow down potential causes for diagnostic testing.7 The rationale for revising the original dimensions for classification are outlined below.
Figure 1.
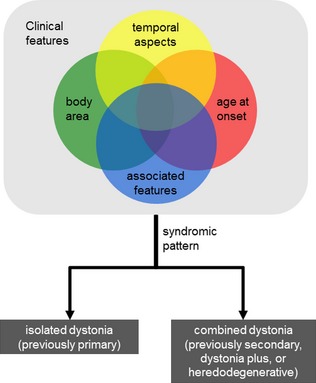
Axis I guides on the recognition of the phenomenology of dystonia and of the prevalent syndromic pattern.
Revising Classification According to Body Region Affected (Axis I)
The classification of the dystonias by the body region affected has enormous practical value in guiding treatment decisions. Patients with focal or segmental dystonias are more likely to be treated with botulinum toxins, and those with broader distributions are more likely to require oral medications or surgery. This approach for classification was retained, with the only recommendation being more‐specific guidelines regarding the diagnosis of generalized dystonia. Previous classifications relied on involvement of one or both legs, but the new classification requires involvement of the trunk and at least two other sites.
Revising Classification According to Age at Onset (Axis I)
Classification by age at onset also has practical clinical value for counseling regarding prognosis and guiding diagnostic testing. Childhood‐onset dystonias are much more likely to become generalized and much more likely to have a discoverable cause. Whereas earlier classifications recognized two groups (early and late onset) with an age cutoff of 26 years, the new classification system has five age groups recognizing a spectrum of onset from pediatric to adult.
The reason for this change was that the previous concept of two groups divided at 26 years of age was based on a single study that focused on discriminating patients with DYT1 dystonia from those who did not.8 This age discriminator is not suitable for application to all dystonias. For example, metabolic disorders with dystonia generally begin before 3 years of age, and the median age for development of DYT6 dystonia is 28 years.9 Patients with dopa‐responsive dystonia and Wilson's disease usually develop symptoms before 20 years of age, but most investigators encourage testing for these diagnoses at least through the age of 40, because they are treatable.
Rather than have different age discriminators for each type of dystonia, the new classification system is based on five standard age groups that are used for other neurological disorders. In this classification system, metabolic disorders will fall mostly in the first two age groups, generalized dystonias will fall mostly in the middle age groups, and the adult‐onset focal dystonias will fall in last two groups. This is more complicated than the original two‐group classification system, but it also is much more broadly applicable.
Adding Classification According to Temporal Aspects (Axis I)
Temporal aspects include manner of onset (acute or insidious), short‐term variations in symptoms (diurnal, intermittent, or action induced), and longer‐term variations in overall severity (static or progressive). These features had not been considered by any previous classification, but have important implications for diagnostic testing, counseling, and treatment options. Acute‐onset dystonia is relatively uncommon and often signifies a conversion disorder or less‐common conditions, such as rapid‐onset dystonia parkinsonism or glutaric aciduria. Diurnal patterns are suggestive of dopa‐responsive dystonia, and intermittent patterns suggest one of the paroxysmal dyskinesias. Relentlessly progressive dystonia often signifies a degenerative process. The importance of these aspects is now formally recognized by a distinct dimension relating to temporal aspects.
Resolving Ambiguities in the Classification According to Etiology Into Associated Clinical Features (Axis I) Versus Causes (Axis II)
The classification of the dystonias according to etiology has seen the most changes over the years, in part because of enormous progress in understanding their many potential causes. The earliest etiological classification was based on two groups (Table 1). One group was idiopathic, and the other was secondary to a known cause. As more was learned about the many causes of dystonia over the years, additional subgroups were introduced. However, these additions often were defined by clinical phenomenology, not etiology, creating a hybrid classification system that mixed etiology with phenomenology. Because this hybrid system lacked a logical organizational plan according to etiology (Table 2), it was not ideal for organizing our growing knowledge of biological causes.
For example, the commonly used terms primary and secondary have led to confusion because they have been used by different investigators to mean different things over the years (Table 2). In early classifications, the term primary was used to refer to idiopathic dystonia, but more recently to phenotypically isolated dystonia, regardless of etiology. The term dystonia‐plus was introduced to highlight disorders where dystonia was a prominent feature, but combined with other neurological abnormalities, again regardless of etiology. Exactly how dominant the dystonia had to be was arbitrary, leading different investigators to include different disorders in this group. The term heredodegenerative was the only category that was based strictly on etiology. Unfortunately, this group became a wastebasket list for an enormous number of unrelated disorders. As a result, it did not provide a plan that organized etiologies in a meaningful way.
The problem of a hybrid dimension for classification was addressed by creating a clinical axis to address associated clinical features and creating a distinct etiological axis for biological causes. The associated neurological and non‐neurological features provide critical information for diagnosis and treatment, but previously they were divided across categories such as “dystonia‐plus” or “heredodegenerative.” Because these aspects relate to clinical phenomenology and not etiology, it was necessary to introduce a new dimension of the clinical axis to emphasize the importance of these associated features (Table 3).
Associated features may include a related movement disorder, such as parkinsonism or myoclonus. They may also include other neurological abnormalities, such as dementia, seizures, or neuropathy. Finally, they may include non‐neurological abnormalities, such as liver disease or hematological abnormalities. A complete assessment of all of the clinical features is essential for delineating syndromic patterns that can aid in diagnosis.7
A separate axis for etiology was established to accommodate any evidence for inheritance or acquired causes, as well as idiopathic (genetically unclassified) sporadic or familial cases. In addition, the etiologic classification includes information regarding any associated neuropathology. This last aspect is compatible with earlier classifications, where the terms primary and dystonia‐plus were sometimes used to highlight forms of dystonia without evidence of structural pathology.
Our understanding of the biological bases for dystonia has grown rapidly in the past few years, with an increasing number of shared biological principles being recognized that could be used to subgroup them.10, 11 Included are shared genetic mechanisms,12, 13 shared molecular mechanisms,14 shared anatomical pathways,15, 16, 17 and shared physiological substrates.18, 19 However, there is currently insufficient information regarding how these biological mechanisms should be organized in the classification of the dystonias. Other dimensions may be added in the etiological axis as more is learned in the future.
Past, Present, and Future
Medicine is a living science. In order to thrive and grow, it must adapt as new information is obtained from both clinical and basic research. Sometimes our fundamental medical concepts need only minor revisions, but other times they must be thrown out and replaced by ones that are more in step with modern knowledge and more adaptable to future needs.
Concepts regarding the definition and classification of the dystonias are no exception. The recently proposed changes are some of the most significant ones proposed in recent years. As a result, they are likely to meet resistance from the inertia of traditional habits and beliefs. However, these changes address numerous weaknesses in existing proposals for classification in a more clinically useful and biologically logical manner. It seems likely that these concepts will evolve again in the future as we continue to learn more about this unique group of disorders.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
H.A.J.: 1A, 1B, 1C, 3A
A.A.: 1A, 1B, 1C, 3B
Disclosures
Funding Sources and Conflicts of Interest: The authors' work related to dystonia has been supported, in part, by grants from private foundations, including the Bachmann‐Strauss Dystonia & Parkinson Foundation, the Benign Essential Blepharospasm Research Foundation, and the DMRF, and Tyler's Hope for a Dystonia Cure. It also has been supported, in part, by research grants from the National Institutes of Health (NIH; U54 NS065701, R01 NS040470, and R01 HD053312) and the Emory University Research Council.
Financial Disclosures for previous 12 months: H.A.J. has received research grant support from the NIH, the Atlanta Clinical & Translational Science Institute, the Emory University Research Council, the Lesch‐Nyhan Syndrome Children's Research Foundation, the DMRF, the Bachmann‐Strauss Dystonia & Parkinson's Foundation, and the Benign Essential Blepharospasm Research Foundation; he also is principal investigator for the Dystonia Coalition, which receives the majority of its support through NIH grant NS065701 from the Office of Rare Diseases Research in the National Center for Advancing Translational Sciences and National Institute of Neurological Disorders and Stroke. The Dystonia Coalition receives additional material or administrative support from industry sponsors (Allergan Inc., Ipsen Biopharm, Medtronics Inc, and Merz Pharmaceuticals) as well as private foundations (The American Dystonia Society, The Bachmann‐Strauss Dystonia and Parkinson Foundation, BeatDystonia, The Benign Essential Blepharospasm Foundation, Dystonia Europe, Dystonia Ireland, the DMRF, the Dystonia Society, the Foundation for Dystonia Research, the National Spasmodic Dysphonia Association, and The National Spasmodic Torticollis Association); and he serves on the scientific advisory boards for Cure Dystonia Now, the DMRF, Tyler's Hope for a Dystonia Cure, the Lesch‐Nyhan Syndrome Children's Research Foundation, and Lesch‐Nyhan Action France. A.A. has received research grant support from the Italian Ministry of Health, the Italian Ministry of Education, the Jacques and Gloria Gossweiler Foundation, and the Beneficentia Stiftung; he is principal investigator for the European Dystonia Network, which receives research funds by the European Cooperation in Science and Technology (COST Action BM1101) and also additional support from industry sponsors (Ipsen SA and Merz Pharmaceuticals GmbH); and he serves on the scientific advisory board for Dystonia Europe.
Acknowledgments
The authors thank the other members of the dystonia consensus committee, including Kailash Bhatia, Susan Bressman, Mahlon DeLong, Stanley Fahn, Victor Fung, Mark Hallett, Joseph Jankovic, Christine Klein, Anthony Lang, Jonathon Mink, and Jan Teller. The work of this committee was supported by the DMRF, the Dystonia Coalition, the European Dystonia COST action, and unrestricted educational grants from Ipsen Pharma and Merz GmbH.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Klein C, Fahn S. Translation of Oppenheim's 1911 paper on dystonia. Mov Disord 2013;28:851–862. [DOI] [PubMed] [Google Scholar]
- 2. Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord 2013;28:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fahn S. The varied clinical expressions of dystonia. Neurol Clin 1984;2:541–554. [PubMed] [Google Scholar]
- 4. Marsden CD. Dystonia: the spectrum of the disease In: Yahr MD, ed. The Basal Ganglia. New York, NY: Raven Press; 1976:351–367. [Google Scholar]
- 5. Thompson VB, Jinnah HA, Hess EJ. Convergent mechanisms in etiologically‐diverse dystonias. Expert Opin Ther Targets 2011;15:1387–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jinnah HA, Hess EJ. Experimental therapeutics for dystonia. Neurotherapeutics 2008;5:198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fung VS, Jinnah HA, Bhatia K, Vidailhet M. Assessment of the patient with dystonia: an update on dystonia syndromes. Mov Disord 2013;28:889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bressman SB, Sabatti C, Raymond D, et al. The DYT1 phenotype and guidelines for diagnostic testing. Neurology 2000;54:1746–1752. [DOI] [PubMed] [Google Scholar]
- 9. Xiromerisiou G, Houlden H, Scarmeas N, et al. THAP1 mutations and dystonia phenotypes: genotype phenotype correlations. Mov Disord 2012;27:1290–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Defazio G, Berardelli A, Hallett M. Do primary adult‐onset focal dystonias share aetiological factors? Brain 2007;130:1183–1193. [DOI] [PubMed] [Google Scholar]
- 11. Jinnah HA, Berardelli A, Comella C, et al. The focal dystonias: current views and challenges for future research. Mov Disord 2013;7:926–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lohmann K, Klein C. Genetics of dystonia: what's known? what's new? what's next? Mov Disord 2013;28:899–905. [DOI] [PubMed] [Google Scholar]
- 13. LeDoux MS. The genetics of dystonias. Adv Genet 2012;79:35–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. LeDoux MS, Dauer WT, Warner T. Emerging molecular pathways for dystonia. Mov Disord 2013;15:968–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Neychev VK, Gross R, Lehericy S, Hess EJ, Jinnah HA. The functional neuroanatomy of dystonia. Neurobiol Dis 2011;42:185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lehericy S, Tijssen MA, Vidailhet M, Kaji R, Meunier S. The anatomical basis of dystonia: current view using neuroimaging. Mov Disord 2013;28:944–957. [DOI] [PubMed] [Google Scholar]
- 17. Zoons E, Booij J, Nederveen AJ, Dijk JM, Tijssen MA. Structural, functional and molecular imaging of the brain in primary focal dystonia–a review. Neuroimage 2011;56:1011–1020. [DOI] [PubMed] [Google Scholar]
- 18. Quartarone A, Hallett M. Emerging concepts in the physiological basis of dystonia. Mov Disord 2013;28:958–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hallett M. Neurophysiology of dystonia: the role of inhibition. Neurobiol Dis 2011;42:177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]