

Abstract
Objectives
A major signaling pathway that regulates cellular aging is the Insulin/IGF-1/Pl3k/Akt/forkhead-box class O (FOXO) transcription factor axis. Previously, we observed that FOXO factors are dysregulated in aged and OA cartilage. The objective of this study was to investigate the impact of downregulated FOXOs on chondrocytes.
Methods
Small interference RNAs (siRNAs) for FOXO1 and FOXO3 were transfected into human articular chondrocytes. Cell viability following treatment with the oxidant tert-Butyl hydroperoxide (t-BHP) was measured by MTT assay. Caspase-3/7 activation and apoptotic cell were examined. Gene and protein expression of antioxidant proteins and autophagy related proteins and changes in inflammatory mediators following treatment with IL-1β were analyzed. Cells transfected with FOXO plasmids were also analyzed.
Results
Cell viability was significantly reduced by siFOXO under treatment with t-BHP. Apoptosis accompanied by caspase activation was significantly induced in FOXO-siRNA transfected chondrocytes. Knock-down of FOXO1 and FOXO1+3 resulted in significant reductions of GPX-1, catalase, LC3, Beclin1, and SIRT1 proteins following treatment with t-BHP. In contrast, constitutive active form of FOXO 3 increased cell viability while inducing GPX1, Beclin1, and LC3 in response to t-BHP. Expression and production of ADAMTS-4 and Chemerin were significantly increased in FOXO-siRNA transfected chondrocytes.
Conclusions
Reduced expression of FOXO transcription factors in chondrocytes increased susceptibility to cell death induced by oxidative stress. This was associated with reduced antioxidant proteins and autophagy related proteins. Our data provide evidence for a key role of FOXO transcription factors as regulators of chondrocyte oxidative stress resistance and tissue homeostasis.
Keywords: FOXO, osteoarthritis, chondrocytes, oxidative stress
INTRODUCTION
Aging is known to be an important risk factor for the development of osteoarthritis (OA) and metabolic and cellular changes in aging have been extensively investigated (1). Genetic analyses have demonstrated that the insulin/insulin-like growth factor-1 (IGF-1)/phosphatidylinositol-3 kinase (PI3K)/Akt signal transduction pathway is involved in aging of many organisms, including nematodes, fruit flies, and mammals (2). In addition, the forkhead-box class O (FOXO) transcription factors, such as DAF-16 in Caenorhabditis elegans (3), and its mammalian homologues, FOXO1, FOXO3, and FOXO4, play an essential role in the IGF-1/Pl3K/Akt signal transduction pathway and can be modulated to reduce age-related diseases (4).
A key function of FOXO transcription factors is in controlling oxidative stress resistance through regulating antioxidants and protein quality control. Dysregulation of FOXO expression or activation contributes to the pathogenesis of age-related diseases affecting bone (5), muscle (6, 7), and the central nervous system (8). Chondrocytes produce reactive oxygen species in response to cytokines (9) and mechanical stress (10, 11). Antioxidant defenses are also compromised in OA-affected and aged cartilage (12–14), leading to changes in chondrocyte phenotype (15–17), senescence (18, 19) and cell death (20), key mechanisms involved in disease onset and progression. Increased vulnerability to reactive oxygen species (ROS)-induced cell death was reported in aging articular cartilage and was related to reduced antioxidants (14). Autophagy is an important mechanism to maintain protein quality under oxidative stress and its activity decreases with aging and in OA (21).
Previously, we reported that FOXO factors are dysregulated in aged and OA cartilage, indicating age-related reduction of FOXO protein expression and increased phosphorylation of FOXO (inactive form of FOXO) in OA cartilage (22). In the present study, we investigated the impact of downregulated FOXO on survival and gene expression in human chondrocytes.
MATERIALS AND METHODS
Cell and mRNA isolation from human articular cartilage
Normal human cartilage was obtained at autopsy from a total of 18 adult donors (age 18 to 64 years, mean ± SD = 41.8 ± 14.9) for cell isolation and 4 adult donors (age 17 to 43 years, mean ± SD = 25.2 ± 10.4) for mRNA isolation with no history of joint disease. OA human cartilage was obtained for mRNA isolation from 5 patients (age 52 to 74 years, mean ± SD = 62.2 ± 8.8) undergoing knee replacement surgery. Human tissues were obtained under approval by the Scripps Human Subjects Committee.
Human chondrocytes were isolated and cultured as described previously (23). The cartilage tissue was incubated with trypsin at 37°C for 10 minutes. After the trypsin solution was removed, the tissue slices were treated for 12 to 16 hours with type IV clostridial collagenase in Dulbecco’s modified Eagle’s medium (DMEM) with 5% fetal calf serum (CS). The isolated chondrocytes were plated at high density in DMEM with 10% CS and antibiotics and allowed to attach to the culture flasks. The cells were incubated at 37°C in a humidified gas mixture containing 5% of CO2 balanced with air. The chondrocytes were used in the experiments at confluence (2–3 weeks in primary culture).
For mRNA isolation directly from cartilage tissue, the cartilage slices were frozen in liquid nitrogen. Then, the frozen tissues were crushed and homogenized. Samples were incubated in Qiazol (Qiagen) at room temperature. After addition of chloroform and vortex vigorously, samples were centrifuged for 15 min at 12,000 × g at 4°C. The aqueous phase was collected. mRNA was extracted using RNAqueous (Ambion) according to the manufacturer’s protocol.
siRNA transfection
Human chondrocytes were seeded in 6-well plates at a density of 2.0×105 cells/well in 96-well plates at a density of 3.0 ~ 8.0 ×103 cells/well for each experiment in DMEM supplemented with 10% CS. After 1 day, small interference RNAs (8 nM) for FOXO1, FOXO3, or the combination of FOXO1 and FOXO3, or Beclin1 were transfected into cells for 24 hours using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s suggested protocols. Then, culture medium was changed to DMEM with 1% CS. To confirm gene-specific silencing effects, protein lysates were prepared at 48 hours after transfection and subjected to Western blot analysis. FOXO1, FOXO3, and a scrambled control siRNA, which has no significant homology to any mammalian gene sequences were purchased from Ambion.
Plasmid transfection
Human chondrocytes were plated in 12-well plates at a density of 2.0×105 cells/well and 96-well plates at a density of 1.0×104 cells/well 18 hours before transfection. Lipofectamine 3000 (Invitrogen) reagent was used to transfect either pcDNA3 empty vector or pcDNA3-Flag-FKHR-AAA (Addgene Plasmid 13508) or pcDNA3-Flag-FKHRL1-AAA (Addgene Plasmid 10709), as recommended by the manufacturer. Six hours after transfection, culture medium was changed to DMEM with 10% CS.
Cell viability assessment by MTT assay and Resazurin assay
After transfection with siFOXO or FOXO plasmid, tert-Butyl hydroperoxide (t-BHP) was added in each well to a final concentration of 0, 25, 50, 100, and 250 μM in DMEM with 1% CS and plates were incubated for 24 h. For MTT assay, 25 μl of fresh MTT (5 mg/ml in PBS) was added to each well. Five hours later, supernatant was aspirated, and 100 μl of DMSO was added to each well. The absorbance values were measured at 540 nm. For Resazurin assay, the cells transfected with siBeclin1 were stimulated with t-BHP at a final concentration of 0, 100, and 250 μM with or without 100 μM of Z-VAD-FMK (pan-caspase inhibitor). After 24 h incubation, 10 μL of resazurin solution (500 μM in PBS) was added, and the cells were incubated for 2.5 h at 37°C. The fluorescence resulting from the reduction of resazurin to resorufin was measured using excitation and emission wavelengths of 530 and 590 nm, respectively. Cell viability was calculated as the percentage of absorbance of t-BHP treated cells (25, 50, 100, and 250 μM of t-BHP) with respect to the cells not treated with t-BHP.
Caspase activation assay
For measuring caspase-3/7 activation under siRNA and t-BHP treatment conditions, the siRNA transfected cells were treated with t-BHP at a final concentration of 0, 100, and 250 μM in DMEM with 1% CS and incubated for 24 h. Caspase-3/7 activation was detected using Caspase-Glo 3/7 Assay (Promega) according to the manufacturer’s protocol.
Detection of intracellular reactive oxygen species (ROS) production
Generation of intracellular ROS in siFOXO transfected cells was detected by aminophenyl fluorescein (APF) assay. APF is selectively converted to a strongly fluorescent compound, fluorescein, upon reaction with ROS such as hydroxyl radical, peroxynitrite and hypochlorite, but not other ROS such as nitric oxide, superoxide and hydrogen peroxide (24). At 48 h after siRNA transfection, chondrocytes in 6-well plates were incubated with 5 μM of APF (Cayman Chemical) for 60 min at 37°C. After washing with HBSS, the cells were stimulated with t-BHP at a final concentration of 0, 100, and 250 μM for 1 h. Then, the cells were trypsinized and re-suspended with HBSS and put into 96-well black plate. Fluorescence was measured using excitation and emission wavelengths of 490 and 515 nm, respectively.
Measurement of reduced and oxidized glutathione
After 48 h transfection with siFOXO, the cells were treated with t-BHP at a final concentration of 0, 100, and 250 μM in DMEM with 1% CS and incubated for 24 h. The ratio of GSSG to GSH was measured as an indicator of oxidative stress using GSH/GSSG Ratio Assay Kit (eEnzyme) according to the manufacturer’s protocol.
Quantitative western blotting
Quantitative western blotting was performed with the LiCor immunofluorescence detection system (LiCor). Proteins were separated on 4–20% SDS Page gels and transferred to nitrocellulose. Membranes were blocked with 1X Odyssey blocking buffer, washed in Tris-Buffered Saline with Tween (TBST), and primary antibodies for FOXO1, FOXO3, SOD2, GPX1, Catalase, LC3, Beclin1, SIRT1, ADAMTS-4, and GAPDH (from Cell Signaling) were added in 1/2X Odyssey buffer in PBS with 0.1% Tween 20. After washing again in TBST, secondary antibodies goat anti-rabbit – IRDye 800 (1:5000 dilution) and goat anti mouse – IRDye 680 (1:10000 dilution), diluted in 1/2X Odyssey buffer in PBS with 0.1% Tween 20 and 0.01% SDS were added. Blots were washed in PBS and then water before image acquisition on the LiCor Odyssey. In lane background was removed (Median: Top/Bottom) before analysis with the Odyssey software version 3.0 (LiCor). Protein of interest integrated intensity values (K counts) were normalized to those of GAPDH.
Quantitative real-time PCR
Human chondrocytes (2.0×105 cells/well) transfected with siRNA were stimulated with IL-1β (0.01 ng/ml). Cells were incubated for 6 h and then mRNA was collected. Total RNA was reversed transcribed to complementary DNA using TaqMan® Reverse Transcription Reagents (Applied Biosystems). Quantitative Real-time PCR was carried out using gene-specific primers and LightCycler 480 Probes Master (Roche). Expression levels of GAPDH were also determined as an internal control. Results were obtained using LightCycler® 480 Instrument II (Roche) and evaluated using Excel (Microsoft). Predesigned primers for FOXO1, FOXO3, FOXO4, iNOS, COX2, IL-6, ADAMTS-4, ADAMTS-5, MMP-13, Chemerin, MCP-1, IL-8, and GAPDH were obtained from Applied Biosystems.
Enzyme-linked immunosorbent assay (ELISA)
After transfection with siFOXO, t-BHP at a final concentration of 0, 100, and 250 μM was added and plates were incubated for 24 h. The quantitative determination of cytoplasmic histone-associated DNA fragments after induced cell death was performed using Cell Death Detection ELISAPLUS Kit (Roche) in accordance with manufacturer’s protocol.
Human chondrocytes transfected with siRNA were stimulated with IL-1β (0.1 ng/ml) for 24 h. Culture medium was then collected for ELISA assay. Chemerin in the supernatant were measured using ELISA kits for Chemerin (Human Chemerin Quantikine ELISA, R&D systems) in accordance with the manufacturer’s protocol.
Statistical analysis
Statistically significant differences were determined with repeated-measures ANOVA. When significant difference was observed, statistical significance compared to the control condition was determined by t-tests with a Bonferroni adjustment to correct for Type-I error associated with multiple comparisons. The results are reported as mean ± S.D. P values less than 0.05 were considered significant.
RESULTS
FOXO mRNA is downregulated in human OA cartilage
We compared expression of FOXO mRNAs between normal and OA cartilage. OA samples were separated into 2 different regions depending on the extent of degradation. The results showed that FOXO1 mRNA expression was significantly lower in non-fibrillated cartilage from OA knees compared to cartilage from normal joints (Figure 1A). FOXO3 mRNA was significantly lower in both non-fibrillated and fibrillated OA cartilage compared to normal cartilage. Expression of FOXO4 was significantly lower than FOXO1 and FOXO3 in normal and OA cartilage, and there was no significant difference in FOXO4 expression between normal and OA cartilage.
Figure 1.
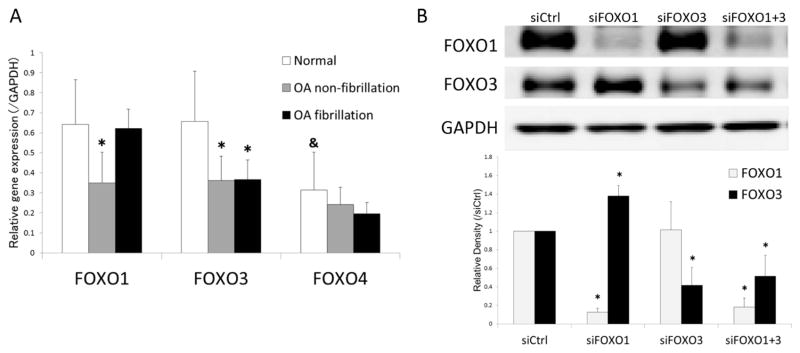
(A) FOXO mRNA expression in human articular cartilage.
RNA samples isolated from articular cartilage were analyzed by real-time PCR using primers for FOXOs as indicated. Results are from a total of 4 normal (N=4) and 5 OA donors (N=5) with separate analysis of non-fibrillated and fibrillated areas. Values are the mean and SD. * = P < 0.05 versus normal; & = P < 0.05 versus FOXO1 and FOXO3.
(B) Transfection with siFOXO in human chondrocytes.
Protein extracts from chondrocytes transfected with siRNAs were analyzed by western blotting using antibodies to FOXOs as indicated. Image is representative of FOXO1 and FOXO3 protein expression in siRNA transfected cells from the same samples. Graph shows the results of a total of 4 donors (N=4). Values are the mean and SD. * = P < 0.05 versus siCtrl.
FOXO downregulation increases cell death in response to oxidative stress in human chondrocytes
To investigate the effect of FOXO downregulation on cell viability under oxidative stress conditions, we applied siFOXO. Transfection with siRNAs reduced FOXO1 protein by 86% and FOXO3 by 60% (Figure 1B). siRNA transfected chondrocytes were exposed to 25 to 250 μM of the exogenous oxidant t-BHP for 24 hours and MTT assay was performed to assess viability. There was no significant difference in cell viability between siCtrl and siFOXO transfected cells in the absence of t-BHP. Following exposure of t-BHP, there was a dose dependent and significant decrease in cell viability, in particular in the cells with knockdown of FOXO1 and both FOXO1 and FOXO3 (Figure 2A). Subsequently, caspase-3/7 activation and apoptosis were detected in order to assess the mechanism of cell death triggered by FOXO down-regulation when exposed to t-BHP. There was a significant increase in caspase activation in cells treated with siFOXO1+3 compared to siCtrl (Figure 2B) and apoptotic cell death was significantly increased in siFOXO1+3 cells treated with 250 μM of t-BHP (Figure 2C).
Figure 2.

(A) Changes in cell viability in siFOXO transfected chondrocytes under oxidative stress with t-BHP.
Chondrocytes were transfected with siRNAs and cell viability under treatment with t-BHP was assessed by MTT assay. Cell viability was calculated as the percentage of absorbance of t-BHP treated cells (25, 50, 100, and 250 μM of t-BHP) with respect cells not treated with t-BHP (0 μM of t-BHP). Graph shows the results of a total of 8 donors (N=8). Values are the mean and SD. * = P < 0.05 versus siCtrl at each concentration of t-BHP.
(B) Caspase activation and (C) induction of apoptosis in response to t-BHP in siRNA treated cells.
Graphs show the results of a total of 5 donors (N=5) for caspase activation and apoptosis detection in siRNA transfected cells treated with t-BHP (100 and 250 μM). Values are the mean and SD as the ratio of t-BHP treated cells (0, 100 and 250 μM of t-BHP) with siCtrl transfected cells not treated with t-BHP (0 μM of t-BHP). * = P < 0.05 versus siCtrl at each concentration of t-BHP.
(D) Changes in ROS generation and (E) redox state of glutathione in response to t-BHP in siRNA treated cells.
Graphs show the results of a total of 8 donors (N=8) for ROS generation and 10 donors for the ratio of GSSG to GSH in siRNA transfected cells treated with t-BHP (100 and 250 μM). Values are the mean and SD as the ratio of t-BHP treated cells (0, 100 and 250 μM of t-BHP) with siCtrl transfected cells not treated with t-BHP (0 μM of t-BHP). * = P < 0.05 versus siCtrl at each concentration of t-BHP. & = P < 0.05 versus siCtrl at 0 μM of t-BHP.
Intracellular ROS generation and oxidized glutathione are increased by FOXO downregulation
To evaluate the changes in redox-sensitive signaling mechanisms by FOXO downregulation under oxidative stress conditions, intracellular ROS generation and reduced and oxidized glutathione were measured. Exogenous t-BHP significantly induced intracellular ROS generation as detected by APF assay (Figure 2D). Moreover, the siFOXO1+3 treated cells showed significant increases in ROS generation compared to the siCtrl. An increased ratio of oxidized (GSSG) to reduced (GSH) state of glutathione was observed under FOXO downregulation and treatment with 250 μM of t-BHP (Figure 2E). These findings suggest that the siFOXO-treated cells were exposed higher levels of oxidative stress compared with siCtrl.
Anti-oxidant proteins and autophagy proteins are decreased by FOXO downregulation
To examine mechanisms related to oxidant induced cell death in chondrocytes with FOXO knock-down, we measured changes in the anti-oxidant proteins SOD2, GPX1, and catalase under the same conditions that were used to analyze cell viability. Knock-down of FOXO resulted in significant reductions in GPX-1 protein with 50 and 250 μM of t-BHP and catalase protein with 250 μM of t-BHP compared to control (Figure 3A). The combination of siFoxO1 and 3 also significantly decreased catalase at 50 μM t-BHP. These results suggest that FOXO downregulation attenuated the anti-oxidative defense by decreasing the expression of anti-oxidant proteins. SOD2 was not changed by downregulation of FOXO at any concentration of t-BHP.
Figure 3.

Changes in anti-oxidant proteins and autophagy proteins in response to t-BHP in siFOXO transfected chondrocytes.
Human chondrocytes transfected with siRNA were stimulated with t-BHP (50 and 250 μM) for 24 h. Protein extracts were analyzed by western blotting using antibodies to (A) anti-oxidant proteins (B) autophagy proteins and Sirt1. Graph shows the results of a total of 4 donors (N=4). Values are the mean and SD. * = P < 0.05 and ** = P < 0.01 versus siCtrl at each concentration of t-BHP. & = P < 0.05 versus siCtrl at 0 μM of t-BHP.
We next investigated whether downregulation of FOXO in chondrocytes affects the regulation of autophagy related genes. LC3-II and Beclin1 were increased in response to t-BHP stimulation in siCtrl treated cells (Figure 3B). This increased LC3-II and Beclin1 expression was significantly reduced in siFOXO1 and siFOXO1+3 treated cells. SIRT1 is an NAD-dependent protein deacetylase that regulates cell cycle, response to DNA damage, apoptosis and autophagy (25) and its expression is strongly reduced in OA cartilage (26). The basal levels of SIRT1 protein were also lower in siFOXO1 and siFOXO1+3 treated cells (Figure 3B) and remained at low levels even in cells treated with t-BHP.
Beclin1 downregulation reduces cell viability in response to oxidative stress in human chondrocytes
Human chondrocytes were treated with t-BHP following transfection of siRNA for Beclin1 in order to test the effect of reduced Beclin1, which is required for the initiation of autophagy, on cell viability under oxidative stress condition. Transfection with siBeclin1 reduced Beclin1 protein by 40% (Figure 4A). There was no significant difference in cell viability between siCtrl and siBeclin1 transfected cells in the absence of t-BHP. Following exposure of t-BHP, there was a significant decrease in cell viability in the cells with knockdown of Beclin1 (Figure 4B). With 250 μM of t-BHP, which is the dose that induces apoptosis, pan-caspase inhibitor (Z-VAD-FMK) significantly increased cell viability in both siCtrl and siBeclin1 whereas there was a significant reduction in siBeclin1 compared to siCtrl. These results suggest that autophagy is a protective mechanism against oxidative stress in human chondrocytes.
Figure 4.

(A and B) The effect of Beclin1 down-regulation on cell viability in response to t-BHP.
Human chondrocytes were transfected with siBeclin1 (A) and cell viability under treatment with t-BHP was examined by Resazurin assay (B). Graph shows the results of a total of 4 donors (N=4). Values are the mean and SD. * = P < 0.05 versus siCtrl. & = P < 0.05 versus siCtrl at 250 μM of t-BHP.
(C and D) The effect of FOXO overexpression on cell viability and anti-oxidant and autophagy proteins.
Image is representative of FOXO1 and FOXO3 protein expression in FOXO plasmid transfected cells. Cell viability under treatment with t-BHP was examined by MTT assay (C). Protein extracts were analyzed by western blotting. Graph shows the results of a total of 4 donors (N=4). Values are the mean and SD. * = P < 0.05 and ** = P < 0.01 versus pcDNA empty vector
FOXO overexpression increases cell viability in response to oxidative stress in human chondrocytes
Transfection with plasmids increased FOXO protein expression (Figure 4C). Both FOXO1 A3 and FOXO3 A3 are constitutive active forms of FOXO mutated at 3 Akt-phosphorylation sites (27). Cell viability in FOXO3 A3 overexpressing cells treated with 100 μM of t-BHP was significantly higher compared to pcDNA3 empty vector (Figure 4C). Overexpression of FOXO3 A3 showed significant increases in GPX-1, SOD2, and LC3 proteins at basal level and GPX1, Beclin1, and LC3 proteins under exposure to 50 μM of t-BHP compared to control vector (Figure 4D). There was no significant difference between FOXO1 A3 and control vector.
ADAMTS-4 and Chemerin gene and protein expression are increased by FOXO downregulation
To determine whether FOXOs are also involved in the regulation of catabolic and inflammatory mediators, we treated chondrocytes with IL-1β and analyzed changes in gene and protein expressions using real-time PCR, western blot, and ELISA.
Expressions of both ADAMTS-4 and Chemerin were significantly induced by siFOXO1 and siFOXO1+3 compared to siCtrl even in the absence of IL-1β. The IL-1β-mediated increases in ADAMTS-4 and Chemerin expression were augmented by siFOXO1 and siFOXO1+3 more than by siCtrl (Figure 5). COX2 was also significantly increased by downregulation of FOXO1, 3, and 1+3 in the absence of IL-1β but there was no difference in COX2 expression with IL-1β stimulation. FOXO knock-down did not significantly alter ADAMTS-5, MMP-13, MCP-1, IL-8, iNOS, and IL-6 expressions regardless of IL-1β stimulation.
Figure 5.

Changes in inflammatory gene expression in response to IL-1β in siFOXO transfected chondrocytes.
Human chondrocytes transfected with siRNA were stimulated with or without IL-1β (0.01 ng/ml) for 6 h. Inflammatory gene expression was analyzed by real-time PCR using primers as indicated. Graph shows the results of a total of 5 donors (N=5). Values are the mean and SD. * = P < 0.05 and ** = P < 0.01 versus siCtrl.
ADAMTS-4 protein was stimulated by IL-1β and significantly increased in siFOXO1 and siFOXO1+3 compared to siCtrl (Figure 6). IL-1β-induced Chemerin release in the supernatant was significantly increased in siFOXO1, siFOXO3, and siFOXO1+3 compared to siCtrl.
Figure 6.

Changes in ADAMTS-4 and Chemerin production in response to IL-1β in siFOXO transfected chondrocytes.
Human chondrocytes transfected with siRNA were stimulated with IL-1β (0.1 ng/ml) for 24 h. ADAMTS-4 protein was analyzed by western blot and Chemerin protein in the supernatant was measured by ELISA. Graph shows the results of a total of 4 donors (N=4). Values are the mean and SD. * = P < 0.05 and ** = P < 0.01 versus siCtrl.
DISCUSSION
The present study demonstrated that down-regulation of FOXO transcription factors in chondrocytes reduced cell viability in response to ROS. This was associated with decreased expression of anti-oxidant proteins and autophagy related proteins, and up-regulated ADAMTS-4 and Chemerin.
Our previous study on FOXO proteins expression in articular cartilage showed age-related reductions of FOXO proteins in human and mouse OA cartilage. In addition, this study demonstrated reduced FOXO1 and FOXO3 mRNA expression levels in human OA cartilage compared to young normal. Age-related reductions in expression of FOXO transcription factors were also reported in other skeletal tissues, resulting in changes in cell functions and extracellular matrix (5, 28). In bone, FoxO1 expression progressively decreased in mice from 2 to 12 months of age whereas FoxO3 and FoxO4 levels remained stable (5). Cell-specific deletion of FoxO resulted in an increase in oxidative stress and osteoblast apoptosis and a decrease in bone mass (5, 29). In mouse muscle, the expression level of FoxO3 and FoxO4 mRNA was significantly lower at 26 months than at 6 and 12 months, but FoxO1 expression showed no significant change with age (28).
FOXO transcription factors have been reported to regulate the expression of ROS scavengers in response to ROS (30, 31). Free radicals are detoxified by ROS scavengers present in chondrocytes and most other cells (32). High concentration of ROS resulted in cell death through necrosis (caused by critical ATP deficiency) or apoptosis. In this study, FOXO down-regulation in human chondrocyte resulted in increased intracellular oxidative stress and ROS-induced apoptosis with reduced ROS scavengers, GPX1 and catalase but not SOD2. These data support the concept that compromised antioxidant defenses related to reduced antioxidants is involved in cartilage aging and OA (14). SOD catalyzes superoxide radicals (O2) to hydrogen peroxide (H2O2), with hydrogen peroxide then being metabolized to H2O by GPX or catalase (33). tert-Butyl hydroperoxide (t-BHP) used in this study is a high thermal stable peroxide that is considered to be detoxified by GPX1 and catalase but not SOD2. On the other hand, Kayal et al. reported that silencing FOXO1 reduced apoptosis and the expression of proapoptotic mediators, caspases and TRAIL, stimulated by TNF-α in murine chondrogenic cell line (ATDC5) (34). Depending on cell context and type of stimulation, FOXO factors regulate diverse cellular functions including ROS detoxification, DNA repair, cell cycle arrest, and apoptosis.
Autophagy is a key mechanism to control protein quality for cellular homeostasis under oxidative stress condition. The rapid activation of autophagy involves post-translational modifications of autophagy proteins that control the autophagy cycle at several levels. In contrast, prolonged autophagy induction requires transcriptional control to replenish autophagy proteins that are destroyed during the fusion of autophagosomes with lysosomes (35–37). FOXO proteins have been reported to regulate autophagy as transcriptional activator for autophagy proteins such as LC3 and Beclin1 (38). In our study, FOXO down-regulation reduced LC3 and Beclin1 that increased in response to t-BHP, suggesting that FOXO proteins support oxidative stress resistance in part via regulating autophagy protein production in human chondrocytes. Down-regulation of Beclin1, which is involved in the initiation of autophagy, also resulted in reduction of cell viability in response to t-BHP. Furthermore, our results demonstrate that FOXO1 down-regulation suppressed SIRT1 which regulates oxidative stress and autophagy by modulating the signaling of FOXO and p53 (25). SIRT1 also could influence autophagy directly via its deacetylation of key components of the autophagy induction network, such as ATG5, 7, and 8 (39).
We observed differences in the effects of FOXO1 and FOXO3 on oxidative stress resistance between two experimental approaches, using siRNA and plasmid transfection. Specifically, FOXO1 siRNA showed greater loss in cell viability compared to FOXO3 siRNA. On the other hand, overexpression study showed that FOXO3 but not FOXO1 increased cell viability in response to t-BHP. Similarly, Rached et al. reported that only FoxO1 was required for resistance to oxidative stress in mouse osteoblasts with conditional FoxO knock out (5). In contrast, Ambrogini et al. reported that overexpression of FoxO3 in osteoblasts increased resistance to oxidative stress and decreased osteoblast apoptosis (29). Thus, although FOXO family members share a common DNA-binding site and regulate overlapping sets of target genes, functional differences between FOXO1 and FOXO3 are observed under different experimental conditions. This study also reproducibly observed that siFOXO1 transfection significantly increased FOXO3 protein expression. In the literature, Essaghir, A et al. reported that FOXO3 upregulates FOXO1 transcription by positive feedback mechanism in human fibroblasts (40). In addition, Al-Mubarak, B et al. reported that FOXO3, and to a lesser extent FOXO1 transactivates the FOXO1 promoter via FOXO binding site in neuronal cells (41). However, there has been no report about FOXO3 regulation by FOXO1. Possible explanation for this FOXO3 upregulation by siFOXO1 is that FOXO1 have negative feedback to FOXO3 expression or FOXO3 is upregulated with compensation for FOXO1 reduction.
FOXO transcription factors are regulated in a stress-dependent manner. Oxidative stress stimuli triggered the translocation of FOXO from the cytoplasm to the nucleus and FOXO acetylation by SIRT1 that activates FOXO transcriptional activity (42, 43). In the presence of t-BHP, GPX1, Beclin1, and LC3 proteins were increased whereas FOXO knock-down prevented these increases. We also found decreased SIRT1 in response to siFOXO, leading to reduced FOXO transcriptional activity. Resveratrol, an activator of SIRT1, was reported to inhibit inflammation and apoptosis and function as an anti-oxidant in chondrocyte by up-regulating SIRT1 expression (44, 45). Collectively, these findings suggest that cells with decreased FOXO and SIRT1 proteins fail to increase anti-oxidant and autophagy proteins in response to t-BHP.
FOXO proteins are also involved in signal transduction pathways related to inflammation (46–48). This study showed that, among inflammatory genes expressed in chondrocytes, ADAMTS-4 and Chemerin gene expression and protein production increased following FOXO1 down-regulation both with and without IL-1β stimulation. Both ADAMTS-4 and Chemerin have no FOXO1 DNA-binding domain at their promoters, however, it has been demonstrated that the direct association of FOXO proteins with diverse transcription factor families such as C/EBP, Smad3/4, and STAT3 can activate or repress diverse target genes and thus mediate the regulation of many cellular processes independently of FOXO DNA-binding (47). Moreover, SIRT1 activation inhibits NF-κB signaling and enhances oxidative metabolism and the resolution of inflammation. SIRT1 inhibits NF-κB signaling directly by deacetylating the p65 subunit of NF-κB complex (49). These findings might explain how the down-regulation of FOXO can affect ADAMTS-4 and Chemerin expression in cells stimulated with IL-1β.
In conclusion, the down-regulation of FOXO transcription factors in chondrocytes attenuated cell viability against oxidative stress, reducing antioxidant proteins and autophagy related proteins. These findings suggest that FOXO dysfunction observed in aging and OA cartilage cause reduced antioxidant defenses and autophagy. We also found that down-regulation of FOXO transcription factors in human chondrocytes induced some genes encoding matrix degrading enzymes and proinflammatory cytokines. Our data provide evidence of the key role of FOXO transcription factor as a mediator of oxidative stress resistance in chondrocytes and cartilage tissue homeostasis.
Acknowledgments
This study was supported by National Institutes of Health grants AG007996 and AR050631, and the Sam and Rose Stein Endowment Fund.
The authors acknowledge Merissa Olmer, Stuart Duffy and Lilo Creighton for technical assistance.
Footnotes
The authors have no conflicts of interest.
Contributors ML had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study design: ML, YA. Acquisition of data: YA, MS, OA, BC. Analysis and interpretation of data: YA, ML. Manuscript preparation and approval: YA, MS, OA, BC, YI, ML.
Competing interests None.
Ethics approval This study was conducted with the approval of the Scripps Human Subjects Committee.
References
- 1.Lotz M, Loeser RF. Effects of aging on articular cartilage homeostasis. Bone. 2012;51:241–8. doi: 10.1016/j.bone.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Longo VD, Finch CE. Evolutionary medicine: from dwarf model systems to healthy centenarians? Science. 2003;299(5611):1342–6. doi: 10.1126/science.1077991. [DOI] [PubMed] [Google Scholar]
- 3.Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278(5341):1319–22. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 4.Maiese K, Chong ZZ, Shang YC. OutFOXOing disease and disability: the therapeutic potential of targeting FoxO proteins. Trends in molecular medicine. 2008;14(5):219–27. doi: 10.1016/j.molmed.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rached MT, Kode A, Xu L, Yoshikawa Y, Paik JH, Depinho RA, et al. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell metabolism. 2010;11(2):147–60. doi: 10.1016/j.cmet.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell metabolism. 2007;6(6):472–83. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117(3):399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manolopoulos KN, Klotz LO, Korsten P, Bornstein SR, Barthel A. Linking Alzheimer’s disease to insulin resistance: the FoxO response to oxidative stress. Molecular psychiatry. 2010;15(11):1046–52. doi: 10.1038/mp.2010.17. [DOI] [PubMed] [Google Scholar]
- 9.Mendes AF, Caramona MM, Carvalho AP, Lopes MC. Differential roles of hydrogen peroxide and superoxide in mediating IL-1-induced NF-kappa B activation and iNOS expression in bovine articular chondrocytes. Journal of cellular biochemistry. 2003;88(4):783–93. doi: 10.1002/jcb.10428. [DOI] [PubMed] [Google Scholar]
- 10.Beecher BR, Martin JA, Pedersen DR, Heiner AD, Buckwalter JA. Antioxidants block cyclic loading induced chondrocyte death. The Iowa orthopaedic journal. 2007;27:1–8. [PMC free article] [PubMed] [Google Scholar]
- 11.Wolff KJ, Ramakrishnan PS, Brouillette MJ, Journot BJ, McKinley TO, Buckwalter JA, et al. Mechanical stress and ATP synthesis are coupled by mitochondrial oxidants in articular cartilage. J Orthop Res. 2013;31(2):191–6. doi: 10.1002/jor.22223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henrotin Y, Kurz B, Aigner T. Oxygen and reactive oxygen species in cartilage degradation: friends or foes? Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society. 2005;13(8):643–54. doi: 10.1016/j.joca.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Scott JL, Gabrielides C, Davidson RK, Swingler TE, Clark IM, Wallis GA, et al. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Annals of the rheumatic diseases. 2010;69(8):1502–10. doi: 10.1136/ard.2009.119966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jallali N, Ridha H, Thrasivoulou C, Underwood C, Butler PE, Cowen T. Vulnerability to ROS-induced cell death in ageing articular cartilage: the role of antioxidant enzyme activity. Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society. 2005;13(7):614–22. doi: 10.1016/j.joca.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 15.Khan IM, Gilbert SJ, Caterson B, Sandell LJ, Archer CW. Oxidative stress induces expression of osteoarthritis markers procollagen IIA and 3B3(−) in adult bovine articular cartilage. Osteoarthritis Cartilage. 2008;16(6):698–707. doi: 10.1016/j.joca.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 16.Kishimoto H, Akagi M, Zushi S, Teramura T, Onodera Y, Sawamura T, et al. Induction of hypertrophic chondrocyte-like phenotypes by oxidized LDL in cultured bovine articular chondrocytes through increase in oxidative stress. Osteoarthritis Cartilage. 2010;18(10):1284–90. doi: 10.1016/j.joca.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 17.Ahmad R, Sylvester J, Ahmad M, Zafarullah M. Involvement of H-Ras and reactive oxygen species in proinflammatory cytokine-induced matrix metalloproteinase-13 expression in human articular chondrocytes. Archives of biochemistry and biophysics. 2011;507(2):350–5. doi: 10.1016/j.abb.2010.12.032. [DOI] [PubMed] [Google Scholar]
- 18.Martin JA, Klingelhutz AJ, Moussavi-Harami F, Buckwalter JA. Effects of oxidative damage and telomerase activity on human articular cartilage chondrocyte senescence. The journals of gerontology Series A, Biological sciences and medical sciences. 2004;59(4):324–37. doi: 10.1093/gerona/59.4.b324. [DOI] [PubMed] [Google Scholar]
- 19.Brandl A, Hartmann A, Bechmann V, Graf B, Nerlich M, Angele P. Oxidative stress induces senescence in chondrocytes. J Orthop Res. 2011;29(7):1114–20. doi: 10.1002/jor.21348. [DOI] [PubMed] [Google Scholar]
- 20.Goodwin W, McCabe D, Sauter E, Reese E, Walter M, Buckwalter JA, et al. Rotenone prevents impact-induced chondrocyte death. J Orthop Res. 2010;28(8):1057–63. doi: 10.1002/jor.21091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis and rheumatism. 2010;62(3):791–801. doi: 10.1002/art.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akasaki Y, Hasegawa A, Saito M, Asahara H, Iwamoto Y, Lotz MK. Dysregulated FOXO transcription factors in articular cartilage in aging and osteoarthritis. Osteoarthritis Cartilage. 2014;22(1):162–70. doi: 10.1016/j.joca.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanco FJ, Ochs RL, Schwarz H, Lotz M. Chondrocyte apoptosis induced by nitric oxide. The American journal of pathology. 1995;146(1):75–85. [PMC free article] [PubMed] [Google Scholar]
- 24.Setsukinai K, Urano Y, Kakinuma K, Majima HJ, Nagano T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. The Journal of biological chemistry. 2003;278(5):3170–5. doi: 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- 25.Salminen A, Kaarniranta K. SIRT1: regulation of longevity via autophagy. Cellular signalling. 2009;21(9):1356–60. doi: 10.1016/j.cellsig.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 26.Fujita N, Matsushita T, Ishida K, Kubo S, Matsumoto T, Takayama K, et al. Potential involvement of SIRT1 in the pathogenesis of osteoarthritis through the modulation of chondrocyte gene expressions. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2011;29(4):511–5. doi: 10.1002/jor.21284. [DOI] [PubMed] [Google Scholar]
- 27.van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nature reviews Molecular cell biology. 2007;8(6):440–50. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 28.Furuyama T, Yamashita H, Kitayama K, Higami Y, Shimokawa I, Mori N. Effects of aging and caloric restriction on the gene expression of Foxo1, 3, and 4 (FKHR, FKHRL1, and AFX) in the rat skeletal muscles. Microscopy research and technique. 2002;59(4):331–4. doi: 10.1002/jemt.10213. [DOI] [PubMed] [Google Scholar]
- 29.Ambrogini E, Almeida M, Martin-Millan M, Paik JH, Depinho RA, Han L, et al. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell metabolism. 2010;11(2):136–46. doi: 10.1016/j.cmet.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419(6904):316–21. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 31.Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295(5564):2450–2. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- 32.Baker MS, Feigan J, Lowther DA. Chondrocyte antioxidant defences: the roles of catalase and glutathione peroxidase in protection against H2O2 dependent inhibition of proteoglycan biosynthesis. The Journal of rheumatology. 1988;15(4):670–7. [PubMed] [Google Scholar]
- 33.Ramakrishnan P, Hecht BA, Pedersen DR, Lavery MR, Maynard J, Buckwalter JA, et al. Oxidant conditioning protects cartilage from mechanically induced damage. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2010;28(7):914–20. doi: 10.1002/jor.21072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kayal RA, Siqueira M, Alblowi J, McLean J, Krothapalli N, Faibish D, et al. TNF-alpha mediates diabetes-enhanced chondrocyte apoptosis during fracture healing and stimulates chondrocyte apoptosis through FOXO1. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2010;25(7):1604–15. doi: 10.1002/jbmr.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandri M. FOXOphagy path to inducing stress resistance and cell survival. Nature cell biology. 2012;14(8):786–8. doi: 10.1038/ncb2550. [DOI] [PubMed] [Google Scholar]
- 36.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Molecular cell. 2010;40(2):280–93. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mammucari C, Schiaffino S, Sandri M. Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy. 2008;4(4):524–6. doi: 10.4161/auto.5905. [DOI] [PubMed] [Google Scholar]
- 38.Ferdous A, Battiprolu PK, Ni YG, Rothermel BA, Hill JA. FoxO, autophagy, and cardiac remodeling. Journal of cardiovascular translational research. 2010;3(4):355–64. doi: 10.1007/s12265-010-9200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng F, Tang BL. Sirtuins’ modulation of autophagy. Journal of cellular physiology. 2013;228(12):2262–70. doi: 10.1002/jcp.24399. [DOI] [PubMed] [Google Scholar]
- 40.Essaghir A, Dif N, Marbehant CY, Coffer PJ, Demoulin JB. The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. The Journal of biological chemistry. 2009;284(16):10334–42. doi: 10.1074/jbc.M808848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Mubarak B, Soriano FX, Hardingham GE. Synaptic NMDAR activity suppresses FOXO1 expression via a cis-acting FOXO binding site: FOXO1 is a FOXO target gene. Channels. 2009;3(4):233–8. doi: 10.4161/chan.3.4.9381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303(5666):2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi Y, Furukawa-Hibi Y, Chen C, Horio Y, Isobe K, Ikeda K, et al. SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. International journal of molecular medicine. 2005;16(2):237–43. [PubMed] [Google Scholar]
- 44.Lei M, Wang JG, Xiao DM, Fan M, Wang DP, Xiong JY, et al. Resveratrol inhibits interleukin 1beta-mediated inducible nitric oxide synthase expression in articular chondrocytes by activating SIRT1 and thereby suppressing nuclear factor-kappaB activity. European journal of pharmacology. 2012;674(2–3):73–9. doi: 10.1016/j.ejphar.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 45.Takayama K, Ishida K, Matsushita T, Fujita N, Hayashi S, Sasaki K, et al. SIRT1 regulation of apoptosis of human chondrocytes. Arthritis Rheum. 2009;60(9):2731–40. doi: 10.1002/art.24864. [DOI] [PubMed] [Google Scholar]
- 46.Watroba M, Maslinska D, Maslinski S. Current overview of functions of FoxO proteins, with special regards to cellular homeostasis, cell response to stress, as well as inflammation and aging. Advances in medical sciences. 2012;57(2):183–95. doi: 10.2478/v10039-012-0039-1. [DOI] [PubMed] [Google Scholar]
- 47.van der Vos KE, Coffer PJ. FOXO-binding partners: it takes two to tango. Oncogene. 2008;27(16):2289–99. doi: 10.1038/onc.2008.22. [DOI] [PubMed] [Google Scholar]
- 48.van der Vos KE, Coffer PJ. The extending network of FOXO transcriptional target genes. Antioxidants & redox signaling. 2011;14(4):579–92. doi: 10.1089/ars.2010.3419. [DOI] [PubMed] [Google Scholar]
- 49.Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cellular signalling. 2013;25(10):1939–48. doi: 10.1016/j.cellsig.2013.06.007. [DOI] [PubMed] [Google Scholar]