

Abstract
The cell-impermeant lidocaine derivative QX-314 blocks sodium channels via intracellular mechanisms. In somatosensory nociceptive neurons, open transient receptor potential vanilloid type 1 (TRPV1) receptors provide a transmembrane passageway for QX-314 to produce long-lasting analgesia. Many cranial primary afferents express TRPV1 at synapses on neurons in the nucleus of the solitary tract and caudal trigeminal nucleus (Vc). Here, we investigated whether QX-314 interrupts neurotransmission from primary afferents in rat brain-stem slices. Shocks to the solitary tract (ST) activated highly synchronous evoked excitatory postsynaptic currents (ST-EPSCs). Application of 300 μM QX-314 increased the ST-EPSC latency from TRPV1+ ST afferents, but, surprisingly, it had similar actions at TRPV1− ST afferents. Continued exposure to QX-314 blocked evoked ST-EPSCs at both afferent types. Neither the time to onset of latency changes nor the time to ST-EPSC failure differed between responses for TRPV1+ and TRPV1− inputs. Likewise, the TRPV1 antagonist capsazepine failed to prevent the actions of QX-314. Whereas QX-314 blocked ST-evoked release, the frequency and amplitude of spontaneous EPSCs remained unaltered. In neurons exposed to QX-314, intracellular current injection evoked action potentials suggesting a presynaptic site of action. QX-314 acted similarly at Vc neurons to increase latency and block EPSCs evoked from trigeminal tract afferents. Our results demonstrate that QX-314 blocked nerve conduction in cranial primary afferents without interrupting the glutamate release mechanism or generation of postsynaptic action potentials. The TRPV1 independence suggests that QX-314 either acted extracellularly or more likely entered these axons through an undetermined pathway common to all cranial primary afferents.
Keywords: NTS, QX-314, solitary tract, TRPV1
lidocaine is a lipophilic, local anesthetic that indiscriminately inhibits neuron excitability by disrupting action potential generation. In contrast, the lidocaine derivative N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium chloride (QX-314) contains a positively charged amino group making it membrane impermeant by passive diffusion and is widely reported to be unable to interact with the intracellular domain of voltage-gated sodium channels (VGSCs) when applied externally (Bischoff et al. 2003). However, intraterminal dialysis of presynaptic terminals of the calyx of Held with low concentrations of QX-314 (200–300 μM) led to a progressive block of VGSCs (Kim et al. 2010b). To overcome the membrane barrier to intracellular entry, one strategy has exploited the expression of the transient receptor potential vanilloid type 1 (TRPV1) receptor to inhibit pain selectively (Binshtok et al. 2007). TRPV1 is a nonselective cation channel activated by high temperature, low pH, and vanilloid ligands (Chung et al. 2008; Premkumar and Abooj 2013). Pairing QX-314 with capsaicin, a TRPV1 agonist, facilitated passage of QX-314 through the TRPV1 channel, which led to QX-314 accumulation and blockade of VGSCs (Binshtok et al. 2007; Brenneis et al. 2013; Puopolo et al. 2013). Since TRPV1 is expressed in a subset of nociceptive peripheral nerves, behavior tests showed that QX-314 coadministered with capsaicin interrupted noxious mechanical and thermal responses, whereas motor function remained unaffected across several somatic targets tested (Binshtok et al. 2007; Kim et al. 2010a). In some behavior studies, however, QX-314 injected alone blocked both sensory and motor function, which raises questions about intracellular access of QX-314 from the extracellular space (Lim et al. 2007; Ries et al. 2009; Shen et al. 2012).
Cranial visceral primary afferents from the heart, lungs, and gastrointestinal system send their axons centrally to the nucleus of the solitary tract (NTS), many of which express TRPV1 (Andresen et al. 2012; Cavanaugh et al. 2011; Patterson et al. 2003). TRPV1 expression divides solitary tract (ST) afferents into two broad classes, TRPV1+ and TRPV1−, and each class contacts different NTS neurons, a segregation that offers unique experimental advantages (Doyle and Andresen 2001; Jin et al. 2004; McDougall et al. 2009). Action potentials evoked in ST axons activate synchronous glutamate release with characteristics that are quite similar for both afferent classes and depend on TTX-sensitive VGSCs and voltage-gated calcium channels (Fawley et al. 2011; Mendelowitz et al. 1995; Schild et al. 1993). However, spontaneous release of glutamate at TRPV1+ afferents relies on a distinct pool of vesicles that are driven by TRPV1 activation and independent from the readily releasable pool responsible for evoked transmission (Fawley et al. 2011, 2014; Peters et al. 2010; Shoudai et al. 2010). This TRPV1-operated pool is highly temperature-sensitive and is responsible for the high basal rates of glutamate release observed at TRPV1+ afferents (Shoudai et al. 2010) and includes caudal trigeminal (Vc) brain-stem second-order sensory neurons (Largent-Milnes et al. 2014).
Here, we report that low concentrations of QX-314 alone delayed and then blocked both ST and trigeminal tract-evoked transmission regardless of TRPV1 expression in NTS and Vc second-order neurons, respectively. The identical timing of QX-314 effects and the temporary “rescue” of evoked ST-excitatory postsynaptic currents (ST-EPSCs) by increased shock intensity suggest that inhibiting the conduction of the ST afferent action potential was the main action of QX-314. Despite this block of evoked release, the spontaneous release of glutamate in NTS neurons remained unaltered. Action potentials evoked by current injection in the postsynaptic neuron were also unaffected by QX-314. These effects were consistent with actions on presynaptic VGSCs within ST afferents while leaving glutamate release mechanisms as well as postsynaptic action potential generation intact. Our results demonstrate that QX-314 specifically blocked cranial afferent transmission similarly for TRPV1+ and TRPV1− afferents by a TRPV1-independent mechanism.
METHODS
All animal procedures were approved by the Institutional Animal Care and Use Committee at Oregon Health and Science University and conformed to animal welfare guidelines issued by the National Institutes of Health (NIH) publication Guide for the Care and Use of Laboratory Animals.
Slice preparations.
NTS brain-stem slices were prepared from adult (>150 g) male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) as described previously (Doyle and Andresen 2001). Under deep isoflurane anesthesia, the brain stem was removed and tilted so that a horizontal brain-stem slice (250 μm thick) contained 1–3 mm of the ST in the same plane as the medial NTS. Slices were cut in ice-cold artificial cerebral spinal fluid (ACSF) using a sapphire blade (Delaware Diamond Knives, Wilmington, DE) mounted on a vibrating microtome (VT1000 S; Leica Microsystems, Bannockburn, IL). Immediately, slices were submerged in a recording chamber containing ACSF composed of, in mM, 125 NaCl, 3 KCl, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3, 10 glucose, and 2 CaCl2, bubbled with 95% O2-5% CO2. ACSF was perfused continuously (1.6–2.0 ml/min) at a bath temperature of 32°C controlled by an in-line heating system (TC2BIP with HPRE2HF and TH-10Km bath probe; Cell MicroControls, Norfolk, VA). For slices to study the Vc, hindbrains of male Sprague-Dawley rat pups (postnatal days 13-23; Charles River Laboratories) were prepared as described previously (Largent-Milnes et al. 2014). Under deep isoflurane anesthesia, the brain stem was removed, blocked, and mounted on a vibrating microtome. Slices were cut with a sapphire blade from the ventral side and then submerged in a recording chamber containing ACSF as above.
Patch-clamp recording.
Recording pipettes (2.5–4.0 MΩ for NTS and 3.5–5.0 MΩ for Vc) were filled with an intracellular solution composed of, in mM, 6 NaCl, 4 NaOH, 130 K-gluconate, 11 EGTA, 1 CaCl2, 10 HEPES, 1 MgCl2, 2 Na2ATP, and 0.2 Na2GTP (pH 7.3). Slices were visualized with infrared differential interference contrast microscopy using a fixed-stage Axioskop 2 FS Plus (Zeiss, Thornwood, NJ) with a digital camera (SPOT Pursuit USB Camera; SPOT Imaging Solutions, Sterling Heights, MI). Neurons were voltage-clamped at −60 mV using a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA), and liquid junction potentials were uncorrected. Data were sampled at 20 kHz and filtered at 10 kHz (Digidata 1321A analog-to-digital converter using Clampex 9; Molecular Devices). Access resistance and input resistance were calculated from current responses to a brief hyperpolarizing step (from −60 to −65 mV, 200 ms) and monitored throughout the experiments. All experiments included the GABAA antagonist gabazine (SR-95531; 3 μM) to isolate EPSCs. Most drugs were purchased from either Tocris (R&D Systems, Minneapolis, MN) or Sigma-Aldrich (St. Louis, MO). QX-314 as the chloride salt was purchased for these studies from three separate vendors, Tocris, Sigma-Aldrich, and Abcam (Cambridge, MA), and dissolved in water. All drugs were bath-applied.
Afferent activation and identification.
After securing the slice in the recording chamber, a concentric bipolar stimulating electrode (200-μm outer diameter; Frederick Haer, Bowdoinham, ME) was placed on the ST 1–3 mm rostral from the cell bodies located in the medial NTS. Once the recording was established, shocks of varied intensity were delivered to the ST in tests using bursts of five shocks at 50 Hz repeated every 3 s to determine the recruitment threshold. Gradual increases in stimulus intensity yielded a recruitment curve for each neuron. Generally, a near-minimum intensity was selected for each study, which triggered the same ST-EPSC without failures to the first shock. Following the recruitment routine, stimulation was switched to the standard protocol that delivered a burst of 5 suprathreshold shocks every 6 s. The latency of the 1st ST-EPSC in each burst was measured as the time from stimulus artifact until onset of the ST-EPSC and analyzed over the 1st 50 iterations. If the standard deviation of the latency (jitter) was <200 μs, the ST-EPSC was considered monosynaptic. ST-EPSCs with jitter >200 μs were considered polysynaptic and not studied further. The presence of a transient increase in frequency of spontaneous EPSCs (sEPSCs) in the 1 s following the evoked ST-EPSCs above the sEPSC rate for the 1 s preceding stimulation was considered an initial indicator that the ST input was TRPV1+ (Fawley et al. 2011; Peters et al. 2010). At the end of each experiment, application of 100 nM capsaicin confirmed the TRPV1+ classification through an increase in the sEPSC frequency and block of the evoked ST-EPSC. TRPV1− afferents showed no synaptic changes during capsaicin.
Postsynaptic excitability assessment.
To test whether QX-314 might have actions on the recorded postsynaptic NTS neurons, a series of tests were conducted in current-clamp. First, we tested the five-shock ST stimulation protocol and evoked excitatory postsynaptic potentials (EPSPs) including action potentials. Second, in the absence of ST stimulation, a series of depolarizing current steps (500 ms) were injected in increments of 20 pA to trigger action potential firing directly. Following these measurements in control conditions, the neuron was placed in voltage-clamp and exposed to QX-314. Once the latency and failure rate of the ST-EPSCs increased during QX-314 exposure, the neuron was switched back to current-clamp to reevaluate the ST stimulation protocol and the current injection protocol in the continued presence of QX-314.
Caudal trigeminal nucleus.
Trigeminal Vc neurons, like NTS, are divided between TRPV1+ and TRPV1− afferents (Fawley et al. 2014; Largent-Milnes et al. 2014). Procedures for Vc neurons followed similar general protocols as for NTS. In Vc slices, the concentric bipolar stimulating electrode was placed on the trigeminal tract 0.5–1 mm rostral from the cell bodies located within the outer lamina (LI/IIo) of the Vc. Neurons were selected for recording from a translucent band ≤200 μm medial to the spinal trigeminal tract. All Vc recordings were done in voltage-clamp to study trigeminal tract-evoked EPSCs. Recruitment curves were generated and analyzed in a fashion similar to NTS neurons, and only synaptic responses with jitter <200 μs were studied in control and in the presence of 300 μM QX-314.
Data analysis and statistics.
All analysis was performed using O-phys (see Hofmann et al. 2011; Gainesville, FL) and Origin 8.6 (OriginLab, Northampton, MA) except for latency measurements (Clampfit 9; Molecular Devices). Most analyses focused on the responses of the first ST-EPSC (EPSC1) in the train of five shocks to avoid the influence of frequency-dependent mechanisms for depression and failures across multiple trials in each condition (McDougall et al. 2009). The latency and the amplitude of the 1st ST-EPSC of the burst of 5 were measured for comparison between control and QX-314. In these comparisons, a sample containing 30 trials (3 min) of the 6-s standard protocol was collected in control and then in the presence of QX-314 over the 3 min (30 trials) before failure onset. For failure rates in QX-314, the 30 trials were collected over the last 3 min of QX-314 application. To evaluate the potential for a systematic difference in the timing of QX-314 responses, the “latency change onset time” was measured as the time from QX-314 application until consecutive latency measurements exceeded 3× the control jitter. The “failure onset time” was measured as the time from QX-314 application until consecutive failures of EPSC1 across successive trials. The frequency and amplitude of sEPSCs were collected during the 1 s before ST stimulation for 30 trials (3 min) during control and at 7 min of exposure to QX-314. Within individual neurons, a Kolmogorov-Smirnov (K-S) test assessed the significance of any changes to interevent times or amplitudes of sEPSCs. Group data for summary purposes were compared using t-tests or 1-way ANOVA with Tukey or Bonferroni post hoc testing (OriginLab).
RESULTS
Shocks to the ST elicited ST-EPSCs with nearly invariant latency indicative of a monosynaptic afferent connection. The burst of shocks evoked marked frequency-dependent depression and latency jitter <200 μs, consistent with second-order neurons. The burst of stimuli often triggered a transient increase in sEPSCs trailing the evoked response, termed the asynchronous release of glutamate (Fig. 1, A and B). The presence of asynchronous release and the high rate of spontaneous release were highly predictive of TRPV1 expression on ST afferents (TRPV1+). Application of capsaicin at the end of the experiment triggered an increase in sEPSC frequency (Fig. 1C) and inhibited the ST-EPSC (data not shown), which verified that ST-EPSCs of this neuron were TRPV1+ (Peters et al. 2010, 2011). In these TRPV1+ neurons, addition of 300 μM QX-314 to the bath progressively increased the latency of the ST-EPSC (Fig. 1, D and E) and ultimately resulted in failures of ST-EPSCs (Fig. 1, D and F). The amplitudes of successful ST-EPSCs also decreased in QX-314. Note that, interspersed with failures (0 amplitude values), there were successful ST-EPSCs with amplitudes similar to those preceding the failures. Across neurons (n = 10), QX-314 increased the latency to 119.5 ± 3.9% of control (Fig. 1G; P = 0.002, paired t-test) and increased the failure rate of evoked ST-EPSCs (Fig. 1H; P < 0.001, paired t-test). Preliminary experiments indicated that lower concentrations of QX-314 (30 μM) were less effective. This lower concentration required significantly longer exposures before the onset of the latency shift (30 μM: 13.84 ± 3.77 min, n = 5; 300 μM: 6.17 ± 0.60 min, n = 10; P = 0.01, t-test) but still increased the latency (to 108.5 ± 3.34%, n = 5; P = 0.045, paired t-test) and the failure rate (n = 3; P = 0.01, paired t-test); thus 300 μM was used for most testing. To guard against the potential for inadvertent, active contaminants, QX-314 from three different vendors (see methods) was tested but yielded similar results. Together, these data suggested that QX-314 acted presynaptically to delay and then ultimately block axonal conduction at ST afferents without explicitly activating TRPV1 (e.g., capsaicin).
Fig. 1.
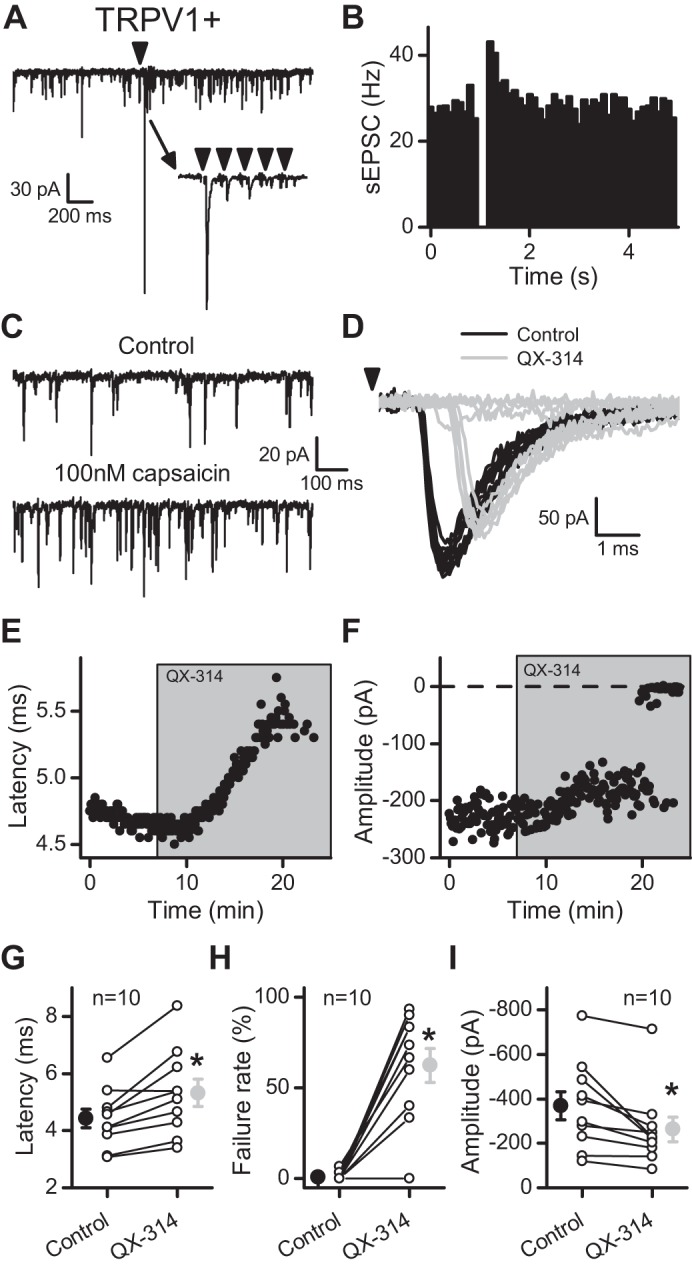
N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium chloride (QX-314) delayed and then blocked evoked solitary tract-excitatory postsynaptic currents (ST-EPSCs) at transient receptor potential vanilloid type 1+ (TRPV1+) afferents. In A–F, the data come from a single representative cell. A: in this single sweep, the arrow indicates stimulation of the ST (5 pulses at 50 Hz), which caused a transient increase in spontaneous EPSCs (sEPSCs). The inset shows the frequency-dependent depression of the evoked response. B: diary plot portrays the transient increase in sEPSC frequency, termed asynchronous release, present after stimulation in TRPV1+ afferents. The events were placed in 100-ms bins and collected over 50 trials (5 min). C: the TRPV1 agonist capsaicin increased the frequency of sEPSCs at neurons expressing TRPV1. D: application of 300 μM QX-314 (light gray) caused an increase in the latency and an ultimate block of the evoked ST-EPSC compared with control (black). The arrow indicates stimulation of the ST. E: exposure to QX-314 resulted in a progressive increase in the latency of the evoked ST-EPSC. F: the amplitude of the evoked ST-EPSC decreased slightly before resulting in failures in the presence of QX-314. G: the latency increased on average over 10 neurons tested (control, black; QX-314, light gray). H: the failure rate of the 1st ST-EPSC (EPSC1) increased in 9/10 neurons (control, black; QX-314, light gray). I: before failures, the amplitude decreased on average over 10 neurons tested (control, black; QX-314, light gray). All asterisks are for P ≤ 0.05.
Given the membrane-impermeant structure of QX-314 and its intracellular site of action (Ragsdale et al. 1994), blockade of synaptic transmission by extracellular exposure without coapplication of a TRPV1 agonist was a surprising result. Entry of QX-314 through tonically active TRPV1 represented one possible explanation. The bath temperature was sufficient to activate TRPV1 within ST afferents and drive spontaneous release of glutamate (Peters et al. 2010; Shoudai et al. 2010). Others suggest that TRPV1 antagonism blocks QX-314 actions (Ries et al. 2009). However, application of 10 μM capsazepine had no effect on latency, amplitude, or failure rate and did not prevent QX-314 from increasing the latency of the evoked ST-EPSC (to 129.3 ± 9.69% of control, n = 5; P = 0.04, 1-way repeated-measures ANOVA) or causing failures (Fig. 2). The existence of TRPV1− afferents provided an additional, natural experimental control for TRPV1 participation in QX-314 entry. We recorded ST-EPSCs from NTS neurons with low basal sEPSC activity and no discernible asynchronous release, suggestive of TRPV1− afferents (Fig. 3, A and B). In such neurons, 300 μM QX-314 increased the latency of the ST-EPSC in a manner quite comparable with TRPV1+ neurons (Fig. 3, D and E). Similar to the TRPV1+ cohort, the latency continued to increase until evoked ST-EPSCs failed (Fig. 3, D and F). Across TRPV1− neurons (n = 6), QX-314 increased the latency to 118 ± 7.0% of control (Fig. 3G; P = 0.02, paired t-test) and increased the failure rate of the ST-EPSC (Fig. 3H; P = 0.003, paired t-test). None of these neurons responded to capsaicin, and thus they were confirmed as TRPV1− (Fig. 3C). Such observations indicated that QX-314 blocked ST transmission irrespective of TRPV1 expression.
Fig. 2.
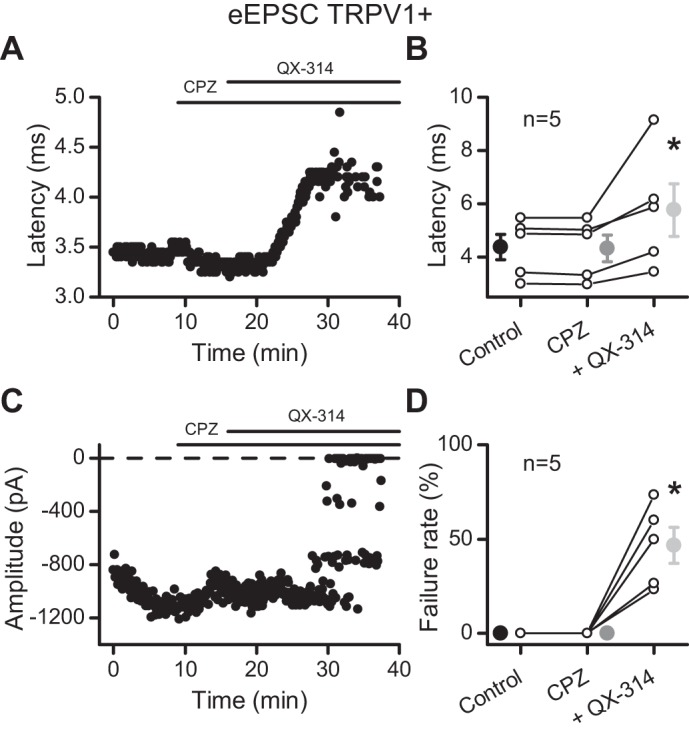
Capsazepine does not prevent the effects of QX-314. In the presence of 10 μM capsazepine (CPZ), a TRPV1 antagonist, QX-314 increased the latency (A) and caused failures (C) of the evoked ST-EPSC in this representative cell. B: summary of the QX-314 effects on latency. D: QX-314 increased the failure rate of EPSC1. All asterisks are for P ≤ 0.05. eEPSC, evoked EPSC.
Fig. 3.
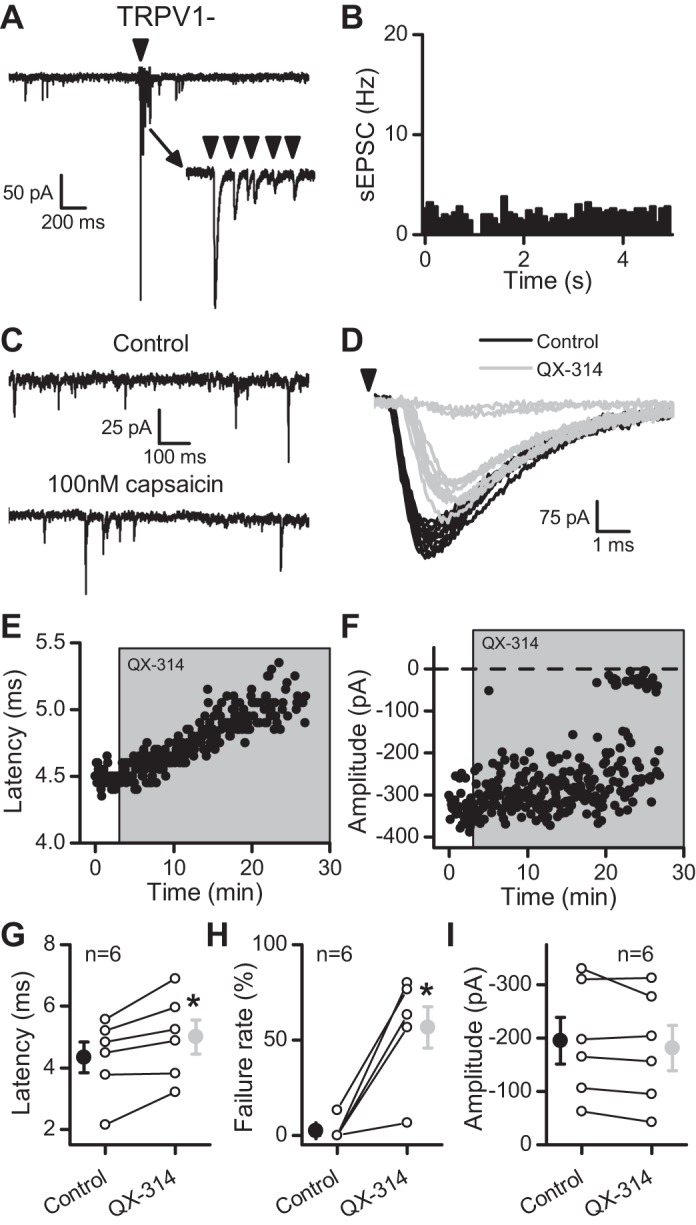
QX-314 delayed and then blocked evoked ST-EPSCs at TRPV1− afferents. In A–F, the data come from a single representative cell. A: in this single sweep, the arrow indicates stimulation of the ST (5 pulses at 50 Hz). Notice the lower rate of sEPSCs before stimulation and the lack of a transient increase in sEPSC frequency following ST stimulation compared with TRPV1+ afferents (Fig. 1). The inset demonstrates the frequency-dependent depression of the evoked response, which is similar to that seen in TRPV1+ afferents. B: TRPV1− afferents lack asynchronous release following stimulation of the ST (compare with Fig. 1B). Events were collected in 100-ms bins over 50 trials (5 min). C: this neuron lacked TRPV1 expression because capsaicin failed to increase the rate of sEPSCs (compare with Fig. 1C). D: similar to TRPV1+ afferents, 300 μM QX-314 increased the latency and caused failure of the evoked ST-EPSCs at a TRPV1− neuron. Black traces are from trials in control, whereas light gray traces are from trials in QX-314. E: the latency progressively increased following exposure to QX-314. F: the evoked ST-EPSC eventually failed in the presence of QX-314. G: the latency increased on average over 6 neurons tested (control, black; QX-314, light gray). H: summary of the failure rate of EPSC1 (control, black; QX-314, light gray). I: before failure, QX-314 did not significantly change the amplitude of ST-EPSCs (control, black; QX-314, light gray). All asterisks represent P ≤ 0.05.
A portion of NTS neurons that received TRPV1− afferents expressed presynaptic P2X3 (Jin et al. 2004; Shigetomi and Kato 2004). The P2X3 ion channel broadly resembles TRPV1 with a wide-pore, nonselective cation channel capable of passing large, organic cations (Virginio et al. 1999). These properties suggest that P2X3 could provide entry for QX-314 at TRPV1− afferents. However, the P2X receptor antagonist PPADS (20 μM) failed to prevent QX-314-induced increases in the latency of the ST-EPSC (121.4 ± 7.3% of control, n = 3; P = 0.17, 1-way repeated-measures ANOVA) or the QX-314 block of the evoked ST-EPSC (Fig. 4), similar to all other ST-EPSCs. Thus neither P2X3 nor TRPV1 was required for QX-314 actions.
Fig. 4.
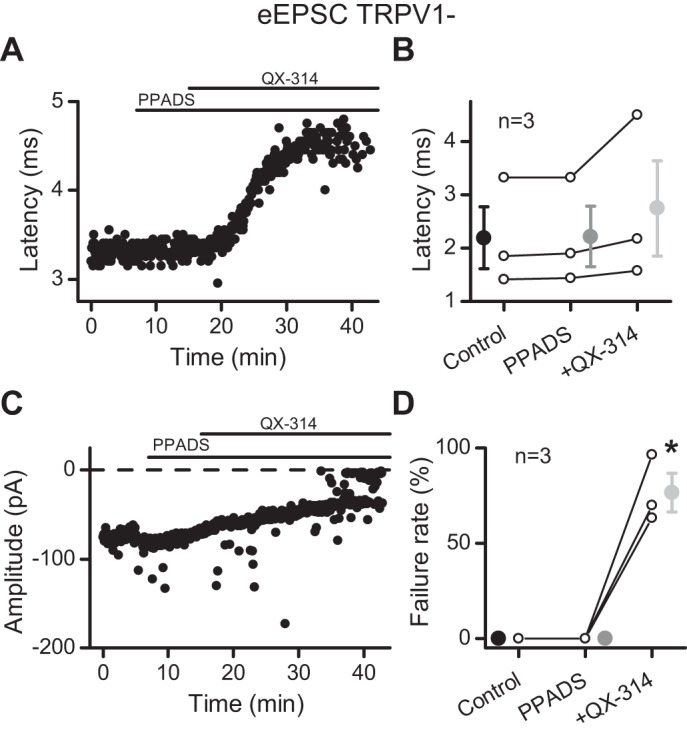
The purinergic receptor antagonist PPADS failed to block the effect of QX-314 at TRPV1− afferents. Application of QX-314 in the presence of 20 μM PPADS does not prevent the increase in latency (A) or the failures (C) of ST-EPSCs in this representative cell. B: summary of the QX-314 effects on latency. D: QX-314 increased the failure rate of EPSC1. All asterisks are for P ≤ 0.05.
QX-314 inhibited evoked release similarly between TRPV1+ and TRPV1− afferents. In both afferent types, the latency of the evoked ST-EPSC progressively increased until failures occurred. The onset of the change in latency was not different in the afferent subsets (Fig. 5A; P = 0.59, t-test), and the time to the start of failures was consistent between afferent types (Fig. 5B; P = 0.95, t-test). These results demonstrate that TRPV1 is not required for the QX-314 effects. The consistency in the timing of these actions suggests that QX-314 acts on a similar process within TRPV1+ and TRPV1− fibers.
Fig. 5.
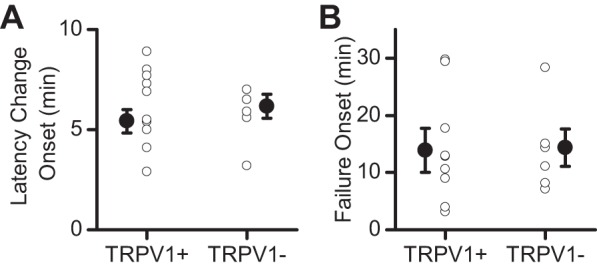
The timing of QX-314 effects is similar between TRPV1+ and TRPV1− afferents. A: the start to latency changes was not different between TRPV1+ and TRPV1− afferents. B: the time to failure onset of the ST-EPSC was similar between TRPV1+ and TRPV1− afferents.
QX-314 block of sodium channels is activity-dependent (Ragsdale et al. 1994). Thus the gradual increase in latency before transmission failure is consistent with a gradual intracellular accumulation of QX-314 that results in concentration-dependent block of VGSCs. We hypothesize that the loss of functioning sodium channels during QX-314 exposure depresses the conduction of action potentials. In support of this idea, the ST shock intensity was increased and succeeded in briefly “rescuing” failed ST-EPSCs (Fig. 6). The restored ST-EPSCs were similar in shape and amplitude to the predrug responses indicating that the higher shock intensity activated the same ST input (Fig. 6C). However, continued exposure to QX-314 resulted in a return to failures of the ST-EPSC (Fig. 6B). The control recruitment curve indicated that higher stimulus intensities failed to recruit added ST inputs in this intensity range, and thus the higher stimulation intensity likely evoked the same ST-EPSC input in the presence of QX-314 (Fig. 6A). The control and QX-314 restored evoked ST-EPSCs (Fig. 6D) had similar rise times (P = 0.83, t-test) and decay time constants (P = 0.5, t-test). These results suggest that neurotransmitter release was similar at both intensities and QX-314 only affected the action potential triggering the release.
Fig. 6.
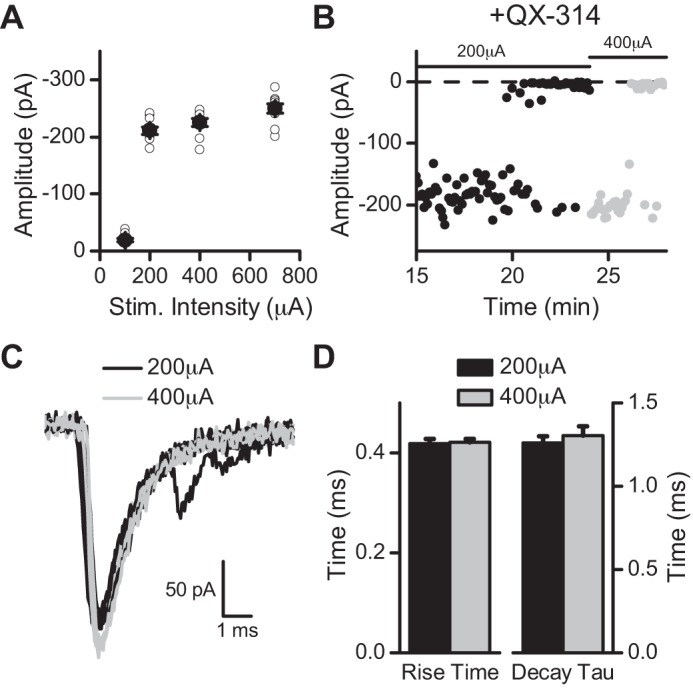
Higher stimulation intensities temporarily rescued ST-EPSCs. A: in control, gradual increases in stimulation intensity (Stim. Intensity) revealed a sharp threshold for obtaining a ST-EPSC. In this recruitment curve, notice that increases in stimulation intensity do not increase the amplitude of the evoked ST-EPSC indicative of a monosynaptic input. B: after failures occurred in the presence of QX-314, an increase in the stimulation intensity from 200 μA (black) to 400 μA (light gray) temporarily rescued the evoked ST-EPSC. Notice the similarity in the amplitudes between the 2 intensities and that the rescued ST-EPSC eventually returned to failures. C: representative traces of the evoked ST-EPSC at 200 μA (black) and 400 μA (light gray). D: the rise time and decay time constant were similar between the 2 intensities.
We attribute the QX-314-induced delay in synaptic timing and the emergence of outright failures to primary effects within the ST afferent fibers, most likely at VGSCs. If QX-314 indiscriminately entered all neurons, then we would expect postsynaptic actions as well. To test this, we triggered action potential responses in current-clamp by injecting current into second-order NTS neurons. In response to our standard ST test protocol, ST shocks elicited EPSPs, which in many neurons evoked a single action potential in response to the first shock in control (Fig. 7A). Addition of QX-314 blocked both the EPSPs as well as the ST-evoked action potentials. In this same cell, injection of a depolarizing current step evoked a train of action potentials, which was similar whether QX-314 was present or not (Fig. 7B). These current injection results were consistent in all NTS neurons tested (TRPV1+: n = 3; TRPV1−: n = 3). QX-314 also had no effect on postsynaptic holding current (n = 22; P = 0.12, paired t-test) or input resistance (n = 22; P = 0.82, paired t-test). Together, these results indicate that external QX-314 targets afferent axons (i.e., presynaptic).
Fig. 7.
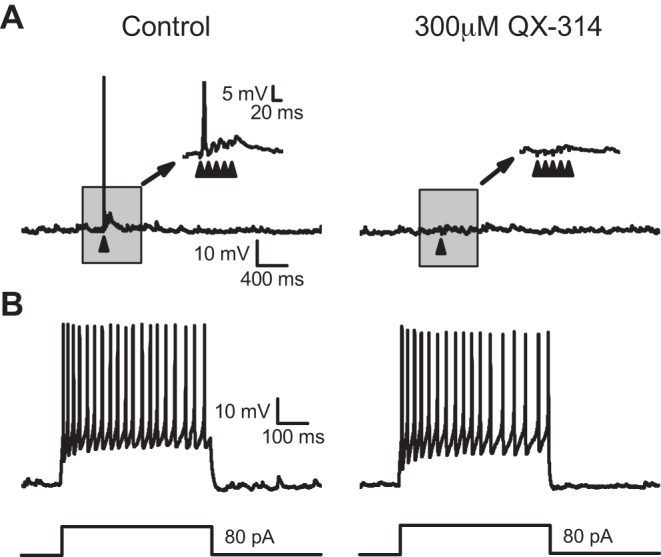
QX-314 does not affect the firing of action potentials in the postsynaptic neuron. A: in current-clamp, stimulating the ST often caused an action potential to fire on the 1st stimulus (left). Notice that in the inset the next 4 stimuli caused excitatory postsynaptic potentials (EPSPs). In the presence of QX-314 (right), the stimuli no longer caused the firing of action potentials or EPSPs. The arrows indicate stimulation of the ST. B: in the same neuron, injecting current (80 pA for 500 ms) into the postsynaptic neuron caused the firing of action potentials in control (left) and in the presence of QX-314 (right). The lack of effect on postsynaptic action potential generation suggests QX-314 is specific to the presynaptic afferents. These results were consistent in all neurons tested (TRPV1+: n = 3; TRPV1−: n = 3).
QX-314 indiscriminately blocked action potential-evoked release from ST afferents. To test whether QX-314 affected glutamate release more broadly, we examined spontaneous release of glutamate (sEPSCs) from either TRPV1+ or TRPV1− afferents. Whereas neurons receiving TRPV1+ ST afferents averaged higher sEPSC rates (TRPV1+: n = 10; TRPV1−: n = 6; P = 0.01, t-test), frequencies were unaltered by 300 μM QX-314 in both afferent types (Fig. 8; TRPV1+: n = 10, P = 0.24; TRPV1−: n = 6, P = 0.12; paired t-test). Similarly, the amplitudes of sEPSCs were unaffected by QX-314 at both afferent types (Fig. 8; TRPV1+: n = 10, P = 0.082; TRPV1−: n = 6, P = 0.85; paired t-test). Thus QX-314 failed to alter the spontaneous release process, a process that does not depend on action potentials or VGSCs.
Fig. 8.
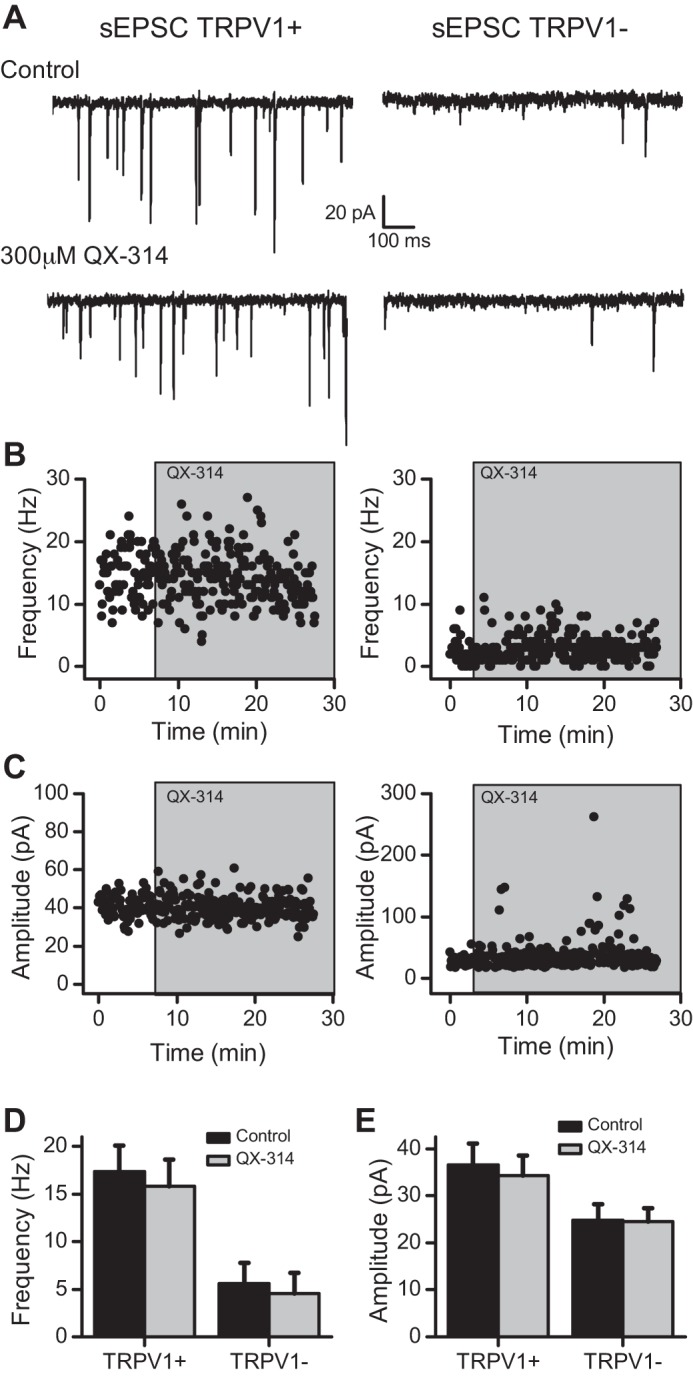
QX-314 does not affect the spontaneous release of glutamate at TRPV1+ or TRPV1− afferents. A: QX-314 failed to affect the frequency or amplitude of sEPSCs at TRPV1+ (left) or TRPV1− (right) afferents. Notice the difference in the rate of release in control conditions between the 2 afferent types. In the same neurons as A, the plots show no change to the frequency (B) or amplitude (C) of the sEPSCs. D: summary of the lack of effect on frequency by QX-314 at TRPV1+ (n = 10) and TRPV1− (n = 6) afferents. E: overall, QX-314 did not change the amplitude of sEPSCs at TRPV1+ (n = 10) or TRPV1− (n = 6) afferents.
The TRPV1-independent effects of QX-314 on action potential conduction could be unique to the NTS, or these effects could be a characteristic of cranial primary afferents. To test this idea, we recorded evoked synaptic responses in trigeminal Vc neurons in horizontal slices triggered by shocks delivered to the spinal trigeminal tract (spVT). Similar to ST stimulation in NTS neurons, shocks to spVT evoked EPSCs with nearly invariant latency (jitter < 200 μs) indicative of a monosynaptic input to Vc neurons (Fig. 9A). QX-314 (300 μM) progressively increased the latency of the evoked EPSC (Fig. 9, B and C) until the evoked EPSCs failed (Fig. 9, B and D). On average (n = 6), the evoked EPSC latency increased by ∼7% (Fig. 9E; P = 0.05, paired t-test), and the failure rate of evoked EPSCs increased nearly 50-fold (Fig. 9F; P = 0.002, paired t-test). Likewise, the evoked EPSC amplitudes decreased by ∼25% (Fig. 9G; P = 0.01, paired t-test). Furthermore, QX-314 did not affect the frequency of sEPSCs (Fig. 9, H and I; P = 0.82, paired t-test). These results in Vc neurons closely parallel the actions of QX-314 at second-order NTS neurons suggesting that the central terminals of all primary afferents may be inhibited by QX-314 in a TRPV1-independent manner.
Fig. 9.
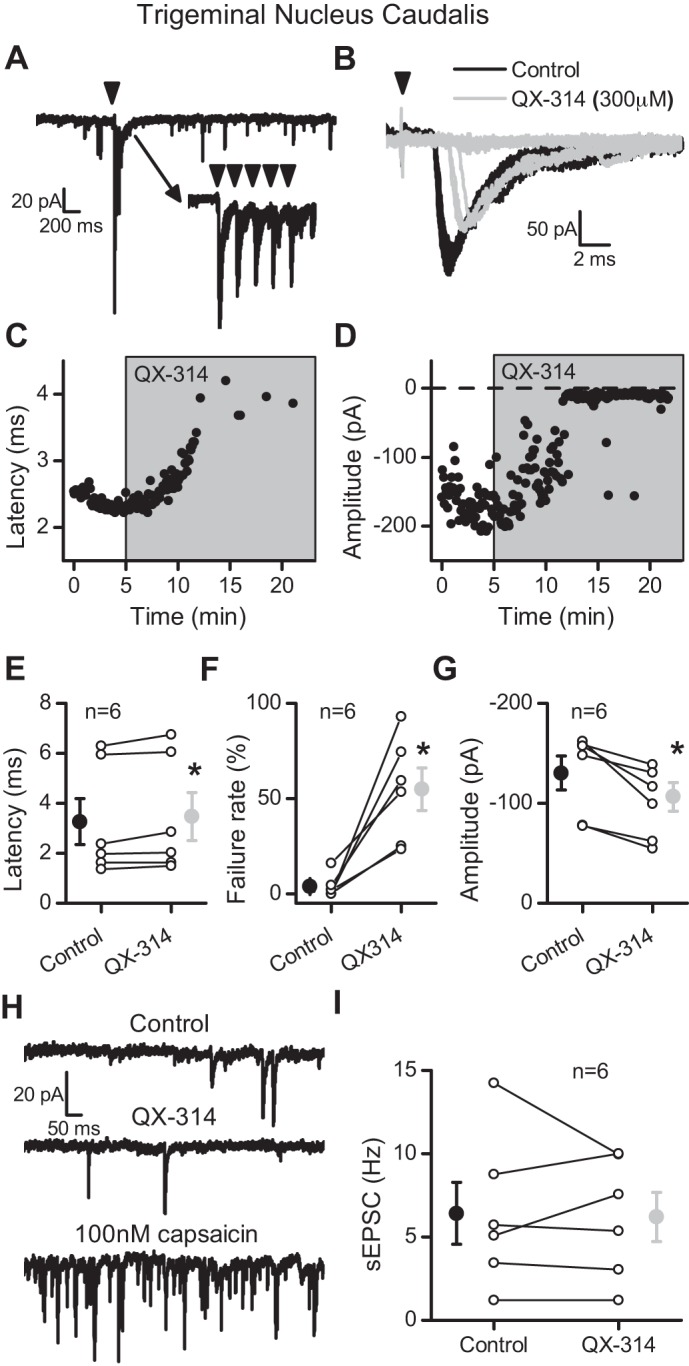
QX-314 increased the latency and then blocked evoked EPSCs at spinal trigeminal tract (spVT) afferents in caudal trigeminal (Vc) neurons. A: representative trace showing evoked EPSCs following stimulation of the trigeminal tract (5 shocks at 50 Hz). Inset traces are of individual sweeps. Arrowheads indicate the time of tract stimulation. B: QX-314 (300 μM, gray traces) prolonged the latency and caused failure compared with control (black traces). The arrowhead indicates stimulation of the trigeminal tract. C: exposure to QX-314 progressively increased the latency of the evoked spVT-EPSC. D: the amplitude of the evoked spVT-EPSC decreased slightly before resulting in failures in the presence of QX-314. E: QX-314 significantly shifted the latency of the evoked EPSCs before failures (n = 6). F: the failure rate of EPSC1 significantly increased following application of QX-314. G: the amplitudes of the evoked EPSCs significantly decreased before failure. H: representative trace of sEPSCs in control (top) and QX-314 (middle). This neuron was also sensitive to capsaicin (100 nM; bottom). I: QX-314 (light gray) did not significantly alter sEPSC rate compared with control values (black). All asterisks are for P ≤ 0.05.
DISCUSSION
Pairing TRPV1 activation with QX-314 at peripheral primary afferents suggests a selective silencing of TRPV1-expressing neurons or axons. Here, we investigated QX-314 block of synaptic transmission from cranial primary afferents to brain-stem neurons. ST afferent transmission offers several unique characteristics well-suited to this investigation (Peters et al. 2010, 2011), including two afferent subtypes segregated to different phenotypes of NTS neurons based on TRPV1 expression and a TRPV1-operated glutamate release mechanism that generates sEPSCs independent from evoked release (Fawley et al. 2014). Surprisingly, external application of QX-314 alone (300 μM) produced afferent synaptic blockade irrespective of TRPV1 expression. QX-314 exposure caused a progressive delay in the latency of action potential-evoked ST-EPSCs followed by complete block of evoked release but failed to alter frequencies or amplitudes of spontaneous release. The indistinguishable response pattern between both afferent types challenges the idea that TRPV1 is necessary to observe QX-314 block in the NTS. Antagonists targeting TRPV1 or P2X3 suggest that neither wide-pore channel was important to the QX-314 effects. QX-314 actions were clearly limited to presynaptic effects since action potential firing in the postsynaptic cell remained unaltered. This finding indicates that properties specific to the afferents make them susceptible to blockade. Similar QX-314 response profiles for spVT-evoked transmission to Vc neurons broadens this finding to central endings of primary afferents in the brain stem.
The two broad ST phenotypes, TRPV1+ and TRPV1−, provide a naturally occurring assay for our studies of QX-314/TRPV1 interactions in ST synaptic transmission (Andresen et al. 2012; Fawley et al. 2014; Jin et al. 2004; Peters et al. 2011). Action potentials evoked by ST stimulation triggered ST-EPSCs, which were indistinguishable between the two primary afferent phenotypes. ST-evoked transmission depends on the activation of both VGSCs and voltage-gated calcium channels (Mendelowitz et al. 1995). In contrast, spontaneous release from these ST afferents occurs even with voltage-gated ion channels blocked, and the sEPSC rate is ∼10-fold higher in TRPV1+ than TRPV1− afferent second-order neurons (Fawley et al. 2011; Jin et al. 2004). For these QX-314 studies, evoked and spontaneous release provided unique assays to compare two forms of synaptic glutamate release, one that depends on voltage-gated ion channels and another form of transmission that does not require VGSC or voltage-gated calcium channel activity, across two afferent phenotypes.
In most studies of TRPV1+ peripheral sensory neurons, extracellular QX-314 alone had no effect on sodium currents or action potential generation (Binshtok et al. 2007; Brenneis et al. 2013; Kim et al. 2010a; Liu et al. 2011). Elegant recent studies measured the current carried by QX-314 as it passed directly through open TRPV1 channels where it interacted with the intracellular domains of VGSCs (Puopolo et al. 2013). Given this strong evidence of direct passage of QX-314 through TRPV1, we were surprised that extracellular application of QX-314 alone blocked evoked ST-EPSCs irrespective of TRPV1 expression. In some previous cases where QX-314 alone had effects, tonic activation of TRPV1 was responsible (Mourot et al. 2012; Ries et al. 2009), and considerable evidence indicates that TRPV1 is tonically active in the NTS (Peters et al. 2010; Shoudai et al. 2010). However, we found no evidence that QX-314 effects differed between TRPV1+ inputs compared with TRPV1− inputs. Addition of the antagonist capsazepine failed to influence the actions of QX-314. Consistent with NTS neurons, application of QX-314 to Vc neurons also resulted in increased latency and failure of evoked EPSCs. Thus the inhibition of evoked synaptic transmission in both ST and trigeminal tract afferents suggests the high likelihood of a common, TRPV1-independent element that would allow QX-314 to enter central afferent axons.
The inhibitory effects of QX-314 on evoked synaptic transmission were indistinguishable between TRPV1+ and TRPV1− afferents. In all NTS neurons tested, the earliest effect of QX-314 was a prolongation in the ST-EPSC latency, which gradually increased until eventual failure of evoked release. Lower doses of QX-314 simply resulted in slower onset and weaker responses. All changes in evoked ST-EPSCs were consistent with a QX-314 block of VGSCs in an activity-dependent manner (Ragsdale et al. 1994). We speculate that the gradual latency increase results from a growing depression of conduction as intracellular QX-314 accumulates and more sodium channels are blocked. In many cases, the amplitude of the ST-EPSC remained unchanged at a time when the latency had increased substantially, which also argues against QX-314 inhibition of voltage-gated calcium channels. The presence of failures interspersed with nearly full-amplitude ST-EPSCs suggests that the response oscillated between failure of conduction and full-amplitude successes. The pattern is incompatible with an accumulating block of voltage-gated calcium channels, which would be graded and progressive in reducing the evoked EPSC amplitude and would not be quickly reversed (jumps from 0 to full-amplitude). We used the chloride salt of QX-314 to avoid the potential for direct action of bromide on calcium currents (Talbot and Sayer 1996). An additional finding consistent with QX-314 inhibition of excitability was that, following the onset of ST-EPSC failures, increasing the stimulus shock intensity briefly restored the ST-EPSC. A titrated reduction of available sodium channels might well appear first as a shift in the threshold for spike generation as QX-314 reduced the available sodium channels and thus reduced the safety factor. The rescue maneuver was only temporarily successful as continuing exposure to QX-314 blocked the restored ST-EPSC, likely due to the stronger inhibition of action potential conduction.
In NTS neurons, evoked and spontaneous release can be independently modulated and in opposite directions (Fawley et al. 2014). Our QX-314 results are consistent with the independence of the evoked and spontaneous pools of glutamate vesicles and their different calcium sources for respective release. The latency and failure data for evoked release were consistent with the known action of QX-314 at VGSCs. This block of the action potentials and evoked release by QX-314 had no effect on spontaneous release, which in TRPV1+ afferents relies on calcium entering through TRPV1 itself and requires neither VGSCs nor voltage-gated calcium channels (Fawley et al. 2011). This suggested that QX-314 did not affect the terminal release machinery but instead inhibited axonal excitability. Activation of certain G protein-coupled receptors located outside of the ST terminals sometimes induced ST transmission failures without affecting the terminal release machinery, and this was interpreted as evidence of suppressed axonal conduction (Bailey et al. 2006). Because QX-314 affected axonal excitability at all afferent types tested, we investigated whether QX-314 had access to the excitability mechanisms in the postsynaptic neuron. At time points when QX-314 had fully blocked ST afferent-evoked responses, it had no effect on postsynaptic firing of action potentials (direct current injection) suggesting that QX-314 did not enter the postsynaptic, second-order neurons. The lack of an effect on action potential firing in the postsynaptic NTS neurons combined with the consistent inhibition of evoked release (TRPV1+/− and NTS/Vc) suggests that this inhibition of excitability by QX-314 was specific to the cranial primary afferents. Substantial differences in ion channel expression are well-known in voltage-gated ion channels across myelinated and unmyelinated nodose primary afferent neurons (Schild et al. 1994; Schild and Kunze 1997). Nothing points to a ready explanation, however, for the common vulnerability of cranial afferents to QX-314. Neither of two well-known wide-pore channels, TRPV1 nor P2X3 (Jin et al. 2004; Virginio et al. 1999), appears to account for QX-314 entry. Other candidates might include TRP ankyrin 1 (TRPA1; Choi et al. 2011; Sun et al. 2009), TRPV2–4 (Nedungadi et al. 2012; Zhao et al. 2010), or TRP melanostatins (TRPMs; Staaf et al. 2010), but these are not universally expressed and thus cannot account for QX-314 vulnerability of all cranial afferents (Zhao et al. 2010). In cardiac cells, a variant of TTX-resistant cardiac sodium channels (Nav1.5) appears to be affected by external binding of QX-314 (Alpert et al. 1989; Qu et al. 1995). A similar external binding seems unlikely at these cranial afferents since washing does not rapidly restore the evoked response at cranial afferents (Brenneis et al. 2013). Alternative genotypes might allow QX-314 passage in mutated neuronal VGSCs (Qu et al. 1995; Ragsdale et al. 1994), but at this point we have no evidence supporting any alternative entry pathway for QX-314.
In summary, our studies demonstrate that QX-314 alone blocked evoked glutamate release from cranial primary afferents. This action is independent of TRPV1 and appears not to affect the vesicular release mechanisms per se. Our studies identify cranial primary afferents as susceptible to QX-314 block of conduction without a role for TRPV1. These findings differ substantially from the TRPV1-dependent inhibition of dorsal root ganglion neurons by QX-314 (Binshtok et al. 2007). It is unclear whether our observations are related to QX-314 reductions in hyperexcitability of injured spinal dorsal horn neurons (Kawamata et al. 2006), inhibition of the compound action potentials in isolated vagus nerve (Omana-Zapata et al. 1997), or sensory-motor nerve block in mice (Lim et al. 2007). Thus cranial primary afferents may be uniquely vulnerable to circulating QX-314 with the potential for disruption of their function.
GRANTS
This work was supported by NIH Grants R01-HL-105703 (M. C. Andresen), F32-DE-022499 (T. M. Largent-Milnes), and F32-HL-112419 (M. E. Hofmann). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute, National Institute of Dental Research, or the NIH.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
M.E.H. and M.C.A. conception and design of research; M.E.H., T.M.L.-M., and J.A.F. performed experiments; M.E.H. and T.M.L.-M. analyzed data; M.E.H., T.M.L.-M., and M.C.A. interpreted results of experiments; M.E.H., T.M.L.-M., and M.C.A. prepared figures; M.E.H. and M.C.A. drafted manuscript; M.E.H., T.M.L.-M., J.A.F., and M.C.A. edited and revised manuscript; M.E.H., T.M.L.-M., J.A.F., and M.C.A. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of T. M. Largent-Milnes: Univ. of Arizona, Dept. of Pharmacology, 1501 N. Campbell Ave., LSN 647, Tucson, AZ 85724-5050.
REFERENCES
- Alpert LA, Fozzard HA, Hanck DA, Makielski JC. Is there a second external lidocaine binding site on mammalian cardiac cells? Am J Physiol Heart Circ Physiol 257: H79–H84, 1989. [DOI] [PubMed] [Google Scholar]
- Andresen MC, Hofmann ME, Fawley JA. The unsilent majority–TRPV1 drives “spontaneous” transmission of unmyelinated primary afferents within cardiorespiratory NTS. Am J Physiol Regul Integr Comp Physiol 303: R1207–R1216, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TW, Jin YH, Doyle MW, Smith SM, Andresen MC. Vasopressin inhibits glutamate release via two distinct modes in the brainstem. J Neurosci 26: 6131–6142, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature 449: 607–610, 2007. [DOI] [PubMed] [Google Scholar]
- Bischoff U, Bräu ME, Vogel W, Hempelmann G, Olschewski A. Local anaesthetics block hyperpolarization-activated inward current in rat small dorsal root ganglion neurones. Br J Pharmacol 139: 1273–1280, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneis C, Kistner K, Puopolo M, Segal D, Roberson D, Sisignano M, Labocha S, Ferreiros N, Strominger A, Cobos EJ, Ghasemlou N, Geisslinger G, Reeh PW, Bean BP, Woolf CJ. Phenotyping the function of TRPV1-expressing sensory neurons by targeted axonal silencing. J Neurosci 33: 315–326, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh DJ, Chesler AT, Bráz JM, Shah NM, Julius D, Basbaum AI. Restriction of transient receptor potential vanilloid-1 to the peptidergic subset of primary afferent neurons follows its developmental downregulation in nonpeptidergic neurons. J Neurosci 31: 10119–10127, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi MJ, Jin Z, Park YS, Rhee YK, Jin YH. Transient receptor potential (TRP) A1 activated currents in TRPV1 and cholecystokinin-sensitive cranial visceral afferent neurons. Brain Res 1383: 36–42, 2011. [DOI] [PubMed] [Google Scholar]
- Chung MK, Guler AD, Caterina MJ. TRPV1 shows dynamic ionic selectivity during agonist stimulation. Nat Neurosci 11: 555–564, 2008. [DOI] [PubMed] [Google Scholar]
- Doyle MW, Andresen MC. Reliability of monosynaptic transmission in brain stem neurons in vitro. J Neurophysiol 85: 2213–2223, 2001. [DOI] [PubMed] [Google Scholar]
- Fawley JA, Hofmann ME, Andresen MC. Cannabinoid 1 and transient receptor potential vanilloid 1 receptors discretely modulate evoked glutamate separately from spontaneous glutamate transmission. J Neurosci 34: 8324–8332, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawley JA, Peters JH, Andresen MC. GABAB-mediated inhibition of multiple modes of glutamate release in the nucleus of the solitary tract. J Neurophysiol 106: 1833–1840, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann ME, Bhatia C, Frazier CJ. Cannabinoid receptor agonists potentiate action potential-independent release of GABA in the dentate gyrus through a CB1 receptor-independent mechanism. J Physiol 589: 3801–3821, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YH, Bailey TW, Li BY, Schild JH, Andresen MC. Purinergic and vanilloid receptor activation releases glutamate from separate cranial afferent terminals. J Neurosci 24: 4709–4717, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata M, Sugino S, Narimatsu E, Yamauchi M, Kiya T, Furuse S, Namiki A. Effects of systemic administration of lidocaine and QX-314 on hyperexcitability of spinal dorsal horn neurons after incision in the rat. Pain 122: 68–80, 2006. [DOI] [PubMed] [Google Scholar]
- Kim HY, Kim K, Li HY, Chung G, Park CK, Kim JS, Jung SJ, Lee MK, Ahn DK, Hwang SJ, Kang Y, Binshtok AM, Bean BP, Woolf CJ, Oh SB. Selectively targeting pain in the trigeminal system. Pain 150: 29–40, 2010a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kushmerick C, Von Gersdorff H. Presynaptic resurgent Na+ currents sculpt the action potential waveform and increase firing reliability at a CNS nerve terminal. J Neurosci 30: 15479–15490, 2010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Largent-Milnes TM, Hegarty DM, Aicher SA, Andresen MC. Physiological temperatures drive glutamate release onto trigeminal superficial dorsal horn neurons. J Neurophysiol 111: 2222–2231, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim TK, MacLeod BA, Ries CR, Schwarz SK. The quaternary lidocaine derivative, QX-314, produces long-lasting local anesthesia in animal models in vivo. Anesthesiology 107: 305–311, 2007. [DOI] [PubMed] [Google Scholar]
- Liu H, Zhang HX, Hou HY, Lu XF, Wei JQ, Wang CG, Zhang LC, Zeng YM, Wu YP, Cao JL. Acid solution is a suitable medium for introducing QX-314 into nociceptors through TRPV1 channels to produce sensory-specific analgesic effects. PLoS One 6: e29395, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougall SJ, Peters JH, Andresen MC. Convergence of cranial visceral afferents within the solitary tract nucleus. J Neurosci 29: 12886–12895, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelowitz D, Yang M, Reynolds PJ, Andresen MC. Heterogeneous functional expression of calcium channels at sensory and synaptic regions in nodose neurons. J Neurophysiol 73: 872–875, 1995. [DOI] [PubMed] [Google Scholar]
- Mourot A, Fehrentz T, Le FY, Smith CM, Herold C, Dalkara D, Nagy F, Trauner D, Kramer RH. Rapid optical control of nociception with an ion-channel photoswitch. Nat Methods 9: 396–402, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedungadi TP, Dutta M, Bathina CS, Caterina MJ, Cunningham JT. Expression and distribution of TRPV2 in rat brain. Exp Neurol 237: 223–237, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omana-Zapata I, Khabbaz MA, Hunter JC, Bley KR. QX-314 inhibits ectopic nerve activity associated with neuropathic pain. Brain Res 771: 228–237, 1997. [DOI] [PubMed] [Google Scholar]
- Patterson LM, Zheng H, Ward SM, Berthoud HR. Vanilloid receptor (VR1) expression in vagal afferent neurons innervating the gastrointestinal tract. Cell Tissue Res 311: 277–287, 2003. [DOI] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Fawley JA, Andresen MC. TRPV1 marks synaptic segregation of multiple convergent afferents at the rat medial solitary tract nucleus. PLoS One 6: e25015, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Fawley JA, Smith SM, Andresen MC. Primary afferent activation of thermosensitive TRPV1 triggers asynchronous glutamate release at central neurons. Neuron 65: 657–669, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premkumar LS, Abooj M. TRP channels and analgesia. Life Sci 92: 415–424, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puopolo M, Binshtok AM, Yao GL, Oh SB, Woolf CJ, Bean BP. Permeation and block of TRPV1 channels by the cationic lidocaine derivative QX-314. J Neurophysiol 109: 1704–1712, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Rogers J, Tanada T, Scheuer T, Catterall WA. Molecular determinants of drug access to the receptor site for antiarrhythmic drugs in the cardiac Na+ channel. Proc Natl Acad Sci USA 92: 11839–11843, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 265: 1724–1728, 1994. [DOI] [PubMed] [Google Scholar]
- Ries CR, Pillai R, Chung CC, Wang JT, MacLeod BA, Schwarz SK. QX-314 produces long-lasting local anesthesia modulated by transient receptor potential vanilloid receptors in mice. Anesthesiology 111: 122–126, 2009. [DOI] [PubMed] [Google Scholar]
- Schild JH, Clark JW, Hay M, Mendelowitz D, Andresen MC, Kunze DL. A- and C-type rat nodose sensory neurons: model interpretations of dynamic discharge characteristics. J Neurophysiol 71: 2338–2358, 1994. [DOI] [PubMed] [Google Scholar]
- Schild JH, Khushalani S, Clark JW, Andresen MC, Kunze DL, Yang M. An ionic current model for neurons in the rat medial nucleus tractus solitarius receiving sensory afferent input. J Physiol 469: 341–363, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild JH, Kunze DL. Experimental and modeling study of Na+ current heterogeneity in rat nodose neurons and its impact on neuronal discharge. J Neurophysiol 78: 3198–3209, 1997. [DOI] [PubMed] [Google Scholar]
- Shen J, Fox LE, Cheng J. Differential effects of peripheral versus central coadministration of QX-314 and capsaicin on neuropathic pain in rats. Anesthesiology 117: 365–380, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Kato F. Action potential-independent release of glutamate by Ca2+ entry through presynaptic P2X receptors elicits postsynaptic firing in the brainstem autonomic network. J Neurosci 24: 3125–3135, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoudai K, Peters JH, McDougall SJ, Fawley JA, Andresen MC. Thermally active TRPV1 tonically drives central spontaneous glutamate release. J Neurosci 30: 14470–14475, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staaf S, Franck MC, Marmigere F, Mattsson JP, Ernfors P. Dynamic expression of the TRPM subgroup of ion channels in developing mouse sensory neurons. Gene Expr Patterns 10: 65–74, 2010. [DOI] [PubMed] [Google Scholar]
- Sun B, Bang SI, Jin YH. Transient receptor potential A1 increase glutamate release on brain stem neurons. Neuroreport 20: 1002–1006, 2009. [DOI] [PubMed] [Google Scholar]
- Talbot MJ, Sayer RJ. Intracellular QX-314 inhibits calcium currents in hippocampal CA1 pyramidal neurons. J Neurophysiol 76: 2120–2124, 1996. [DOI] [PubMed] [Google Scholar]
- Virginio C, MacKenzie A, Rassendren FA, North RA, Surprenant A. Pore dilation of neuronal P2X receptor channels. Nat Neurosci 2: 315–321, 1999. [DOI] [PubMed] [Google Scholar]
- Zhao H, Sprunger LK, Simasko SM. Expression of transient receptor potential channels and two-pore potassium channels in subtypes of vagal afferent neurons in rat. Am J Physiol Gastrointest Liver Physiol 298: G212–G221, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]