

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by skeletal muscle atrophy and weakness, ultimately leading to respiratory failure. The purpose of this study was to assess changes in skeletal muscle excitation-contraction (E-C) coupling and intracellular Ca2+ handling during disease progression in the G93A*SOD1 ALS transgenic (ALS Tg) mouse model. To assess E-C coupling, single muscle fibers were electrically stimulated (10–150 Hz), and intracellular free Ca2+ concentration was assessed using fura-2. There were no differences in peak fura-2 ratio at any stimulation frequency at 70 days (early presymptomatic). However, at 90 days (late presymptomatic) and 120–140 days (symptomatic), fura-2 ratio was increased at 10 Hz in ALS Tg compared with wild-type (WT) fibers (0.670 ± 0.02 vs. 0.585 ± 0.02 for 120–140 days; P < 0.05). There was also a significant increase in resting fura-2 ratio at 90 days (0.351 ± 0.008 vs. 0.390 ± 0.009 in WT vs. ALS Tg; P < 0.05) and 120–140 days (0.374 ± 0.001 vs. 0.415 ± 0.003 in WT vs. ALS Tg; P < 0.05). These increases in intracellular Ca2+ in ALS Tg muscle were associated with reductions in the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase proteins SERCA1 (to 54% and 19% of WT) and SERCA2 (to 56% and 11% of WT) and parvalbumin (to 80 and 62% of WT) in gastrocnemius muscle at 90 and 120–140 days, respectively. There was no change in dihydropyridine receptor/l-type Ca2+ channel at any age. Overall, these data demonstrate minimal changes in electrically evoked Ca2+ transients but elevations in intracellular Ca2+ attributable to decreased Ca2+-clearance proteins. These data suggest that elevations in cellular Ca2+ could contribute to muscle weakness during disease progression in ALS mice.
Keywords: skeletal muscle, intracellular Ca2+, excitation-contraction coupling, amyotrophic lateral sclerosis
amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease or motor neuron disease, is a progressive neurodegenerative disease characterized by muscle weakness, wasting, and paralysis, which ultimately is fatal because of respiratory failure. One of the genetic defects identified in the familial inheritance of ALS is a glycine-to-alanine point mutation at codon 93 (G93A) in the gene encoding superoxide dismutase 1 (SOD1) (26). SOD1 is a metalloprotein that prevents free radical-mediated oxidative damage to the cells by catalyzing the dismutation of superoxide to hydrogen peroxide (H2O2). The G93A mutation is a gain-of-function mutation with increased SOD1 activity and normal dismutase function but enhanced production of reactive oxygen species (ROS) such as H2O2 (4, 31). A mouse model of ALS, the G93A*SOD1 mouse, recapitulates much of the ALS pathology (12). In this model, multiple lines of research have shown the selective vulnerability of motoneurons to the oxidative damage of this SOD1 mutation, leading to skeletal muscle dysfunction (12, 13, 17, 18, 24). However, changes in SOD1 activity, and thus oxidative stress, occur in all cell types and therefore can affect skeletal muscle fiber as well as motoneuron function.
There is increasing interest in the defects in nonneuronal cells for understanding the progressive deterioration in muscle function with ALS. At the level of the skeletal muscle, there are reports of early, presymptomatic adaptations of the muscle fiber itself. In isolated single muscle fibers from G93A*SOD1 transgenic (Tg) mice, depolarization of mitochondria near the neuromuscular junction was observed, and, in response to an osmotic challenge, there were regional increases in Ca2+ release and propagation of Ca2+ waves in the neuromuscular junction region of the fiber (37). These defects were observed in 37-day-old presymptomatic mice as well as in symptomatic mice at 3–4 mo (∼90–120 days) of age, suggesting that these changes in the muscle fiber occur before there is evidence of muscle force loss. More recently, decreased cytosolic Ca2+ buffering by mitochondria was observed in these regions during patch-clamped membrane depolarization (35). This further suggests that primary defects in the muscle fiber are associated with altered intracellular Ca2+ signaling. Unfortunately, it is not currently known whether there are alterations in the physiological processes of excitation-contraction (E-C) coupling that are critical in the normal communication between nerve and muscle.
The purpose of this study was to investigate the underlying mechanisms that regulate muscle force production, specifically the E-C coupling process. We hypothesized that mutant SOD1-induced oxidative stress of the muscle fiber would result in disruptions in E-C coupling and would impair the ability of G93A*SOD1 muscle fibers to generate optimal Ca2+ transients in response to electrically evoked action potentials. In contrast, we observed elevated Ca2+ transients at a low stimulation frequency and increases in resting intracellular Ca2+ starting at a presymptomatic age. These increases were associated with decreases in the Ca2+-clearance/buffering proteins. These data indicate alterations in intracellular Ca2+ regulation during disease progression in the G93A*SOD1 mouse model of ALS but no impairments in the ability to release Ca2+ in response to electrical activation signals.
METHODS
Ethical approval.
All procedures were conducted under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Maryland, College Park.
Animals.
C57BL/6 SJL hybrid female and C57BL/6 SJL-Tg SOD1*G93A male mice were obtained from Jackson Laboratories. Wild-type (WT) and G93A*SOD1 ALS heterozygote mice were bred to establish a colony at our animal care facility at the University of Maryland. Mice were weaned at 21 days and genotyped to determine whether they were WT or G93A*SOD1 Tg (ALS Tg) mice according to protocols previously described by others (12). To monitor disease progression, motor function was assessed using a grip test starting at 63 days and repeated every week until mice were euthanized. The grip test was similar to that previously described (6), with mice being placed on a metal grid (20 cm wide, 40 cm long; grid placed 40 cm above table) and allowed to grip with fore- and hind-limb paws before inverting the grid. The time that mice would grip the grid was recorded, with a maximum of 180 s per trial allowed.
Male and female ALS Tg mice along with WT littermates were euthanized at the presymptomatic ages of 70 days (no visible weakness or mobility limitation) and 90 days (tremors possible but no observable weakness) and symptomatic ages 120–140 days (visible muscle weakness, hind-limb paralysis, and reduced mobility). These age groups were chosen based on previous reports of functional muscle decline becoming evident between 84 and 105 days (22), mice becoming symptomatic between 90 and 120 days, and death at 120–150 days (15, 29). The stages of disease progression were confirmed by use of the grip test (described above). Time of death for the group at 120–140 days was within 24 h of mice being able to move only on forelimbs (i.e., dragging both hindlimbs). At time of use, animals were euthanized by CO2 inhalation followed by cervical dislocation. Tissues were harvested for immediate dissection to obtain single fibers from the flexor digitorum brevis (FDB) muscle or were quick frozen in liquid nitrogen for later analysis of muscle protein levels.
Single muscle fiber isolation and E-C coupling measurements.
Intact single muscle fibers were obtained from the FDB by collagenase digestion as originally described by Bischoff (3). Briefly, strips of FDB muscle were placed in 0.2% collagenase (Worthington, Type 2) in minimal essential media with 10% fetal bovine serum and 1% penicillin/streptomycin (MEM/FBS media) and allowed to digest 4 h in a tissue culture incubator (37°C, 95% O2-5% CO2). After 4 h, fibers were triturated in MEM/FBS media and then left in the incubator overnight. Fibers were assessed for changes in E-C coupling within 24 h of isolation.
For E-C coupling measurements, fibers were loaded with 1 μM fura-2 AM in MEM/FBS media for 15 min at room temperature. Fura-2 AM media were then removed by quick centrifugation, and fibers were resuspended in fresh MEM/FBS media. Fibers loaded with fura-2 were placed in a culture/stimulation chamber (Cell MicroControls) containing parallel electrodes on top of a Nikon TiU microscope stage. Once in the chamber, fibers were continuously perfused with a stimulating tyrode (121 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 0.5 mM MgCl2, 0.4 mM NaH2PO4, 24 mM NaHCO3, and 5.5 mM glucose) pH 7.3 when continuously bubbled with 95% O2-5% CO2 with the use of a Gilson Minipuls 3 peristaltic pump and vacuum pump. Intracellular Ca2+ levels were assessed by fura-2 fluorescence ratio (ratio of excitation at 340 and 380 nm; emission at 510 nm) with the use the IonOptix Hyperswitch system, with the Hyperswitch enabling collection of ratiometric data at a frequency of 250 Hz. To facilitate physiological interpretation of these data, fura-2 ratios were converted to intracellular free Ca2+ concentration ([Ca2+]i) using the methods of Grynkiewicz et al. (11). The fura-2 ratio was calibrated in vitro using the Ca2+ ionophore A23187 and 10 mM EGTA or 1 mM CaCl2, with Rmin and Rmax determined to be 0.33 and 3.50, respectively. The Kd for Ca2+ for fura-2 was 224 nM (11). To rule out a lack of sensitivity of fura-2, a high-affinity Ca2+ dye (27) for detecting increases in Ca2+ levels at high stimulation frequencies, we also evaluated resting and 100-Hz Ca2+ levels using both fura-2 and fura-4F, a low-affinity dye, in a subset of fibers (n = 11 WT; n = 15 ALS Tg) from one symptomatic-age mouse (132 days old). In this set, the response to caffeine was also assessed (n = 7 ALS Tg fibers).
Single muscle fibers isolated by collagenase digestion retain an intact, polarized sarcolemmal membrane, which can be depolarized by electrical field stimulation to activate the normal physiological process of E-C coupling and muscle contraction. We stimulated these fibers with a range of stimulation frequencies to cover the physiological firing frequencies for slow motor units with type I fibers (10 and 30 Hz), fast-fatigue-resistant motor units with type IIa fibers (50 and 70 Hz), and fast-fatigable motor units with type IIb fibers (100, 120, and 150 Hz) (16). Fura-2-loaded single fibers were initially assessed for their ability to respond to stimulation. Fibers that elicited a robust contraction at a 100-Hz test stimulus were utilized for subsequent analyses. These fibers were stimulated for 350 ms with 0.5-ms pulse duration at 10, 30, 50, 70, 100, 120, and 150 Hz (S48 Square Pulse Stimulator, GRASS Technologies) with 1-min rest between frequencies. In this preparation, single fibers were unloaded and underwent shortening during electrical activation. However, there was no movement artifact attributable to the ratiometric nature of the fura-2 signal. All single fiber data were collected at room temperature (23°C). Data were collected on fibers from one WT and one ALS Tg mouse of the same age on the same day to minimize the influence of day-to-day variability in E-C coupling measurements.
Peak fura-2 ratio at each frequency was determined by the average fura-2 ratio in the last 100 ms of the 350-ms tetanus, when Ca2+-fura-2 should be at a steady state. For fibers where the peak fura-2 levels were not maintained for 350 ms, separate analyses were completed to determine the percent of fibers failing to maintain peak Ca2+ levels (i.e., failing fibers) at each stimulation frequency. The number of failing fibers, relative to the number of fibers assessed (% failing), was tabulated for both WT and ALS Tg fibers. These analyses were only completed for muscle fibers from 120–140-day-old mice.
Muscle protein analyses.
The superficial gastrocnemius (GAS) muscle was used for analysis of Ca2+-regulatory protein levels by Western blot. This white/glycolytic superficial portion was utilized because it has a high level of expression of the fast-type Ca2+-regulatory proteins sarcoplasmic reticulum (SR)/endoplasmic reticulum (ER) Ca2+ ATPase 1 (SERCA1) and parvalbumin (PV) but low expression of the slow isoform SERCA2. To confirm that superficial GAS reflected the relative changes in the FDB where Ca2+ transients were assessed, we also confirmed changes in SERCA1 protein in a subset (n = 4) of FDB muscles at 90 and 120–140 days. Muscle samples were homogenized in lysis buffer (20 mM HEPES, pH 7,5, 100 mM NaCl, 1.5 mM MgCl2, 0.1% Triton X-100, and 20% glycerol) containing 1 mM dithiothreitol and a protease inhibitor cocktail (cOmplete mini EDTA-free Protease Inhibitor Cocktail, Roche). Homogenates were incubated on ice for 20 min, followed by centrifugation at 20,000 g for 5 min, and then the supernatant was removed, quick frozen in liquid nitrogen, and stored at −80°C for subsequent analysis. Protein levels were determined using a BCA protein kit (Thermo Scientific). Samples were then solubilized in loading buffer and denatured (5 min at 100°C). Western blot conditions were optimized for each protein: fast SERCA1 and slow SERCA2 isoforms (15 μg protein; 8% gels), PV (2.5 μg protein; 15% gels), and the dihydropyridine receptor (DHPR) subunit α1 (DHPR-α1) (5 μg protein; 8% gels). Proteins were transferred to PVDF membrane and probed with antibodies for SERCA1 (Thermo Scientific; 1:1,000), SERCA2 (Santa Cruz Biotechnology; 1:1,000), PV (Swant; 1:1,000), and DHPR-α1 (Thermo Scientific MA3–920; 1:1,000). The level of each respective protein was quantified by chemiluminescence using SuperSignal West Dura Chemiluminescent Substrate (Thermo Scientific) and imaged using a chemiluminescence imaging system (GeneGnome, Syngene). Protein expression levels are expressed as arbitrary units (AU) using ImageJ software for densitometry analysis. Membranes were stained with MemCode Reversible Protein Stain Kit (Thermo Scientific) to confirm equal loading. Initially, β-actin was assessed as a loading control, but we noted variability in β-actin protein levels that were inconsistent with total protein stain on the membrane. Because of known effects of the G93A*SOD1 mutation on protein misfolding and formation of protein aggregates (19, 29) and the challenges with using standard housekeeping proteins (i.e., β-actin and β-tubulin) for loading controls for samples from neurological disease models (21), total protein stain was determined to be the best loading control for skeletal muscle from these mice. We confirmed that GAPDH showed equal loading similar to total protein stain in a subset of membranes (see Fig. 4C).
Fig. 4.

Protein levels for SERCA1 in superficial gastrocnemius (GAS) and flexor digitorum brevis (FDB) muscle of WT and ALS Tg mice. Protein levels for SERCA1 were determined by Western blot analysis and quantified by chemiluminescence. A: raw data for superficial GAS muscle for representative samples at 70, 90, and 120–140 days. Membrane staining for total protein is shown to confirm equal loading. B: average luminescence values obtained by densitometric analysis and expressed as arbitrary units (AU) for GAS muscle. C: raw data for FDB muscle for representative samples at 90 and 120–140 days. Membrane staining for GAPDH and total protein confirms equal loading. D: average luminescence values obtained by densitometric analysis shown as AU for FDB muscle. Data shown represent means ± SE for WT and ALS Tg mice at 70 days (n = 3), 90 days (n = 3), and 120–140 days (n = 5) for GAS and at 90 days (n = 4) and 120–140 days (n = 4) for FDB; *P < 0.05 vs. WT.
Study design and statistical analysis.
To assess the changes in E-C coupling in G93A*SOD1 muscle, we analyzed 10–15 fibers per mouse for a total of 30–50 fibers for each genotype at each time point. Values are expressed as means ± SE. To evaluate differences in protein expression between WT and ALS Tg mice, data were analyzed using one-way ANOVA with between-subject design and genotype as the independent variable. Comparisons were made for each age group separately. Tukey's post hoc analyses were used for all significant ANOVAs, and P < 0.05 used to determine statistical significance. To assess whether the percent of failing fibers was different between ALS Tg and WT mice, χ2 analysis was used.
RESULTS
The average ages and body masses of mice used in the study are shown in Table 1. At 120–140 days, body mass for ALS Tg mice was reduced to 40% WT level (P < 0.05). There were also age-dependent decreases in motor function, with significant reductions in grip time to 42%, 16 and 7% of WT levels at 98, 105, and 112 days, respectively (Fig. 1; P < 0.05). At 91 days, there was no significant decline in grip time (P = 0.122). Thus the 90-day time point reflects a late presymptomatic age just before onset of overt symptoms, consistent with previous reports (29). On the basis of grip function, 70 days represents an early presymptomatic phase, 90 days a late presymptomatic phase, and 120–140 days a symptomatic phase of disease progression.
Table 1.
Age and body mass of WT and G93A*SOD1 ALS Tg mice
| Age Group | Age at Time of Use, days | Body Weight, g |
|---|---|---|
| 70 days | ||
| WT | 69 ± 3 (3) | 25.0 ± 4.9 |
| ALS Tg | 70 ± 3 (3) | 20.8 ± 3.0 |
| 90 days | ||
| WT | 94 ± 3 (3) | 21.8 ± 3.2 |
| ALS Tg | 95 ± 4 (3) | 19.3 ± 1.6 |
| 120–140 days | ||
| WT | 134 ± 6 (5) | 29.4 ± 2.8 |
| ALS Tg | 134 ± 6 (5) | 17.7 ± 1.3* |
Data represent means ± SE; no. of mice per time point shown in parentheses.
P < 0.01 vs. wild-type (WT). ALS, amyotrophic lateral sclerosis; Tg, transgenic.
Fig. 1.
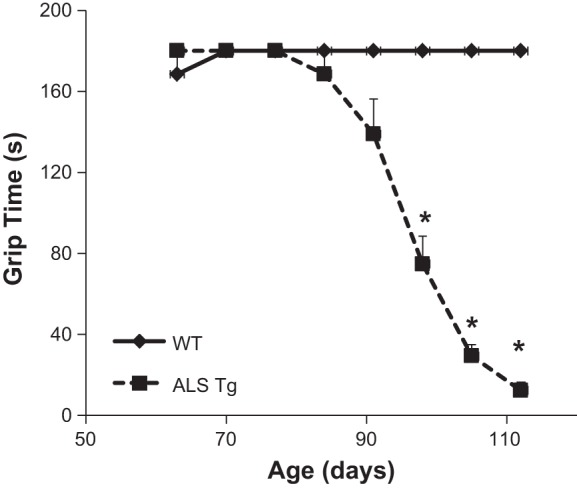
Changes in grip function in wild-type (WT) and amyotrophic lateral sclerosis transgenic (ALS Tg) mice with age. Grip function measured as time in seconds that mice could grip a wire grid. Data represent means ± SE for mice at 63–77 days (n = 11), 84–98 days (n = 8), and 105–112 days (n = 5); *P < 0.05.
Alterations in E-C coupling in intact single muscle fibers of ALS Tg mice.
In response to electrical stimulation, there were no differences in peak fura-2 ratio between single fibers isolated from 70-day-old WT and ALS Tg mice at any of the frequencies tested. However, at 90 and 120–140 days, there were increases in fura-2 ratio at the lowest frequency assessed (see Fig. 2A). The average steady-state fura-2 ratio was significantly higher in ALS Tg fibers at 10 Hz (0.637 ± 0.027 vs. 0.578 ± 0.024 at 90 days and 0.670 ± 0.030 vs. 0.585 ± 0.022 at 120–140 days for ALS Tg vs. WT, respectively; P < 0.05) with the difference at 30 Hz not quite reaching statistical significance (0.868 ± 0.030 vs. 0.791 ± 0.042 for ALS Tg vs. WT at 120–140 days; P = 0.135). There were no differences in fura-2 ratio at the higher frequencies (50–150 Hz). There was no difference in response to 100-Hz stimulation between WT and ALS Tg using either fura-2 or fura-4F (Fig. 2B). However, when ALS Tg fibers were stimulated at 100 Hz with and without caffeine, there was a 26% increase in fura-2 ratio in the presence of caffeine (1.318 ± 0.13 vs. 1.044 ± 0.10 in ALS Tg vs. WT P > 0.05; see Fig. 2C). Although most fibers were able to respond to electrical stimulation, we did observe fibers that failed to maintain a plateau in the fura-2 peak, with an increase in the percentage of fibers failing at higher stimulation frequencies. This phenomenon is observed in a fraction of all muscle fibers and was not greater in ALS Tg compared with WT fibers of the 120–140-day age group (see Table 2).
Fig. 2.

Fura-2 ratio in response to electrical stimulation in single muscle fibers from WT and ALS Tg mice. Average changes (A) in steady-state fura-2 ratios for WT (⧫; solid line) and ALS Tg (■; dashed line) mice at 120–140 days. Data represent means ± SE for all fibers: 70 days (n = 28), 90 days (n = 30), and 120–140 days (n = 42); *P < 0.05. Average change in fura-2 and fura-4F ratio at 100-Hz stimulation frequency (B) in a subset of fibers from symptomatic (132-day-old) mouse (WT n = 11); ALS Tg (n = 15). Average fura-2 response with 100-Hz stimulation with and without 5 mM caffeine (C) for ALS Tg fibers (n = 7). Resting fura-2 ratio (D) for WT (solid bars) and ALS Tg mice (hatched bars) at 70, 90, and 120–140 days; means ± SE for 70 days (n = 28), 90 days (n = 30), and 120–140 days (n = 42); *P < 0.05.
Table 2.
Percentage of fibers failing to maintain peak Ca2+ transients in WT and G93A*SOD1 ALS Tg mice
| Stimulation Frequency, Hz | WT | ALS Tg |
|---|---|---|
| 10 | 0/18 (0%) | 1/21 (5%) |
| 30 | 1/18 (6%) | 3/21 (14%) |
| 50 | 2/18 (12%) | 5/21 (24%) |
| 70 | 3/18 (17%) | 7/21 (33%) |
| 100 | 4/18 (23%) | 9/21 (43%) |
A subset of muscle fibers from 120–140-day-old mice was assessed for the ability to sustain peak Ca2+ transients over the final 100 ms of the 350-ms tetanus. The number of fibers that failed to maintain the peak Ca2+ transient at each stimulation frequency is shown and presented as a % failing fibers.
In contrast to the minimal changes in peak fura-2 ratio, there was a consistent increase in resting fura-2 ratio in fibers from ALS Tg compared with WT mice. Significant differences were observed as early as 90 days and persisted at 120–140 days of age (see Fig. 2D). At 90 days resting, fura-2 ratio increased from 0.351 ± 0.008 (WT) to 0.390 ± 0.009 (ALS Tg) (P < 0.05), equivalent to an increase in [Ca2+]i from 35 nM to 104 nM. At 120–140 days, fura-2 ratio increased from 0.374 ± 0.001 (WT) to 0.415 ± 0.003 (ALS Tg) (P < 0.05), equivalent to an increase from 78 nM to 152 nM. Overall, these data show an increase in resting Ca2+ in ALS Tg fibers, suggesting impaired Ca2+ handling in skeletal muscle.
To assess whether the rise in resting fura-2 ratio was attributable to an impairment of Ca2+-clearance mechanisms, we examined the time for the fall in fura-2 ratio during the return to baseline after contraction (5, 10). Figure 3A shows raw data traces of fura-2 transients in response to 10-Hz tetani. There is a notably slower return to baseline in the ALS Tg fibers (traces b and c) compared with the WT fiber (trace a). In 120–140-day-old mice, the average time for fura-2 to return to 75% of the prestimulation baseline was significantly longer in fibers from ALS Tg mice (Fig. 3B; P < 0.05). This 23% increase in time for Ca2+ to be removed from the cytoplasm suggests an impairment of Ca2+-clearance mechanisms.
Fig. 3.
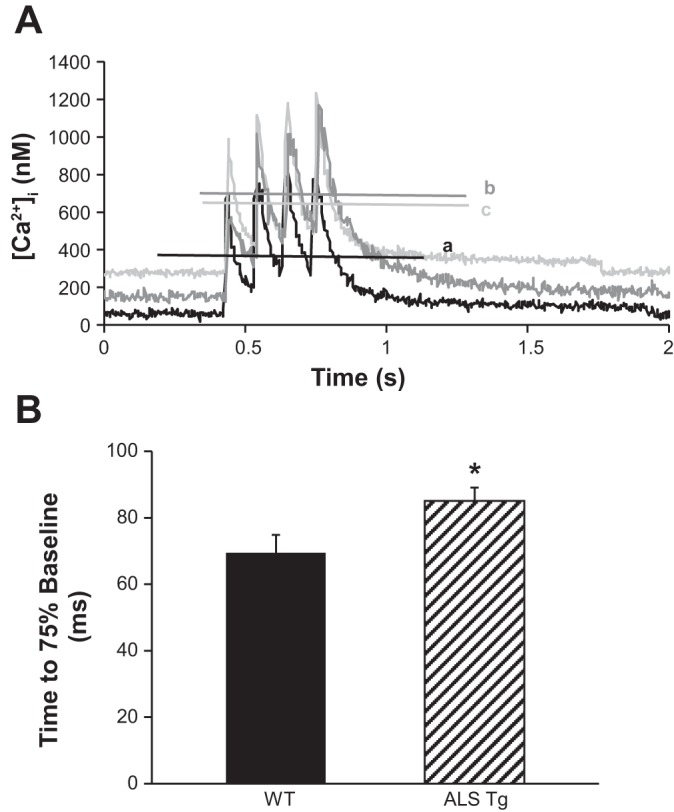
Slowing of return to baseline of fura-2 ratio following 10-Hz electrical stimulation in single muscle fibers from WT and ALS Tg mice. Sample 10-Hz Ca2+ transients (A) showing prolonged time taken for Ca2+ to return to baseline levels in muscle fibers from a 130-day-old ALS Tg mouse (gray lines) compared with a 130-day-old WT mouse (black line). Horizontal lines show steady-state [Ca2+]i over last 100 ms. B: average time taken for the fura-2 ratio to return to 75% of its resting level for single fibers from 120–140-day-old mice shown for WT (solid bars) and ALS Tg (hatched bars) (n = 42); *P < 0.05.
Alterations in the skeletal muscle Ca2+-handling proteins.
To investigate the mechanisms of the elevated intracellular Ca2+ in the single fibers, we examined the key Ca2+-regulatory proteins of skeletal muscle. SERCA1 protein levels were dramatically reduced in superficial GAS muscle of ALS Tg compared with WT mice as early as 90 days (see Fig. 4, A and B). In ALS Tg mice, SERCA1 protein level was reduced to 54% and 19% of WT levels (P < 0.05) at 90 and 120–140 days, respectively, with expression at 70 days not different. In the FDB muscle, SERCA1 was not different at 90 days but was reduced to 9% of WT levels at 120–140 days (Fig. 4, C and D). Note that, in some ALS Tg mice, SERCA1 cannot even be detected (lane 4) at 120–140 days. Because SERCA1 is the primary isoform in fast fibers and SERCA2 in slow fibers, we assessed whether there was an adaptive increase in SERCA2 isoform expression in GAS of ALS Tg mice. SERCA2 protein, however, was also dramatically reduced, reaching 56% and 11% WT levels at 90 and 120–140 days, respectively (Fig. 5, A and B). There was no difference in SERCA2 level in ALS Tg compared with WT mice at 70 days. These data suggest that there is no compensatory increase in SERCA2 in the larger limb muscles when SERCA1 has decreased protein expression.
Fig. 5.
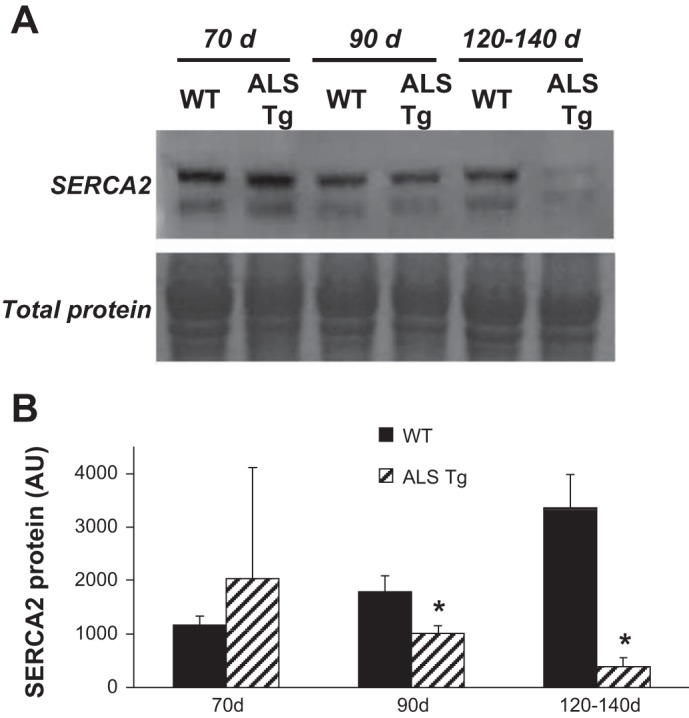
Protein levels for SERCA2 in skeletal muscle of WT and ALS Tg mice. A: SERCA2 protein level for superficial GAS muscle for representative samples at 70, 90, and 120–140 days. Membrane staining for total protein is shown to confirm equal loading. B: average SERCA2 protein levels quantified by densitometric analysis and expressed as arbitrary units are shown for superficial GAS muscle. Data shown represent means ± SE for WT and ALS Tg mice at 70 days (n = 3), 90 days (n = 3), and 120–140 days (n = 5); *P < 0.05 vs. WT.
Changes in cytoplasmic free Ca2+ concentration are also regulated by the Ca2+-buffering protein PV. In ALS Tg mice, PV protein content was significantly reduced in superficial GAS at 90 and 120–140 days to 80% and 62% of WT levels (P < 0.05), respectively (see Fig. 6, A and B).
Fig. 6.
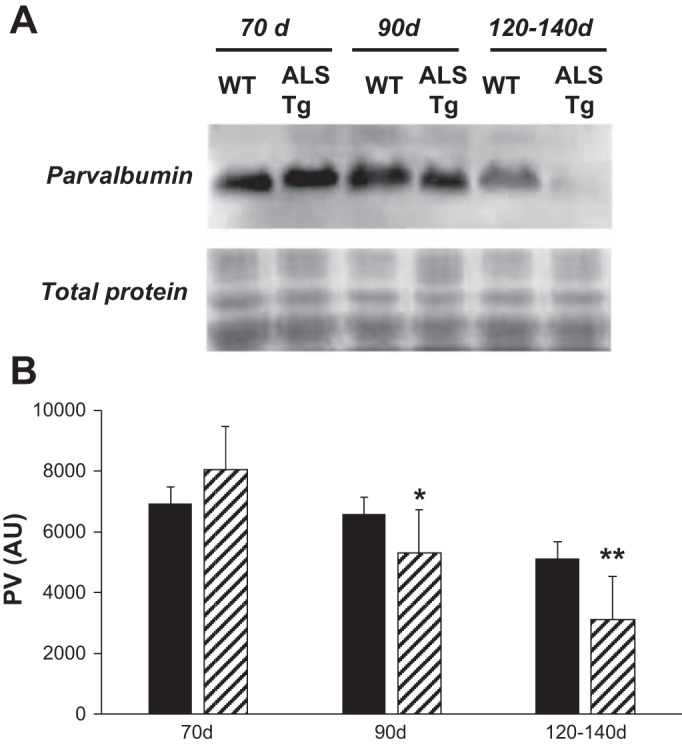
Protein levels for parvalbumin (PV) in superficial GAS muscle of WT and ALS Tg mice. Protein levels for PV were determined by Western blot analysis and quantified by chemiluminescence. A: Western blot image for representative samples at 70, 90, and 120–140 days from superficial GAS muscle. B: average luminescence values are obtained by densitometric analysis, expressed as arbitrary units, and are shown for superficial GAS muscle. Data shown represent means ± SE for WT and ALS Tg mice at 70 days (n = 3), 90 days (n = 3), and 120–140 days (n = 5); *P < 0.05 vs. WT, **P < 0.01 vs. WT.
To determine whether the increase in peak fura-2 ratio at 10 Hz was associated with changes in Ca2+-release proteins, we measured the expression level of DHPR-α1. There was no difference in DHPR-α1 subunit in ALS Tg compared with WT superficial GAS muscle. There were no differences at the early presymptomatic ages (70 and 90 days; data not shown) or at the symptomatic age (120–140 days; Fig. 7). These data indicate that there are no changes in the content of voltage-sensing Ca2+ channel in ALS Tg mice.
Fig. 7.
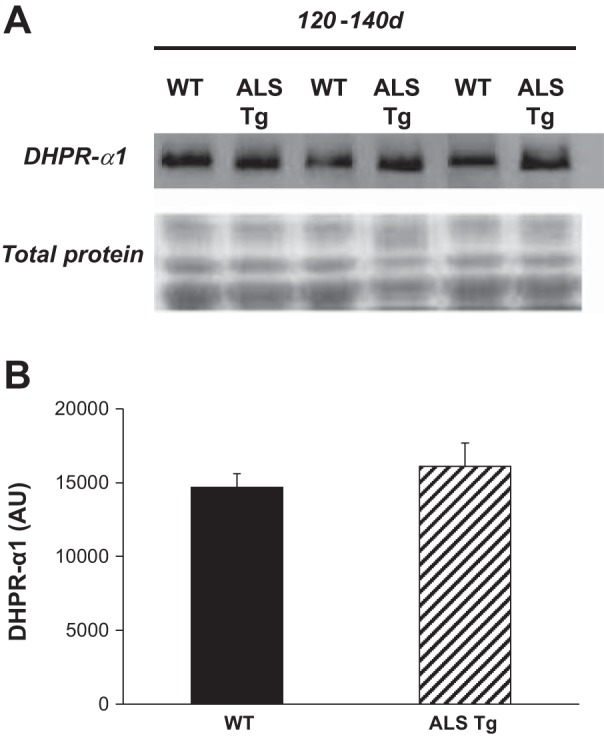
Protein levels for dihydropyridine receptor α1 subunit (DHPR-α1) in superficial GAS muscle of WT and ALS Tg mice. Protein levels for DHPR-α1 were determined by Western blot analysis and quantified by chemiluminescence. A: raw data shown for superficial GAS for 3 representative samples at 120–140 days. Membrane staining for total protein is shown to confirm equal loading. B: average luminescence values obtained by densitometric analysis and expressed as arbitrary units are shown for superficial GAS muscle. Data shown represent means ± SE for WT and ALS Tg mice at 120–140 days (n = 5); *P < 0.05 vs. WT.
DISCUSSION
There is increasing interest in the skeletal muscle-specific defects in neuromuscular diseases and their role in neuromuscular junction dismantling and disease progression (7, 8, 32). Muscle-specific mutations in SOD1 recapitulate many of the defects observed in the G93A*SOD1 model of ALS (32). Therefore, there is a need to more fully understand the mechanisms by which nerve-muscle communication is altered in disease states with respect to E-C coupling mechanisms that regulate muscle force-producing capacity. On the basis of our current findings, we propose a model whereby the G93A*SOD1 mutation results in increased resting and peak intracellular Ca2+ at low firing frequencies in skeletal muscle. This increase in intracellular Ca2+ is associated with a decrease in SR Ca2+ pump capacity and decreased levels of the Ca2+-uptake/buffering proteins SERCA1, SERCA2, and PV but no change in l-type Ca2+ channel. Alterations in these Ca2+-regulatory mechanisms reiterate the impairments in intracellular Ca2+ regulation in the muscle itself in ALS.
The primary focus of the current study was to investigate E-C coupling defects in the G93A*SOD1 mouse model of ALS. In contrast to our initial hypothesis, there were no decreases in intracellular Ca2+ transients or in the ability of single muscle fibers from the G93A*SOD1 mice to respond to electrical activation. Rather, there was a 26–37% increase in steady-state Ca2+ at the lowest stimulation frequency (10 Hz) starting at the 90-day late presymptomatic stage, which persisted at the symptomatic stage. These data are consistent with a recent report of an ∼10% increase in the patch-clamp depolarization-evoked Ca2+ transient in single fibers from 3–4-mo-old (∼90–120 days) G93A*SOD1 mice (35). Although this increase in the clamped depolarization-induced [Ca2+]i was attributed to a decrease in Ca2+ buffering by mitochondria (35), our data suggest that decreased SERCA protein levels as well as downregulation of the Ca2+-buffering protein PV also contribute to this increase in cytoplasmic Ca2+ in muscles with the G93A*SOD1 mutation. This may be a direct result of the increased generation of ROS from the G93A gain of function mutation (36) and the decreased SERCA function attributable to ROS production (34). It is known that in vitro SR Ca2+ ATPase activity is reduced up to 50% by oxidation of sulfhydryl groups on the Ca2+ ATPase, increased protein carbonyl groups, and lipid peroxidation (30). Thus increased free radicals in skeletal muscle of G93A*SOD1 mice could lead to impaired SR Ca2+ pump function, which, combined with reduction in SERCA1/2 expression, impair the ability of the cell to remove Ca2+ after contractile activity. This Ca2+ overload, beginning in the late presymptomatic stage, may contribute to the cellular pathology in skeletal muscle fibers during disease progression.
In the current study, we observed marked downregulation of the SERCA1 pump protein (91% decrease in FDB at 120–140 days) with only a minor (23% at 120–140 days) reduction in Ca2+-clearance capacity assessed using fura-2 intracellular Ca2+ transients. These data appear at odds but may reflect the high affinity of fura-2 for Ca2+ and thus an underestimation of SR Ca2+ pump function in the fibers. Interestingly, the decrease in SERCA1 protein content in FDB lagged behind the changes in GAS, being reduced only by the 120–140-day time point compared with a reduction in GAS to 54% of WT by 90 days and 19% of WT by 120–140 days. This may reflect the order of degeneration of their respective motoneuron branches or the greater relative impairment of GAS with a higher type IIb fiber composition. Given that splayed feet are an early sign of disease onset and progression from distal to proximal limb muscles (29), the greater and earlier reduction in SERCA1 expression in GAS compared with FDB muscle may be due to fiber type differences. Collectively, however, these assessments support the notion that the SR Ca2+ pump mechanism is unable to tightly regulate intracellular Ca2+ after physiological activation of muscle fibers.
In addition to impaired Ca2+-removal mechanisms, it is possible that alterations in other Ca2+-regulatory proteins contribute to the rise in intracellular Ca2+ in muscle fibers from the G93A*SOD1 mice. We cannot rule out the potential increase in Ca2+ release through the ryanodine receptor or flux through Ca2+-leak channels, which are also altered by ROS (9, 33), or alterations in function of the store-operated Ca2+-entry channels. Pathological conditions can also activate Ca2+ leak through inositol 1,4,5-trisphosphate receptors, which are activated during oxidative stress (28). We did not assess the expression level or function of these proteins so cannot rule out these as contributing mechanisms. We did assess changes in DHPR-α1 subunit, which is also susceptible to changes with muscle pathology (25). The lack of change in DHPR-α1 indicates that the content of the voltage-sensing Ca2+ release mechanism is not altered. Although this suggests that the DHPR is not altered, our data do not preclude changes in voltage-sensing function or altered Ca2+ current of the DHPR as contributors to elevated resting or low-frequency intracellular Ca2+. Given the focus of this study on electrically induced Ca2+ transients and measurement of Ca2+-clearance/removal proteins, a limitation of this study is the lack of insight into possible alterations in other mechanisms of Ca2+ entry in explaining the increase in resting and low-frequency Ca2+.
Maintenance of single muscle fiber E-C coupling under high-frequency stimulation in vitro is surprising given the dramatic reduction in muscle force-producing capacity that begins as early as 60 days in tibialis anterior muscle of ALS Tg mice (14). In electrophysiological studies involving stimulation of motoneurons in the ventral root, Hegedus et al. (14) showed decreases in maximum force output of 80% in 60-day-old G93A*SOD1 mice. In contrast, in stimulating muscle directly in vitro, Dobrowolny et al. (7) showed decreases in both maximum tetanic and specific force of 37% in extensor digitorum longus and 39% in soleus at 16 wk in a muscle-specific G93A*SOD1 mouse. A limitation in our study is that we were unable to measure muscle contractile function in the same muscles or from the same mice from which we obtained single fibers. However, on the basis of previously published data, we hypothesized that there would be an impairment in E-C coupling that would result in reduced Ca2+ transients with electrical activation in the ALS Tg mice. Surprisingly, we did not observe a decrease. This could be due to a bias in our single fiber selection criteria, where we select fibers that have robust contractions, thus biasing toward an analysis of the healthiest fibers in the pool of fibers from an FDB muscle. However, the change we did observe was actually an increase in the Ca2+ transients in the healthiest fibers. The observed increase in Ca2+ was only detected under resting conditions and at low stimulation frequencies. This low-frequency-specific effect could be due to technical reasons given that fura-2 is a high-affinity dye and would be saturating at high stimulation frequencies, possibly limiting the ability to detect increases at the higher frequencies.
Another adaptation in skeletal muscle of the G93A*SOD1 mice is the shift in muscle fiber type attributable to neural remodeling. In the fast tibialis anterior muscle of 60-day-old mice, the 80% reduction in maximum force output (14) was due to a 60% decrease in the total number of motor units and primarily to a 65% decrease in innervated type IIb muscle fibers and loss of fast-fatigable motor units. Preferential loss of these high-force-producing motor units is thought to be due to the increased susceptibility of the fast motoneurons to the oxidative damage or other protein-processing changes that lead to motoneuron death (20). Most fibers, however, remain innervated attributable to the sprouting of motoneurons from adjacent fast-fatigue-resistant (type IIa and IIx) and slow (type I) motoneurons and then reinnervate and transform the old type IIb fibers to type IIa or the intermediate type IIb/x and IIx/a fibers. This muscle remodeling enables maintenance of force production at submaximum levels. The current study shows that force would, if anything, be increased at the slow motor-unit-firing frequencies (10–30 Hz) attributable to higher peak Ca2+ transients. The increase in average cytoplasmic Ca2+ at low-frequency stimulation, however, cannot be explained by any muscle fiber type shift per se (i.e., fast to slow fiber type transition) because slow fibers have a lower peak Ca2+ upon stimulation than fast fibers (1, 2).
Given the shift in fiber type from fast IIb to IIa or slow type I fibers, it is possible that the decrease in SERCA1 and PV protein expression is due to a muscle fiber type difference. This would not, however, explain the decrease in SERCA2 protein, which is a slow/cardiac-specific isoform and would be expected to increase rather than decrease. Interestingly, the shift in muscle fiber type is different in an equine motor neuron disease where there is an increase in fast type IIa/x hybrid and pure type IIx fibers and a decrease in slow type I fibers as well as a shift to increased SERCA1 associated with the muscle atrophy (23). Equine motor neuron disease is a sporadic acquired neuromuscular disorder and thus suggests that the fiber-type shift and decrease in SERCA1 and PV observed in the G93A*SOD1 mice may be a specific result of the G93A*SOD1 mutation and resulting increase in oxidative stress in skeletal muscle.
Summary.
In summary, we report increased resting Ca2+ and increased peak cytosolic Ca2+ at low stimulation frequencies but overall preserved stimulation-induced Ca2+ transients in skeletal muscle from G93A*SOD1 mice. These altered Ca2+ dynamics are due to reductions in Ca2+-clearance mechanisms, including decreased SR Ca2+ pump capacity, decreased PV protein content, and decreased SERCA1/2 protein as disease progresses. These changes are associated with decreased motoneuron function and disease progression in ALS mice and thus may contribute to the ongoing pathophysiology in ALS.
GRANTS
This work was supported by the University of Maryland, College Park new investigator funds to E. Chin. D. Mázala is supported by NIH Predoctoral Institutional Training Grant T32-AG000268 to J. M. Hagberg.
DISCLOSURES
Eva R. Chin is the Founder and Chief Scientific Officer of MyoTherapeutics. No other conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: E.R.C. conception and design of research; E.R.C., D.C., K.B., and D.A.G.M. performed experiments; E.R.C., K.B., and D.A.G.M. analyzed data; E.R.C. interpreted results of experiments; E.R.C., D.C., and D.A.G.M. prepared figures; E.R.C. drafted manuscript; E.R.C. and D.A.G.M. edited and revised manuscript; E.R.C., D.C., and K.B. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. James Hagberg, Dr. Espen Spangenburg, and Dr. MaryAnn Ottinger for critical reading of this manuscript. We also thank Brittany Jacobs, Samuel A. English, and Spencer Bonar for contributions to data collection and analyses.
Current address for Dr. K. Bobyk: National Institute of Diabetes and Digestive and Kidney Diseases, The National Institutes of Health, Bethesda, MD 20892.
REFERENCES
- 1.Baylor SM, Hollingworth S. Intracellular calcium movements during excitation-contraction coupling in mammalian slow-twitch and fast-twitch muscle fibers. J Gen Physiol 139: 261–272, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baylor SM, Hollingworth S. Sarcoplasmic reticulum calcium release compared in slow-twitch and fast-twitch fibers of mouse muscle. J Physiol 551: 125–138, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bischoff R. Proliferation of muscle satellite cells on intact myofibers in culture. Dev Biol 115: 129–139, 1986. [DOI] [PubMed] [Google Scholar]
- 4.Bogdanov MB, Ramos LE, Xu ZS, Beal MF. Elevated “hydroxyl radical” generation in vivo in an animal model of amyotrophic lateral sclerosis. J Neurochem 71: 1321–1324, 1998. [DOI] [PubMed] [Google Scholar]
- 5.Capote J, DiFranco M, Vergara JL. Excitation-contraction coupling alterations in mdx and utrophin/dystrophin double knockout mice: a comparative study. Am J Physiol Cell Physiol 298: C1077–C1086, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deforges S, Branchu J, Biondi O, Grondard C, Pariset C, Lecolle S, Lopes P, Vidal PP, Chanoine C, Charbonnier F. Motoneuron survival is promoted by specific exercise in a mouse model of amyotrophic lateral sclerosis. J Physiol 587: 3561–3572, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, Boncompagni S, Belia S, Wannenes F, Nicoletti C, Del Prete Z, Rosenthal N, Molinaro M, Protasi F, Fano G, Sandri M, Musaro A. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab 8: 425–436, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Dupuis L, Gonzalez de Aguilar JL, Echaniz-Laguna A, Eschbach J, Rene F, Oudart H, Halter B, Huze C, Schaeffer L, Bouillaud F, Loeffler JP. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS One 4: e5390, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Favero TG, Webb J, Papiez M, Fisher E, Trippichio RJ, Broide M, Abramson JJ. Hypochlorous acid modifies calcium release channel function from skeletal muscle sarcoplasmic reticulum. J Appl Physiol 94: 1387–1394, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Goonasekera SA, Lam CK, Millay DP, Sargent MA, Hajjar RJ, Kranias EG, Molkentin JD. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J Clin Invest 121: 1044–1052, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440–3450, 1985. [PubMed] [Google Scholar]
- 12.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264: 1772–1775, 1994. [DOI] [PubMed] [Google Scholar]
- 13.Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SODIG93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 28: 154–164, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Hegedus J, Putman CT, Tyreman N, Gordon T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol 586: 3337–3351, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heiman-Patterson TD, Deitch JS, Blankenhorn EP, Erwin KL, Perreault MJ, Alexander BK, Byers N, Toman I, Alexander GM. Background and gender effects on survival in the TgN(SOD1–G93A) 1 Gur mouse model of ALS. J Neurol Sci 236: 1–7, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Hennig R, Lomo T. Firing patterns of motor units in normal rats. Nature 314: 164–166, 1985. [DOI] [PubMed] [Google Scholar]
- 17.Higgins CMJ, Jung CW, Xu ZS. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci 4: 16, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V, Cleveland DW. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 67: 575–587, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kabuta T, Kinugawa A, Tsuchiya Y, Kabuta C, Setsuie R, Tateno M, Araki T, Wada K. Familial amyotrophic lateral sclerosis-linked mutant SOD1 aberrantly interacts with tubulin. Biochem Biophys Res Commun 387: 121–126, 2009. [DOI] [PubMed] [Google Scholar]
- 20.Larsson L, Ansved T. Effects of ageing on the motor unit. Prog Neurobiol 45: 397–458, 1995. [DOI] [PubMed] [Google Scholar]
- 21.Li R, Shen Y. An old method facing a new challenge: re-visiting housekeeping proteins as internal reference control for neuroscience research. Life Sci 92: 747–751, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mead RJ, Bennett EJ, Kennerley AJ, Sharp P, Sunyach C, Kasher P, Berwick J, Pettmann B, Battaglia G, Azzouz M, Grierson A, Shaw PJ. Optimised and rapid pre-clinical screening in the SOD1(G93A) transgenic mouse model of amyotrophic lateral sclerosis (ALS). PLoS One 6: e23244, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palencia P, Quiroz-Rothe E, Rivero JL. New insights into the skeletal muscle phenotype of equine motor neuron disease: a quantitative approach. Acta Neuropathol (Berl) 109: 272–284, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Raimondi A, Mangolini A, Rizzardini M, Tartari S, Massari S, Bendotti C, Francolini M, Borgese N, Cantoni L, Pietrini G. Cell culture models to investigate the selective vulnerability of motoneuronal mitochondria to familial ALS-linked G93ASOD1. Eur J Neurosci 24: 387–399, 2006. [DOI] [PubMed] [Google Scholar]
- 25.Renganathan M, Messi ML, Delbono O. Dihydropyridine receptor ryanodine receptor uncoupling in aged skeletal muscle. J Membr Biol 157: 247–253, 1997. [DOI] [PubMed] [Google Scholar]
- 26.Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364: 362, 1993. [DOI] [PubMed] [Google Scholar]
- 27.Struk A, Szucs G, Kemmer H, Melzer W. Fura-2 calcium signals in skeletal muscle fibers loaded with high concentrations of EGTA. Cell Calcium 23: 23–32, 1998. [DOI] [PubMed] [Google Scholar]
- 28.Szlufcik K, Missiaen L, Parys JB, Callewaert G, De Smedt H. Uncoupled IP3 receptor can function as a Ca2+-leak channel: cell biological and pathological consequences. Biol Cell 98: 1–14, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Progr Neurobiol 85: 94–134, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Voss P, Engels M, Strosova M, Grune T, Horakova L. Protective effect of antioxidants against sarcoplasmic reticulum (SR) oxidation by Fenton reaction, however, without prevention of Ca-pump activity. Toxicol In Vitro 22: 1726–1733, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Wiedau-Pazos M, Goto JJ, Rabizadeh S, Gralla EB, Roe JA, Lee MK, Valentine JS, Bredesen DE. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science 271: 515–518, 1996. [DOI] [PubMed] [Google Scholar]
- 32.Wong M, Martin LJ. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum Mol Genet 19: 2284–2302, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia R, Stangler T, Abramson JJ. Skeletal muscle ryanodine receptor is a redox sensor with a well defined redox potential that is sensitive to channel modulators. J Biol Chem 275: 36556–36561, 2000. [DOI] [PubMed] [Google Scholar]
- 34.Xu KY, Zweier JL, Becker LC. Hydroxyl radical inhibits sarcoplasmic reticulum Ca2+-ATPase function by direct attack on the ATP binding site. Circ Res 80: 76–81, 1997. [DOI] [PubMed] [Google Scholar]
- 35.Yi J, Ma C, Li Y, Weisleder N, Rios E, Ma J, Zhou J. Mitochondrial calcium uptake regulates rapid calcium transients in skeletal muscle during excitation-contraction (E-C) coupling. J Biol Chem 286: 32436–32443, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yim MB, Kang JH, Yim HS, Kwak HS, Chock PB, Stadtman ER. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: An enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc Natl Acad Sci USA 93: 5709–5714, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou J, Yi J, Fu R, Liu E, Siddique T, Rios E, Deng HX. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J Biol Chem 285: 705–712, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]