

Abstract
Cigarette smoke (CS) exposure increases the frequency and severity of respiratory tract infections. Despite this association, the mechanisms underlying the increased susceptibility to respiratory virus infection are poorly understood. Retinoic acid-inducible gene I (RIG-I) is an important regulator of influenza virus-induced expression of antiviral cytokines, mainly interferons (IFNs), which are necessary to clear viral infections. In this study, we compared the innate cytokine responses of two mouse CS exposure models following a challenge with influenza A virus (IAV): 1) exposure of the mice to cigarette smoke extract (CSE) intratracheally and 2) exposure of the mice to CS in a whole body exposure chamber. Both intratracheal CSE treatment and whole body CS exposure caused antiviral immunosuppression in these mice, and both CS exposure methods inhibited RIG-I induction. CS attenuated influenza-induced antiviral IFNs and IP-10 expression in vivo. However, we did not find that CS inhibited induction of the proinflammatory cytokines IL-6 and TNF-α, whose expression was induced by IAV. Interestingly, IAV infection also increased Toll-like receptor 3 (TLR3) expression in mouse lung, but CS exposure did not impact TLR3 induction in these mice. Together, the results support our previous finding in a human lung organ culture model that the suppression of RIG-I induction and antiviral cytokine responses by CS are likely important in the enhanced susceptibility of smokers to influenza infection in the lung.
Keywords: lung, cytokine, retinoic acid-inducible gene I, Toll-like receptor 3, mouse
cigarette smoke (CS), as well as sidestream smoke, increases the susceptibility to pulmonary infection with pathogens, including influenza A virus (IAV), rhinovirus, respiratory syncytial virus, and various bacterial pathogens (2, 5, 30). In particular, epidemiological studies have shown that influenza infection is seven times more frequent and is much more severe in smokers than nonsmokers (1). Both active and passive CS exposure increase the risk of infections (23). A cohort study of female military recruits showed that smoking was a risk factor for severe influenza-like illness during an outbreak of influenza A (H1N1) subtype infection (17). Thailand's National Avian Influenza Surveillance system reported that current or former smoking was among the several risk factors associated with a fatal outcome from human influenza infection (11). In the spring of 2013, the high mortality of avian influenza H7N9 in humans caused great concern in China and the world. Age and a history of smoking were the most significant risk factors that predicted a fatal outcome in human H7N9 infection (21). The cause of increased susceptibility to infections in smokers is likely multifactorial but clearly includes alterations of immunological host defenses through unknown mechanisms. Because adaptive humoral immunity, which is measured by influenza-specific humoral antibody production, was unaffected in CS-exposed, influenza-infected human and animal models (22, 25, 26), CS more likely affected the innate immune system during influenza infection.
Innate immunity, an evolutionarily older defense strategy, is the first line of host defense against invading microorganisms. Although recognizing and responding to pathogens in a nonspecific manner, the innate immune system provides immediate protection against infection. IAV is first mechanistically identified through pattern recognition receptors (PRRs), with recognition of 5′-triphosphorylated single-stranded RNA by the cytoplasmic RNA helicase, retinoic acid-inducible gene 1 (RIG-I), and detection of double-stranded RNA (dsRNA) by the endosomal TLR3. RIG-I is critical for recognition of IAV and initiation of the early antiviral cytokine response, mainly interferons (IFNs), to prevent further viral infection in the lung (24). Using an in vitro human lung organ culture model, we have demonstrated that cigarette smoke extract (CSE) suppresses viral-mediated induction of the major RNA virus sentinel RIG-I, and CSE treatment inhibits influenza-induced antiviral cytokine expression in human lung, particularly IFN-β and IP-10 (35). We have also found that antioxidants prevent CSE-mediated suppression of RIG-I induction by IAV and that immunosuppression by CSE is not due to nonspecific cytotoxicity.
Animal models have been used to demonstrate impairments in host defense against IAV infection in CS-exposed animals. Consistent with increased risk of influenza infection among smokers, in vivo studies of experimentally induced pulmonary infection have found increased susceptibility to IAV in CS-exposed animals by other investigators (7, 9). To expand our findings from the human lung organ culture model, we have developed two CS exposure animal models to study CS and IAV infection, an intratracheal CSE treatment mouse model and a whole body CS exposure model. In this report, we used the two models to determine whether inhibition of the antiviral RIG-I response is responsible for the immunosuppressive effects of CS that we found earlier and could be responsible for the resultant increased mortality in mice during IAV infection observed earlier (7). We have demonstrated that CS suppressed RIG-I induction by IAV infection, the subsequent antiviral cytokine responses in both animal models, and that the immunosuppressive effect of CS may play a critical role in the enhanced susceptibility of CS-exposed mice to serious IAV infection.
MATERIALS AND METHODS
Preparation of influenza virus stock and plaque assays.
H1N1 influenza virus, A/PR/34/8 (PR8), was passaged in Madin-Darby canine kidney (MDCK) cells. Virus was grown in MDCK cells in DMEM-F-12 with ITS+ (BD Biosciences, Franklin Lakes, NJ) and trypsin, harvested at 72 h postinfection, and titered by plaque assay in MDCK cells. There was no detectable endotoxin in the final viral preparations used in the experiments as determined by limulus amebocyte lysate assay (Cambrex, Walkersville, MD). The lower limit of detection of this assay is 0.1 EU/ml or ∼20 pg/ml lipopolysaccharide. For plaque assay, whole mouse lungs were collected and homogenized in 1 ml of ice-cold PBS. Solid debris was pelleted by centrifugation, and viral titer was determined using a standard plaque assay technique on MDCK cells. Results were expressed in plaque-forming units (pfu) per milliliter of extract.
Animals.
Specific pathogen-free 6- to 8-wk-old female C57BL/6 mice were purchased from Taconic Farms (Hudson, NY). Mice were housed at 20°C on a 12:12-h light-dark cycle in sterile microisolator cages and fed ad libitum with sterile chow and water. The Institutional Animal Care and Use Committee of the University of Oklahoma Health Sciences Center approved all of the protocols for the animal experiments. After arrival, mice were acclimatized for at least 7 days.
CSE preparation.
CSE was prepared by a modification of the method developed by Carp and Janoff (3). Briefly, one (100-mm) cigarette without filter was combusted with a pump. The smoke was bubbled through 25 ml of PBS at a speed of 50 ml/min. The resulting suspension was filtered through a 0.22-μm pore filter (Lida Manufacturing, Kenosha, WI) to remove bacteria and large particles. This solution, considered to be 100% CSE, was diluted and applied to the mice within 30 min of preparation. The nicotine concentration of 100% CSE was 73.48 ± 1.08 μg/ml. Mice were anesthetized by exposure to isoflurane (Min Rad, Bethlehem, PA) in a bell jar until effect. Mice were administrated 50 μl of CSE intratreacheally as described below for 5 days/wk for 6 wk.
Intratracheal CSE inoculation model.
Intratracheal inoculation did not involve surgery or open wounds. After receiving isofluorane anesthesia, the animal was then suspended by its teeth on a wire on an inclined board. The animal's mouth was opened, and the tongue was gently extended out of the mouth with forceps. The animal was positioned to hold open the epiglottis and trachea while the nose was sealed by the gloved finger. The sterile CSE solution (50 μl) was instilled directly into the trachea using a micropipette fitted with a sterile tip. In this manner, most of the solution was inhaled into the lungs. In our trial experiment, 50 μl of methylene blue (0.2% in saline) were used as tracer solution to ensure that our intratracheal inoculation procedure was appropriate and that we had delivered the liquid into the alveolar space. During and upon completion of each procedure, the animals were observed closely to ensure adequate recovery.
Whole body CS exposure model.
Mice were exposed to the smoke of 3R4F reference cigarettes (University of Kentucky, Lexington, KY) for 5 h/day. Mice receiving CS were gradually brought up to the target exposure over a period of 2 wk and treated 5 days/wk for 6 wk (7). Treatment was administered by placing mice in a Plexiglas smoking chamber (Teague Enterprises, Davis, CA). Smoke exposure was standardized to total suspended particles = 90 mg/m3, carbon monoxide = 350 parts/min, and 11% mainstream and 89% sidestream smoke in the chamber of the machine. These parameters were verified by determining carboxyhemoglobin levels using an IL-682 CO-Oximeter (Instrumentation Laboratories, Lexington, MA). Machine noise controls consisted of mice in the same room, outside the chamber. “No smoke” treatment groups (NS) were conducted for the same periods of time, but mice were exposed to filtered room air.
Influenza virus infection.
The last smoke exposure by either method occurred the day before the mice were infected with IAV PR8. IAV infection was performed after isofluorane anesthesia, as above. For intratracheal inoculation, IAV PR8 solution (50 μl) was instilled directly into the trachea using a micropipette fitted with a sterile tip. Because this resulted in 5–10% mortality after the first 6 wk of intratracheal CSE exposure, we elected to use intranasal PR8 exposure for the subsequent whole body CS exposure experiments. For intranasal inoculation, IAV PR8 solution (50 μl) was administered by intranasal instillation directly into the nares as the animal was held in a vertical position, with a dose that was 20% of the intranasal does that resulted in 50% mortality at 11 days (LD50-11d). Control animals received PBS.
Bronchoalveolar lavage.
Mice were killed by isofluorane overdose. Bronchoalveolar lavage was performed using a closed thorax technique by exposing the trachea, nicking the bottom of the larynx, and inserting a 3/4-in. 22-gauge cannula in the proximal trachea. The proximal end of the trachea was tied off, and 0.6 ml of sterile PBS was gently introduced in the lungs and recovered. This was repeated three times for a total volume of 1.8 ml. Return volume varied by <10% between samples. Bronchoalveolar lavage fluid (BALF) was centrifuged to remove cells. Cells obtained were placed on slides for determination of cell populations using a Cytopro Cytocentrifuge (Wescor, Logan, UT) and stained with DiffQuik (Dade Behring, Newark, DE). Differential counts were made with ≥400 cells/sample from 2 slides/mouse. The BALF was pooled and frozen.
ELISA and multiplex immunoassay.
ELISAs of IP-10 and IL-6 cytokine protein levels in the BALF were accomplished by ELISA kits (R&D Systems, Minneapolis, MN). IFN-γ, IP-10, IL-6, and TNF-α cytokine protein levels in the BALF were determined by multiplex immunoassay (Affymetrix, Santa Clara, CA). The assay was run on a Bio-Plex 100 multiplex system (Bio-Rad, Hercules, CA).
RIG-I protein determination by immunoblotting.
The mouse lungs were harvested, homogenized, and then lysed in 500 μl of cold lysis buffer [150 mM NaCl, 50 mM Tris (pH 8.0), 10 mM EDTA, NaF, sodium pyrophosphate, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and 10 μg of leupeptin/ml]. Lung homogenates were clarified by centrifugation at 10,000 g, at 4°C for 10 min, and the clarified lysates were mixed with SDS-PAGE sample buffer [60 mM Tris (pH 6.8), 10% glycerol, and 2.3% SDS] and heated to 95°C for 5 min. The samples were separated by 4–15% gradient gel and electrophoretically transferred to polyvinylidene fluoride membranes. For the detection of proteins, the membranes were immunoblotted with rabbit polyclonal antibody specific for RIG-I (Abcam, Cambridge, MA) and GAPDH (R&D Systems). The membranes were viewed with horseradish peroxidase-labeled goat anti-rabbit IgG (Cell Signaling Technology, Beverly, MA) and chemiluminescent reagents (Pierce Biotechnology, Rockford, IL). Blots were developed using the Syngene G:box Bioimaging System and GeneTools software (Syngene, Frederick, MD) and quantified using ImageQuant software (BD/Molecular Dynamics, Bedford, MA).
Measurement of mRNA expression by quantitative real-time PCR.
Total RNA from lung was extracted using a modified TRIzol (Invitrogen, Carlsbad, CA) protocol and spectrophometrically quantitated, and the integrity was verified by formaldehyde agarose gel electrophoresis. Equal amounts (1 μg) of RNA from each sample were used with oligo(dT) as primers for production of cDNA (SuperScript II First-Strand Synthesis System for RT-PCR; Invitrogen). Gene-specific primers for the PRRs, cytokines, and the GAPDH housekeeping genes were used. The primer sequences were as follows: RIG-I forward 5′-ATTGTCGGCGTCCACAAAG-3′; RIG-I reverse 5′-GTGCATCGTTGTATTTCCGCA-3′; IFN-β forward 5′-CCATCAACTATAAGCAGCTCCAGC-3′; IFN-β reverse 5′-CCACCATCCAGGCGTAGCTGTTG-3′; β-actin forward 5′-CAGAAGGACTCCTATGTGGGTG-3′; β-actin reverse 5′-GGATCTTCATGAGGTAGTCTGTC-3′; TLR3 forward 5′-CTGGAGCCAGAACTGTGCC-3′; TLR3 reverse 5′-GTTCTTGGAGGTTCTCCAG-3′; IL-6 forward 5′-CCGGAGAGGAGACTTCACAG-3′; IL-6 reverse 5′-GGTACTCCAGAAGACCAGAGG-3′; IFN-λ 2/3 forward 5′-AGCTGCAGGCCTTCAAAAAG-3′; IFN-λ 2/3 reverse 5′- TGGGAGTGAATGTGGCTCAG-3′; and nucleoprotein (NP) forward 5′-AGGACAAGAGCTCTTGTTCG-3′; NP reverse 5′-CTCTTGTGTGCTGGATTCTC-3′. Quantitative real-time PCR (qRT-PCR) was performed using 100 ng sample RNA and SYBR Green (Quanta Biosciences, Gaithersburg, MD) in a Bio-Rad CFX96 Touch Real-Time PCR Detection System. Results were calculated and graphed from the ΔCT of target gene and normalizer.
Histological and immunohistochemistry analysis of mouse lung tissue.
At day 7 after IAV infection, whole body CS-exposed mice were killed, and lungs were fixed in 4% paraformaldehyde in PBS at room temperature for 30 min and then embedded in paraffin. Fixed tissue was hematoxylin and eosin (H & E) stained to assess inflammation and fibrosis. Sections (3–5 μm) were mounted on glass slides and immunoprobed with a rabbit anti-mouse polyclonal antibody for RIG-I (Abcam) or an anti-NP polyclonal antibody (36). After being washed, the sections were probed with a donkey anti-rabbit secondary antibody conjugated to Alexa Fluor 546 (all from BD/Molecular Probes). Transmitted light and fluorescent microscopy images were obtained from an Olympus BX51 microscope running Cellsens imaging software (Olympus, Center Valley, PA).
Statistical analysis.
Where applicable, the data have been expressed as means ± SE. Statistical significance was determined by one-way ANOVA with Student-Newman-Keuls post hoc correction for multiple comparisons. Significance was considered as P < 0.05.
RESULTS
IFN-β, IL-6, IP-10, and RIG-I are induced by IAV infection in mice.
To determine the RIG-I and cytokine induction profile in IAV-infected mice, we infected C57BL/6 mice by intratracheal inoculation with a single, nonlethal dose of the IAV PR8 strain (300 pfu). The mock group was sham treated by intratracheal inoculation with a single dose of an equal volume of PBS. Lung tissues and BALF were collected at 3, 5, 7, and 10 days after infection and tested by qRT-PCR or ELISA, respectively. Both RIG-I and IFN-β mRNA were induced by virus in mouse lung at 3 days after PR8 infection. Induction was the highest at 7 days after infection (Fig. 1, A and B). We also determined antiviral cytokine IP-10 and proinflammatory cytokine IL-6 protein induction in BALF by ELISA (Fig. 1, C and D). IL-6 peaked early at 5 days, whereas IP-10, whose expression is known to be directly regulated by RIG-I (15, 32), was the highest at 7 days after infection, as was RIG-I. Body weight of individual animals was monitored for 15 days. The mice lost a substantial amount of their initial body weight, and the lowest body weights were recorded at day 8 after viral infection. The weight started to recover on day 9 (Fig. 1E). Plaque assay showed that viral titers were elevated by 3 days of infection and remained elevated through 7 days of infection with a two-log decline in titer by 10 days after infection (Fig. 1F). Therefore, we found day 7 to be the optimum end point for determining the antiviral response in subsequent mouse experiments.
Fig. 1.

Interferon (IFN)-β, IL-6, IP-10, and retinoic acid-inducible gene I (RIG-I) are induced after influenza A virus (IAV) infection in mouse lung. Mice were intratracheally infected with 300 plaque-forming units (pfu) of the H1N1 influenza virus A/PR/34/8 (PR8) strain. IFN-β (A) and RIG-I (B) mRNA were assessed by quantitative real-time PCR (qRT-PCR), normalized against β-actin mRNA. IP-10 (C) and IL-6 (D) proteins in BALF were assessed by ELISA. Body weights were measured daily. The data were normalized to each mouse's starting body weight (E). Lung viral titers were determined at the indicated time points postinfection by plaque assay on Madin-Darby canine kidney (MDCK) cells (F). Data are expressed means ± SE (6 mice/group for A–D, 15 mice for E, and 3 mice/group for F). *Significant difference between the indicated groups, P < 0.05.
Intratracheal CSE treatment attenuates influenza-mediated induction of the antiviral cytokines IFN-β and IP-10 mRNA, but not of the proinflammatory cytokines IL-6 and TNF-α.
The effect of CS on the mouse lung innate immune cytokine response to IAV was first examined in an intratracheal CSE-treated mouse model. We chose this modified mouse model to expose CSE to the lungs and upper airways of the animals because mouse intratracheal inoculation does not involve surgery or open wounds (6). Briefly, after anesthesia, 50 μl of sterile CSE solution were instilled directly in the trachea. Mice were treated with CSE or PBS (mock) for 6 wk and then infected with nonlethal 300 pfu of the IAV PR8 strain by the same procedure. The effects of CSE treatment followed by IAV infection on the lung inflammatory response were measured by qRT-PCR of lung tissue and ELISA of BALF at day 7 after infection. As expected, PR8 influenza exposure induced a vigorous antiviral response at the mRNA level, since IP-10 and IFN-β mRNA levels were increased 105- and 2,500-fold, respectively, over baseline by virus. However, consist with our earlier results in the human lung model, CSE exposure before infection suppressed the host antiviral response with inhibition of the IP-10 and IFN-β mRNA response by 20 and 25%, respectively (Fig. 2, A and B). Proinflammatory cytokines IL-6 and TNF-α mRNA were also induced by IAV. However, CSE did not inhibit induction of these two cytokines (Fig. 2, C and D).
Fig. 2.

Cigarette smoke extract (CSE) suppresses lung antiviral IFN-β and IP-10 but not proinflammatory IL-6 and TNF-α responses in influenza-infected mice. Mice were treated with CSE for 6 wk and then infected with 300 pfu of the IAV PR8 strain. All treatments, infections, and appropriate control exposures were administered intratracheally. At day 7, the mice were killed, and lung tissues were collected for RNA extraction. Endogenous mRNA levels of the antiviral cytokines IFN-β (A) and IP-10 (B) and the proinflammatory cytokines IL-6 (C) and TNF-α (D) were assessed by qRT-PCR. Each expression was normalized against β-actin mRNA. Data are expressed as means ± SE (4 mice/group). *Significant difference between CSE-treated PR8-infected and non-CSE-treated PR8-infected groups, P < 0.05.
The effect of CSE on the host response at the level of translation was determined by ELISA. PR8 infection induced IP-10 protein 6-fold and IL-6 protein 110-fold over baseline in BALF (Fig. 3). Similar to the qRT-PCR mRNA results, the host antiviral IP-10 protein response was decreased by 19% in CSE-exposed mice. Interestingly, the proinflammatory IL-6 protein was not significantly decreased (Fig. 3B).
Fig. 3.

CSE exposure inhibits IAV induction of the antiviral cytokine protein IP-10 but not the proinfammatory cytokine IL-6 in bronchoalveolar lavage fluid (BALF). Mice were treated with CSE for 6 wk and then infected with 300 pfu of the IAV PR8 strain. All treatments, infections, and appropriate control exposures were administered intratracheally. Antiviral IP-10 (A) and proinfammatory IL-6 (B) cytokine protein levels in BALF harvested 7 days after infection were determined by ELISA. Mock animals were inoculated with PBS. Data are expressed as means ± SE (4 mice/group). *Significant difference between CSE-treated PR8-infected and non-CSE-treated PR8-infected groups, P < 0.05.
Intratracheal CSE treatment attenuates influenza-mediated induction of RIG-I in vivo, and may enhance viral replication.
To determine if the immunosuppression of the host antiviral response to influenza by CSE could be due to RIG-I inhibition, mRNA and protein levels of RIG-I in mouse lung were assessed by qRT-PCR and Western blot. In a similar manner to the effects in our human lung model, CSE exposure decreased the antiviral RIG-I response by 65% in mouse lung (Fig. 4A). At the level of translation, Western blot showed a similar suppressive effect of CSE on RIG-I protein induction in infected lung (Fig. 4C). To assess viral containment, the effect of CSE exposure on IAV replication was also determined by measuring viral NP levels in lung tissue by qRT-PCR. NP mRNA levels were increased in CSE-exposed animals compared with unexposed controls, although the difference was not significant (Fig. 4B). This suggests that the consequence of CSE-mediated immunosuppression of antiviral responses in mice may be enhanced viral replication, consistent with the increased mortality seen in these animals and in human smokers.
Fig. 4.
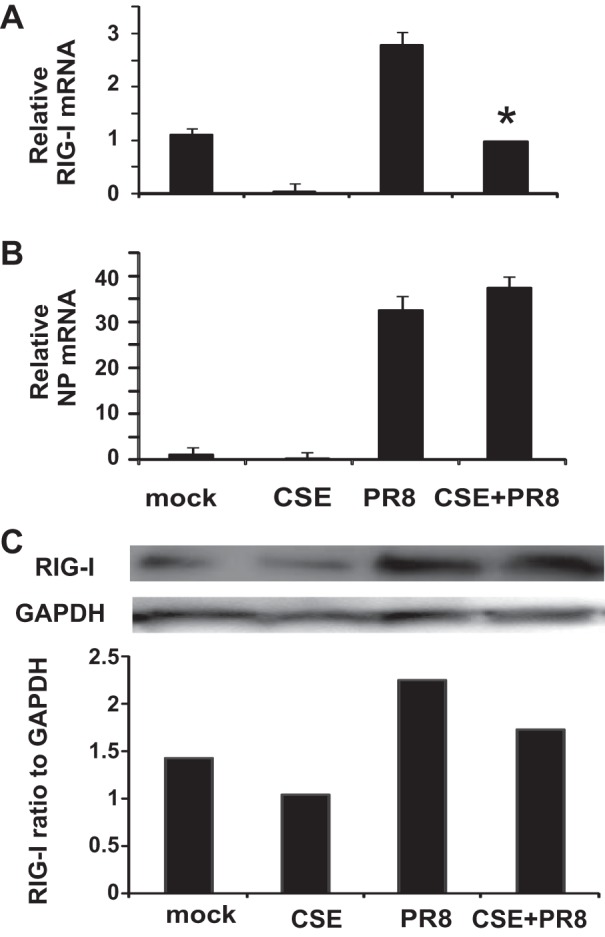
RIG-I mRNA and protein induction is impaired by CSE exposure during influenza infection, and whole lung viral replication may be enhanced. Mice were intratracheally treated with CSE for 6 wk and then infected with 300 pfu of the IAV PR8 strain. At day 7, the mice were killed, and lung tissues were collected for RNA and protein extraction. The RIG-I mRNA (A) levels were assessed by qRT-PCR normalized against β-actin, and RIG-I protein was determined by Western blot normalized to GAPDH (C). Influenza replication was assessed by measurement of viral nucleoprotein (NP) mRNA (B) levels normalized against β-actin mRNA. Mock-treated mice were inoculated with PBS. Data are expressed as means ± SE (4 mice/group). *Significant difference between CSE-treated PR8-infected and non-CSE-treated PR8-infected groups, P < 0.05.
These results demonstrate that CSE exposure suppresses the RIG-I-mediated antiviral host response in our intratracheal CSE exposure animal model. Immunosuppression may lead to enhanced viral replication and increased mortality in both humans and mice (7, 28).
Whole body CS exposure enhances influenza-induced lung injury.
Although the intratracheal model is quick and inexpensive, it is not the organic route of CS inhalation that is the gold standard of such studies. Therefore, our next set of experiments used a whole body smoke exposure model to determine if this method of CS delivery would show similar attenuation of the RIG-I and antiviral cytokine inductions during IAV infection. A smoke exposure system from Teague Enterprises was used. C57BL/6 mice were exposed to CS for 6 wk in the chamber and then intranasally infected with a nonlethal (low) dose of the IAV PR8 strain, and the samples were taken after 7 days. This infectious dose was 20% of LD50–11d determined experimentally in our laboratory using C57BL/6 mice. We included a “no smoke, no handling” (NS) control that gave a normal lung baseline. PBS sham treatment was used as the negative (no virus) control. We also included a “noise control” (NC) group to account for the noise produced by the Teague system. The whole body model was used to measure CS effects on the lung antiviral and proinflammatory responses.
As expected, H & E-stained sections of IAV-infected-only lung showed typical viral pneumonia, capillary and small vessel thromboses, interstitial edema and inflammatory infiltration, varying degrees of acute intra-alveolar edema and/or hemorrhage, and necrotizing bronchitis and bronchiolitis. IAV infection also led to neutrophilic exudates that were characteristic of massive infiltrates of neutrophils/lymphocytes that filled the alveolar air spaces (Fig. 5C). CS-exposed lung without virus infection showed mild inflammation and immunoreactive cell influx (Fig. 5B). However, in lung exposed to both CS and IAV, complete loss of the epithelial layer can be seen, often associated with the formation of hyaline membranes at these sites. Some areas showed organizing diffuse alveolar damage and development of pulmonary granulation tissue and fibrosis (Fig. 5D). In both CS and NS mouse lungs, IAV infection led to neutrophilic exudates that were characteristic of massive infiltrate of neutrophils/lymphocytes that filled the alveolar air spaces (Fig. 5).
Fig. 5.

Whole body cigarette smoke (CS) exposure enhances lung tissue damage after IAV infection. Mice were exposed to CS for 6 wk in a smoke exposure chamber (Teague Enterprises) and then intranasally infected with a nonlethal dose of IAV PR8 strain. Samples were harvested after 7 days. Top, lung sections from mice that were PBS mock infected: normal-appearing tissue from mice not exposed to virus or CS treatment (A); mild inflammation and influx in tissue exposed to CS treatment only alone (B). Middle, sections from mice that were IAV PR8 infected: viral pneumonia, vessel thrombosis, edema, infiltration, intra-alveolar hemorrhage, and bronchitis in tissue from mice infected with IAV alone (C); diffuse alveolar damage in tissue from mice with both CS and influenza treatment (D). The scale bar denotes 200 μm. Insets E and F with higher magnification are showing the loss of epithelial layer and alveolar damage of corresponding panels C and D. The lungs of 3 mice from each treatment group were processed for histology, and results shown were typical for the group.
Whole body CS suppresses the antiviral but not the proinflammatory immune response by significantly attenuating RIG-I.
We measured RIG-I and cytokine mRNA levels in lung tissue after whole body smoke exposure and IAV treatment using qRT-PCR. PR8 virus induced a vigorous RIG-I, antiviral, and proinflammatory cytokine response. As with CSE treatment, CS exposure before infection suppressed the antiviral IFN-β and IFN-λ 2/3 cytokine response but did not significantly affect the proinflammatory cytokine IL-6 mRNA response (Fig. 6, A, B, and C). CS also inhibited the RIG-I mRNA response (Fig. 6D), consistent with our hypothesis that CS worsens IAV infection through immunosuppression of the RIG-I-mediated antiviral response. The inhibition of RIG-I was also confirmed at the protein level by immunoblotting (Fig. 6E). Of note, we examined the other PRR, TLR3, which has been shown to be involved in anti-influenza virus response. There was a 4.2-fold induction of TLR3 by IAV in the noise control group (NC + PR8, Fig. 6F), which was less than that seen with induction of RIG-I by virus (18-fold, Fig. 6D). However, CS did not suppress TLR3 induction by virus and may have instead enhanced TLR3 induction, although the difference was not significant.
Fig. 6.

Whole body CS treatment suppresses IAV-stimulated RIG-I and antiviral cytokine mRNA induction but not proinfammatory cytokine mRNA induction. Mice were exposed to CS or not (NS) for 6 wk in a smoke exposure chamber and then intranasally infected with low-dose IAV PR8. Lung was harvested after 7 days of infection. Noise controls (NC) were mice in the same room, outside the chamber. Intranasal PBS was used as a negative control for infection. RIG-I (D), antiviral cytokines IFN-β (A) and IFN-λ 2/3 (B), proinfammatory cytokine IL-6 (C), and the PRR, Toll-like receptor 3 (TLR3) (F) mRNA expressions were assessed by qRT-PCR, normalized against β-actin. RIG-I protein expression (E) in tissue was detected by immunoblotting, normalized to GAPDH. Data are expressed as means ± SE (3 mice/group). *Significant difference between CS + PR8 and NC + PR8 groups, P < 0.05.
To confirm that inhibition of antiviral cytokine protein induction was reflected at the protein level, we measured cytokine amounts in BALF from mice exposed to IAV with or without CS treatment using multiplex immunoassay. IAV PR8 induced a 496-fold IFN-γ and a 179-fold IP-10 increase (Fig. 7, A and B) in antiviral cytokine release at 7 days postinfection. Adding whole body smoke exposure before infection decreased IAV-stimulated IFN-γ and IP-10 release by 80 and 68%, respectively (Fig. 7, A and B). On the contrary, CS did not change the levels of the proinflammatory cytokine proteins IL-6 and TNF-α in BALF from virus-infected mice (Fig. 7, C and D). The fact that differential antiviral and proinflammatory cytokine attenuation occurred may be due to direct regulation of antiviral cytokine by RIG-I, whereas proinflammatory cytokines were controlled by other pathways, possibly by TLR3.
Fig. 7.

Whole body CS treatment suppresses IAV-stimulated antiviral cytokine IFN-γ and IP-10 protein induction in BALF but does not affect the proinflammatory cytokine IL-6 and TNF-α protein response. Mice were exposed to CS or not for 6 wk in a smoke exposure chamber and then intranasally infected with low-dose IAV PR8. BALF was harvested after 7 days of infection. NC were mice in the same room, outside the chamber. Intranasal PBS was used as a negative control for infection. Antiviral cytokine IFN-γ (A) and IP-10 (B) and proinflammatory cytokine IL-6 (C) and TNF-α (D) protein levels in the BALF were determined by multiplex immunoassay. Data are expressed as means ± SE (3 mice/group). *Significant difference between CS + PR8 and NC + PR8 groups, P < 0.05.
CS treatment does not alter the composition of the inflammatory cell population in the BALF of infected mice.
We measured the lung inflammatory cell response in BALF in IAV-infected mice with/without whole body smoke exposure. Influenza significantly increased the percentage of lymphocytes (Fig. 8B) and neutrophils (Fig. 8C) in BALF on day 7 after infection but decreased the percentage of alveolar macrophages almost by half (Fig. 8A). However, CS exposure did not significantly alter the levels of these cells during viral infection. These results indicate that, although CS increased mortality and decreased antiviral responses, this effect was not likely mediated through inhibition of inflammatory cell recruitment to the lung. Furthermore, the associated unchanged proinflammatory cytokine levels did not appear to alter inflammatory cell recruitment.
Fig. 8.

The BALF immune cell differential in IAV-infected mice is not influenced by CS treatment. C57BL/6 mice were left unexposed or were exposed to CS for 6 wk in a smoke exposure chamber and then intranasally infected with low-dose IAV PR8 or treated with PBS. BALFs were collected from mice 7 days after infection. Cells were stained with Diff-Quik and counted. Data are expressed as means ± SE (3 mice/group).
RIG-I induction by influenza is suppressed by CS as detected by immunohistochemistry.
Last, we examined CS suppression of RIG-I induction by IAV in mouse lung by immunostaining. This was performed since whole lung quantification may underestimate the effects of cell-specific alterations in RIG-I. Mice exposed to CS for 6 wk were infected with IAV and killed after 7 days. Lungs were processed for immunohistochemistry for detection of IAV NP and RIG-I using an anti-NP polyclonal antibody (36) and an anti-RIG-I monoclonal antibody (Abcam), respectively. The mice exposed to PBS were used to demonstrate baseline fluorescence. The PBS mock control showed minimal background immunofluorescence when stained for NP and RIG-I (Fig. 9, left), with comparable signal to the nonspecific signal seen in virus-exposed animals stained with secondary antibody alone (Fig. 9, right). RIG-I was highly expressed in infected cells morphologically identified as the cuboidal type II alveolar epithelial cells. CS-treated lung, as supported by previous qRT-PCR, ELISA, and Western blot techniques, exhibited decreased RIG-I protein induction. Viral NP expression appeared to be in more discrete areas in control infected mice, whereas it was more evenly distributed in CS mice (Fig. 9, middle). The results suggested that virus replicated rapidly in mouse lung during 7 days of infection, with concurrent induction of RIG-I. However, CS exposure suppressed the induction of RIG-I by PR8 in the animals compared with ones that did not receive CS treatment.
Fig. 9.

Whole body CS exposure suppresses RIG-I induction by IAV and may alter distribution of IAV replication. Mice were exposed to CS or not for 6 wk in a smoke exposure chamber and then intranasally infected with low-dose IAV PR8 or PBS. Mouse lungs were processed for immunohistochemistry for detection of RIG-I protein (represented in green) or IAV NP (red) using an anti-NP polyclonal antibody and an anti-RIG-I monoclonal antibody, respectively, labeled with Alexa Fluor 546. The lungs of 3 mice from each treatment group were processed for histology, and results shown were typical for the group. The bar represents 100 μm for all images.
DISCUSSION
Although the link between CS exposure and susceptibility to influenza has been recognized for many years, the mechanisms underlying this association are poorly understood. Recently we reported that CSE-mediated immunosuppression of antiviral responses is by attenuation of the protective RIG-I response to IAV in our human lung organ culture model. In the current study, we investigated the effects of CS exposure on the production of antiviral cytokines concurrent with IAV infection using intratracheal and whole body delivery models. Both the intratracheal CSE treatment model and the whole body CS exposure model caused antiviral immunosuppression, and CSE/CS exposure inhibited the RIG-I-initiated antiviral response.
Signaling events regulated by RIG-I during IAV infection are important in the antiviral IFN response and for modulation of other cytokine responses. An earlier publication showed that infection with influenza resulted in the reduced expression of transcription factor IFN regulatory factor 7 (IRF7) in nasal epithelial cells (NEC) from smokers (16). It is known that stimulation of RIG-I activates specific signaling pathways that lead to activation of nuclear transcription factors such as NF-κB, IFN regulatory factor 3, and IRF7, which are then translocated to the nucleus where they act as transcription factors for type I and III IFNs (8, 19). Because IRF7 is located downstream of RIG-I, reduced expression of IRF7 is most likely the subsequent effect of RIG-I suppression by CS observed here.
Animal models have been widely used as human substitutes to study CS-related diseases (4, 14, 27, 31). In the present study, mice were exposed to CS using two distinct smoke exposure systems. The first, an intratracheal CSE instillation exposure system, has been used to study smoke-induced inflammation. The second system, a whole body exposure system, has also been developed and used for many years (7, 29). For comparison, CSE administration resulted in similar innate immunosuppressive effects as the whole body exposure system. Thus, the CSE model could be an inexpensive alternative CS exposure model to the whole body smoking machine system. The findings also suggest that both mouse models are not only useful in understanding the detrimental consequences of CS but also for delineating the mechanisms underlying the deleterious effects of CS in respiratory diseases.
Local inflammation and elevated cytokine production are the hallmarks of host innate immune responses that act to protect against infection by viruses and are essential in local control of invading microbes. There are two types of innate immune cytokine responses to infection with IAV, the antiviral response and the proinflammatory response. Our results demonstrated that CS inhibits IAV-induced RIG-I mRNA expression and subsequent antiviral cytokine mRNA responses in the lung and that the proinflammatory cytokine response was intact, if not increased, after CS exposure. A similar effect was observed at the level of translation as BALF antiviral cytokine protein induction, but not proinfammatory cytokine induction was decreased by CS exposure in influenza-infected mice. Lung inflammatory cell populations were altered by IAV infection but not by CS treatment during infection. CS poses an extreme danger to body surveillance systems, since antiviral cytokines, mainly IFNs, are necessary for the initial innate response that assists with control of viral infections. However, we did not find CS inhibition of the proinflammatory cytokine response represented by IL-6 and TNF-α. The proinflammatory cytokine response, when uncontrolled, may increase the severity of primary viral pneumonia or it may predispose to the subsequent development of serious secondary bacterial infections. Our results agreed with an earlier report that showed that high-dose IAV infection induced significant airway inflammation, and CS increased levels of IL-6 and TNF-α compared with sham-exposed animals (26). Interestingly, we found that CS exposure did not impact cell expression of TLR3 in the lungs of the mice, which was also demonstrated by Robbins et al. (26). The results suggested proinflammatory cytokines might be controlled by TLR3 in response to IAV in mice. dsRNA is produced during viral replication and is recognized by endosomal TLR3 (10). Because the increased TLR3 might cause uncontrolled inflammation, our results provide an explanation why TLR3-deficient mice appear to be more resistant to influenza infections than WT mice, in terms of mortality (20). In in vitro studies, CSE enhances rhinovirus-induced TLR3 expression and IL-8 secretion in A549 cells (34). In vivo, CS augments the expression and responses of TLR3 in human macrophages (18) and in murine lung tissue. However, two research groups have reported that smoking decreased TLR3 induction in human lung macrophages and NEC (13, 33). In our previous study, we also found a modest increase in TLR3 in human lung during IAV infection that was inhibited by CSE (35). It is known that TLR3 is highly expressed in mouse innate immune cells but shows a low level of expression in human monocytes, macrophages, and dendritic cells (12). This might lead to some conflicting results in studies of TLR3 expression in human and mouse models. More work needs to be done to address whether TLR3 is involved in increased proinflammatory cytokines and plays a role in the increased vulnerability of smokers to influenza.
Taken together, our results confirm that RIG-I is an important PRR that senses IAV infection and activates antiviral cytokine defenses in mouse lung. Both CS exposure animal models support our previous finding in a human model that the depression of RIG-I induction and antiviral cytokine response by CS are likely important in the enhanced susceptibility of smokers to influenza infection in the lung. These results provide new insight into the mechanisms by which CS exposure compromises pulmonary host defenses against influenza infections.
GRANTS
The research described in this work was partially supported by a pilot grant from the National Institute of Allergy and Infectious Diseases, project 5U19-AI-062629 (to W. Wu), by a Clinical Innovator Award from the Flight Attendant Medical Research Institute (to J. P. Metcalf), by the Merit Review Program of the Department of Veterans Affairs, the National Institutes of Health (NIH) (1 I01 BX001937), by the National Institute of Allergy and Infectious Diseases, project 5U19-AI-62629 (to J. P. Metcalf), by NIH Grant R01-HL-116876, a grant from the Oklahoma Center for Adult Stem Cell Research, and NIH Grant P20-GM-103648 Core Facility (to L. Liu).
DISCLOSURES
The authors have no financial conflicts of interest.
AUTHOR CONTRIBUTIONS
Author contributions: W.W. and L.L. conception and design of research; W.W., W.Z., and Y.D.Z. analyzed data; W.W. drafted manuscript; W.Z., S.M., and J.L.B. performed experiments; E.S.D. and J.P.M. edited and revised manuscript; J.P.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge the assistance from the Oklahoma Medical Research Foundation Imaging Analysis core facility and Yang Song from the Oklahoma Medical Research Foundation for help on histology evaluation. We also thank Dr. Dennis Burian and David Hutchings from the Civil Aerospace Medical Institute, Federal Aviation Administration, Oklahoma City for technical assistance on this project.
REFERENCES
- 1.Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med 164: 2206–2216, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Bagaitkar J, Demuth DR, Scott DA. Tobacco use increases susceptibility to bacterial infection (Abstract). Tob Induc Dis 4: 12, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carp H, Janoff A. Possible mechanisms of emphysema in smokers. In vitro suppression of serum elastase-inhibitory capacity by fresh cigarette smoke and its prevention by antioxidants. Am Rev Respir Dis 118: 617–621, 1978. [DOI] [PubMed] [Google Scholar]
- 4.Churg A, Cosio M, Wright JL. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am J Physiol Lung Cell Mol Physiol 294: L612–L631, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Eddleston J, Lee RU, Doerner AM, Herschbach J, Zuraw BL. Cigarette smoke decreases innate responses of epithelial cells to rhinovirus infection. Am J Respir Cell Mol Biol 44: 118–126, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elliott MK, Sisson JH, West WW, Wyatt TA. Differential in vivo effects of whole cigarette smoke exposure versus cigarette smoke extract on mouse ciliated tracheal epithelium. Exp Lung Res 32: 99–118, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng Y, Kong Y, Barnes PF, Huang FF, Klucar P, Wang X, Samten B, Sengupta M, Machona B, Donis R, Tvinnereim AR, Shams H. Exposure to cigarette smoke inhibits the pulmonary T-cell response to influenza virus and Mycobacterium tuberculosis. Infect Immun 79: 229–237, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4: 491–496, 2003. [DOI] [PubMed] [Google Scholar]
- 9.Gualano RC, Hansen MJ, Vlahos R, Jones JE, Park-Jones RA, Deliyannis G, Turner SJ, Duca KA, Anderson GP. Cigarette smoke worsens lung inflammation and impairs resolution of influenza infection in mice (Abstract). Respir Res 9: 53, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, Si-Tahar M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem 280: 5571–5580, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Hanshaoworakul W, Simmerman JM, Narueponjirakul U, Sanasuttipun W, Shinde V, Kaewchana S, Areechokechai D, Levy J, Ungchusak K. Severe human influenza infections in Thailand: oseltamivir treatment and risk factors for fatal outcome. PLoS One 4: e6051, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinz S, Haehnel V, Karaghiosoff M, Schwarzfischer L, Muller M, Krause SW, Rehli M. Species-specific regulation of Toll-like receptor 3 genes in men and mice. J Biol Chem 278: 21502–21509, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Horvath KM, Brighton LE, Zhang W, Carson JL, Jaspers I. Epithelial cells from smokers modify dendritic cell responses in the context of influenza infection. Am J Respir Cell Mol Biol 45: 237–245, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang J, Okuka M, McLean M, Keefe DL, Liu L. Effects of cigarette smoke on fertilization and embryo development in vivo. Fertil Steril 92: 1456–1465, 2009. [DOI] [PubMed] [Google Scholar]
- 15.Imaizumi T, Kumagai M, Taima K, Fujita T, Yoshida H, Satoh K. Involvement of retinoic acid-inducible gene-I in the IFN-γ/STAT1 signalling pathway in BEAS-2B cells. Eur Respir J 25: 1077–1083, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Jaspers I, Horvath KM, Zhang W, Brighton LE, Carson JL, Noah TL. Reduced expression of IRF7 in nasal epithelial cells from smokers after infection with influenza. Am J Respir Cell Mol Biol 43: 368–375, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kark JD, Lebiush M. Smoking and epidemic influenza-like illness in female military recruits: a brief survey. Am J Public Health 71: 530–532, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koarai A, Yanagisawa S, Sugiura H, Ichikawa T, Akamatsu K, Hirano T, Nakanishi M, Matsunaga K, Minakata Y, Ichinose M. Cigarette smoke augments the expression and responses of toll-like receptor 3 in human macrophages. Respirology 17: 1018–1025, 2012. [DOI] [PubMed] [Google Scholar]
- 19.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 4: 69–77, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si-Tahar M. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog 2: e53, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S, Sun J, Cai J, Miao Z, Lu M, Qin S, Wang X, Lv H, Yu Z, Amer S, Chai C. Epidemiological, clinical and viral characteristics of fatal cases of human avian influenza A (H7N9) virus in Zhejiang Province, China. J Infect 16: 00239–00239, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Mancini DA, Mendonca RM, Mendonca RZ, do Prado JA, Andrade Cde M. Immune response to vaccine against influenza in smokers, non-smokers and, in individuals holding respiratory complications. Boll Chim Farm 137: 21–25, 1998. [PubMed] [Google Scholar]
- 23.Peat JK, Keena V, Harakeh Z, Marks G. Parental smoking and respiratory tract infections in children. Paediatr Respir Rev 2: 207–213, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314: 997–1001, 2006. [DOI] [PubMed] [Google Scholar]
- 25.Pyhala R, Takala J, Turunen A, Aho K. Smoking and influenza in the elderly: a sero-epidemiological study. Eur J Respir Dis 64: 212–216, 1983. [PubMed] [Google Scholar]
- 26.Robbins CS, Bauer CM, Vujicic N, Gaschler GJ, Lichty BD, Brown EG, Stampfli MR. Cigarette smoke impacts immune inflammatory responses to influenza in mice. Am J Respir Crit Care Med 174: 1342–1351, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Sayers NM, Drucker DB. Animal models used to test the interactions between infectious agents and products of cigarette smoked implicated in sudden infant death syndrome. FEMS Immunol Med Microbiol 25: 115–123, 1999. [DOI] [PubMed] [Google Scholar]
- 28.Sherman CB. The health consequences of cigarette smoking. Pulmonary diseases. Med Clin North Am 76: 355–375, 1992. [DOI] [PubMed] [Google Scholar]
- 29.Simet SM, Sisson JH, Pavlik JA, Devasure JM, Boyer C, Liu X, Kawasaki S, Sharp JG, Rennard SI, Wyatt TA. Long-term cigarette smoke exposure in a mouse model of ciliated epithelial cell function. Am J Respir Cell Mol Biol 43: 635–640, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singanayagam A, Joshi PV, Mallia P, Johnston SL. Viruses exacerbating chronic pulmonary disease: the role of immune modulation (Abstract). BMC Med 10: 27, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slotkin TA. Nicotine and the adolescent brain: insights from an animal model. Neurotoxicol Teratol 24: 369–384, 2002. [DOI] [PubMed] [Google Scholar]
- 32.Taima K, Imaizumi T, Yamashita K, Ishikawa A, Fujita T, Yoshida H, Takanashi S, Okumura K, Satoh K. Expression of IP-10/CXCL10 is upregulated by double-stranded RNA in BEAS-2B bronchial epithelial cells. Respiration 73: 360–364, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Todt JC, Freeman CM, Brown JP, Sonstein J, Ames TM, McCubbrey AL, Martinez FJ, Chensue SW, Beck JM, Curtis JL. Smoking decreases the response of human lung macrophages to double-stranded RNA by reducing TLR3 expression (Astract). Respir Res 14: 33, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang JH, Kim H, Jang YJ. Cigarette smoke extract enhances rhinovirus-induced toll-like receptor 3 expression and interleukin-8 secretion in A549 cells. Am J Rhinol Allergy 23: e5–e9, 2009. [DOI] [PubMed] [Google Scholar]
- 35.Wu W, Patel KB, Booth JL, Zhang W, Metcalf JP. Cigarette smoke extract suppresses the RIG-I-initiated innate immune response to influenza virus in the human lung. Am J Physiol Lung Cell Mol Physiol 300: L821–L830, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Air GM. Expression of functional influenza virus A polymerase proteins and template from cloned cDNAS in recombinant vaccinia virus infected cells. Biochem Biophys Res Commun 200: 95–101, 1994. [DOI] [PubMed] [Google Scholar]