

Abstract
Exposure to relatively high levels of chlorine (Cl2) gas can occur in mass-casualty scenarios associated with accidental or intentional release. Recent studies have shown a significant postexposure injury phase to the airways, pulmonary, and systemic vasculatures mediated in part by oxidative stress, inflammation, and dysfunction in endogenous nitric oxide homeostasis pathways. However, there is a need for therapeutics that are amenable to rapid and easy administration in the field and that display efficacy toward toxicity after chlorine exposure. In this study, we tested whether nitric oxide repletion using nitrite, by intramuscular injection after Cl2 exposure, could prevent Cl2 gas toxicity. C57bl/6 male mice were exposed to 600 parts per million Cl2 gas for 45 min, and 24-h survival was determined with or without postexposure intramuscular nitrite injection. A single injection of nitrite (10 mg/kg) administered either 30 or 60 min postexposure significantly improved 24-h survival (from ∼20% to 50%). Survival was associated with decreased neutrophil accumulation in the airways. Rendering mice neutropenic before Cl2 exposure improved survival and resulted in loss of nitrite-dependent survival protection. Interestingly, female mice were more sensitive to Cl2-induced toxicity compared with males and were also less responsive to postexposure nitrite therapy. These data provide evidence for efficacy and define therapeutic parameters for a single intramuscular injection of nitrite as a therapeutic after Cl2 gas exposure that is amenable to administration in mass-casualty scenarios.
Keywords: halogen, acute lung injury, nitric oxide, inflammation
exposure to chlorine gas (Cl2) and resultant toxicity are concerns in civilian and military settings. Several examples pertaining to the use of Cl2 as a chemical weapon and large-scale accidental exposures attributable to the derailment of trains transporting Cl2 through populated areas have been documented (3, 8, 21, 31, 34). The potential for future incidents remains because of the demand for Cl2 in multiple industrial processes (8). Several studies have provided insight into the pathogenesis of Cl2 toxicity, which is characterized by injury to the airways during the initial halogen insult. This is followed by a postexposure injury phase occurring over a time span ranging from days to months, which affects the airways and pulmonary and systemic vasculature (9, 11, 20, 25, 26, 30, 37). Clinically, this presents as reactive airway syndrome, hypoxemia, noncardiogenic edema leading to acute lung injury, and development of acute respiratory distress syndrome, which chronically leads to reactive airway disease, increased sensitivity to pulmonary infections, fibrosis, and bronchiolitis obliterans, as well as dermal injury (9, 10, 12, 17, 24, 26, 28, 30). Exciting recent studies are providing insights into the postexposure mechanisms that include increased oxidative stress, neurogenic and airway inflammation, fibrosis, loss of nitric oxide (NO) signaling homeostasis, and loss of endogenous airway repair mechanisms (2, 9–11, 15, 20, 22, 25, 26, 36). This information has led to the testing and demonstration of efficacy for antioxidant therapy (9, 16, 22, 23, 37), anti-inflammatory (using corticosteroids) treatment (5), β2-agonists (29), phosphodiesterase inhibitors (4), transient receptor potential channel inhibitors (1, 2), and strategies that improve NO bioavailability (28, 36).
An important consideration for Cl2 toxicity is that potential therapies are effective when administered postexposure. They must also be amenable to stockpiling with a rapid and easy method of administration. These are all key features for mass-casualty scenarios. In this context, intramuscular (IM) injection is a preferred modality. Given the multitude of mechanisms involved in postexposure toxicity, it is unlikely that one therapeutic will prevent all aspects of pathogenesis of Cl2 toxicity. Moreover, in some cases, therapeutics targeted to specific aspects of Cl2 toxicity may not be amenable to rapid administration in a mass-casualty scenario. For example, inhaled therapies, although being the most efficient means of delivering large amount of therapeutic agents to lung epithelia, are logistically more challenging as a first-line therapeutic in a mass-casualty scenario but more feasible if given in a hospital setting. Thus therapeutics that improve immediate/short-term survival after Cl2 exposure are also critical to develop, the goal being that this will allow for administration of other targeted therapies that can be administered in a more controlled and clinical environment.
NO is a critical mediator of homeostasis in all organ systems. In addition to its generation by one of three nitric oxide synthases, multiple studies over the last decade have shown that enzymatic production of NO is complemented by nonenzymatic mechanisms for NO generation from nitrite (18). A functional role for the latter pathway has been demonstrated in both physiological and pathophysiological contexts, and perhaps the most evidence is in therapeutics where the goal is to improve or replete NO bioactivity in ischemic and inflammatory diseases (14, 19, 33). Consistent with this, our recent studies showed that nitrite administered by intraperitoneal (IP) or IM injections, after Cl2 exposure prevented acute lung injury and development of reactive airways in rats (28, 36). Importantly, nitrite also meets the therapeutic criteria outlined above for mass-casualty scenarios and has an established safety profile for use in humans in the context of cyanide antidotes, which is further supported by more recent phase 1/2 studies. In this study, using a model of lethal Cl2 exposure, we demonstrate the therapeutic potential for nitrite administered postexposure to improve survival and provide insights into underlying mechanisms of protection.
MATERIALS AND METHODS
Materials.
Unless stated otherwise all reagents were purchased from Sigma (St. Louis, MO). Quantitative chemokine (CXCL2 and CXCL1) ELISA kits were purchased from R&D Systems (Minneapolis, MN). Bio-Rad Protein Assay Reagent Kits were purchased from Bio-Rad (Hercules, CA). Male (25–27 g, 8–11 wk of age) and female (18–20 g, 8–11 wk of age) C57bl/6 mice were purchased from Harlan (Indianapolis, IN) and kept on 12-h:12-h light/dark cycles with access to standard chow and water ad libitum before and after chlorine gas exposure.
Mouse exposure to chlorine gas.
Whole body exposures of male and female mice to Cl2 gas were performed as previously described (16, 37). Exposures were performed with either two or five mice in the same chamber at any one time, and all exposures were performed between 8:00 AM and 12:00 PM. Exposure conditions were either 400 parts per million (ppm) Cl2 in air for 30 min or 600 ppm for 45 min using 400-ppm and 600-ppm Cl2 tanks, respectively. Tanks were replaced when the pressure in the tanks reached 500 psi. In each case, immediately following exposure, mice were returned to room air. The mice were monitored hourly for 12 h and every 6 h thereafter for 24 h. All experiments involving animals were conducted according to protocols approved by the UAB IACUC.
IM nitrite administration.
Mice received a single injection of sodium nitrite in PBS (0.1–100 mg/kg final concentration) in the gluteus maximus region at either 30 min or 60 min after cessation of Cl2 exposure. In addition, a multiple-dose nitrite therapy protocol (10 mg/kg at 60 min, 180 min, and 300 min) was also tested. Nitrite stocks were prepared daily in sterile PBS with injection volumes of 25 μl.
Acute lung injury assessment.
Mice were euthanized with intraperitoneal ketamine and xylazine (100 and 10 mg/kg body wt, respectively). An incision was made at the neck to expose the trachea, and a 3-mm endotracheal cannula inserted. Lungs were lavaged with 3 × 1 ml of PBS; ∼1.8–1.9 ml was recovered in all groups. Mice were then exsanguinated by cardiac puncture for collection of blood. Recovered aliquots of lavage fluid were kept on ice and centrifuged immediately at 300 g for 10 min to pellet cells. Supernatants were removed and stored on ice for protein analysis using the Bio-Rad Protein Assay Reagent Kit compared with BSA standards. Cells were resuspended in 100 μl PBS and counted using a Neubauer hemocytometer. Cells were then placed on slides using a cellspin (Tharmac, Drosselweg, Germany) and stained using a two-stain set consisting of eosin Y and a solution of thiazine dyes (Quik-Stain; Siemens, Washington, DC). Differential counts (specifically monocytes, neutrophils, and lymphocytes) were then performed on slides via light microscopy.
Chemokine measurement.
Mouse CXCL1 (keratinocyte-derived chemokine, KC) and CXCL2 (macrophage inflammatory protein 2, MIP-2) were measured in the plasma and bronchoalveolar lavage fluid (BALF) by ELISA per manufacturer protocols.
Inducing neutropenia.
C57Bl/6 mice were rendered neutropenic as described previously (35) by IP injection with 200 μg of either anti Ly-6G (clone 1A8) (cat. no. BE0075-1; Bxcel, London, UK) or IgG2a Isotype control (cat. no. BE0089, Bxcel) 24 h before Cl2 gas exposure. Mice were exposed to Cl2 gas and received nitrite (10 mg/kg, 30 min after Cl2 exposure) therapy as described above.
Survival analysis.
Survival was assessed using the Log rank (Mantel-Cox) test. For each exposure, mice were preassigned to either be exposed to Cl2 only or Cl2 followed by nitrite therapy. In some cases, mice assigned to a nitrite therapy group died before receiving nitrite, i.e., during or within 30 min or 60 min postexposure. These animals were excluded from the study because they did not receive therapy and constituted n = 2 for 1 mg/kg nitrite, n = 5 for 10 mg/kg nitrite at 30 min postexposure; n = 4 for 10 mg/kg nitrite, n = 1 for 100 mg/kg nitrite at 60 min postexposure; n = 1 for 10 mg/kg nitrite at 60 min, 180 min, and 300 min postexposure. The numbers of replicates are indicated in the figure legends. All analyses were performed using GraphPad Prism (La Jolla, CA). Significance was set at P < 0.05.
RESULTS
Nitrite administration after chlorine exposure improves 24-h survival.
The goals of this study were to test whether nitrite administered by IM injection postexposure is an effective postexposure therapy in a lethal model of Cl2 exposure. To date, the beneficial effects of nitrite therapy on sublethal Cl2 injury have been established (28, 36). Using male mice, initial studies determined exposure conditions leading to >75% mortality over 24 h. When five mice at a time were exposed to 600 ppm of Cl2 for 45 min, <10% mortality was observed over 24 h; however, mortality significantly increased (>85% by 24 h) if only two mice at a time were exposed (not shown). Consistent with our previous studies, 400 ppm Cl2 for 30 min did not cause any mortality despite causing significant lung injury (29, 36, 37). We therefore chose exposure conditions of 600 ppm, 45 min with two mice being exposed at any given time.
Figure 1A shows the effects of nitrite (0.1, 1.0, and 10.0 mg/kg) administered by IM injection 30 min after Cl2 exposure. Neither 0.1 mg/kg nor 1.0 mg/kg nitrite affected mortality. However, a single administration of 10.0 mg/kg at 30 min postexposure significantly improved 24-h survival by ∼50%. Figure 1B shows that nitrite (10 mg/kg) also afforded protection when administered 60 min after Cl2 exposure, which was no different compared with nitrite administered at 30 min. The protective effect of nitrite injection at 60 min after Cl2 exposure was lost, however, if a higher dose (100 mg/kg) was used, consistent with previous studies showing that nitrite has an optimum therapeutic concentration (7, 28). Because the circulating half-life of nitrite is ∼20–40 min, we also tested a multiple-dose regimen to establish whether a single injection of nitrite was sufficient to afford protection or whether multiple doses would provide better protection. Nitrite (10 mg/kg) administered at 1 h, 3 h, and 5 h after Cl2 exposure did not improve survival relative to Cl2 alone although a trend toward protection was noted (P = 0.15, Figure 1B). We also noted that, when combined, data from Fig. 1, A and B, indicated that the greatest degree of survival benefit was observed over the first 12–16 h postexposure.
Fig. 1.
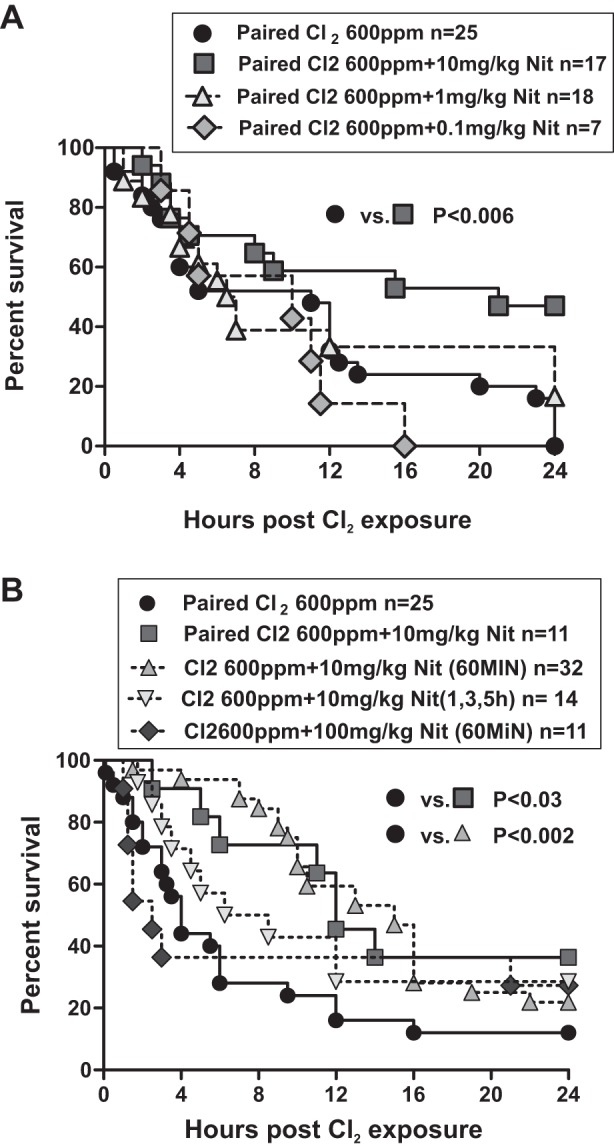
Nitrite-dependent protection against chlorine-induced mortality in mice. A: male mice were exposed 2 at a time to Cl2 600 parts per million (ppm), 45 min and then brought back to room air. 30–60 min thereafter, mice received either saline or nitrite (Nit) at 0.1 mg/kg, 1.0 mg/kg, or 10.0 mg/kg at 30 min after Cl2 exposure by intramuscular injection. Data show Kaplan-Meier survival curves. *P < 0.006 between 10 mg/kg nitrite 30 min and saline by the Log-rank test. No significant difference between 0.1 mg/kg nitrite (P = 0.33) or 1.0 mg/kg nitrite (P = 0.26) and saline was observed. B: male mice were exposed to Cl2 at 600 ppm, 45 min and then brought back to room air, and nitrite was administered as indicated in figure by intramuscular (IM) injection. Data show Kaplan-Meier survival curves. P < 0.03 between saline and nitrite (10 mg/kg, 30 min) and P < 0.002 between saline and nitrite (10 mg/kg, 60 min). No significant difference was observed between saline and either multiple nitrite administration (P = 0.15) or nitrite (100 mg/kg, 60 min) (P = 0.75) groups.
Role of polymorphonuclear leukocytes in nitrite-dependent protection.
To evaluate the potential mechanisms by which IM nitrite afforded survival benefit, mice were exposed to Cl2 gas 600 ppm, 45 min and administered saline or IM nitrite (10 mg/kg) 30 min postexposure. Mice were continually observed over the next 6 h, and, if they died during this time, BALF and blood were collected immediately for assessment of MIP-2 or KC and accumulation of protein and inflammatory cells. At 6 h, all mice were killed, and similar measurements of acute lung injury were made (5/8 mice in the Cl2 gas-only group and 9/9 mice in the nitrite-treatment group survived to 6 h). Figure 2A shows that BAL protein increased to similar extents in both Cl2 and Cl2 + nitrite groups. However, BAL levels of inflammatory cells were markedly lower in nitrite-treated mice compared with Cl2-alone-exposed mice (Fig. 2B); this effect was specific for polymorphonuclear leukocytes (PMN) (Fig. 2C). Figure 2D shows that, in addition to decreasing PMN infiltration, nitrite therapy also improved viability of cells collected from the BALF. To gain insights into how nitrite prevented PMN trafficking into the lungs, BAL and plasma levels of the proneutrophilic chemokines MIP-2 (CXCL2) and KC (CXCL1) were measured (Fig. 2, E and F, respectively). Surprisingly, highest chemokine levels were observed in the nitrite group relative to air, with trends toward increased levels relative to Cl2 also noted.
Fig. 2.

Effect of nitrite therapy on indices of acute lung injury. Male mice were exposed to Cl2 600 ppm, 45 min and then brought back to room air and 30 min thereafter received saline or nitrite 10 mg/kg by IM injection. Mice were continually observed over the next 6 h, and, if they died during this time, bronchoalveolar lavage fluid (BALF) and blood were collected immediately. At 6 h, all mice were killed, and BALF and plasma were collected. A and B: BALF levels of protein and total cell accumulation. *P < 0.01 relative to air, #P < 0.05 relative to Cl2 alone by 1-way ANOVA with Tukey's posttest. C: differential cell analysis. D: viability of BALF inflammatory cells. *P < 0.01 relative to respective + nitrite group by t-test. E: BALF and plasma macrophage inflammatory protein 2 (MIP-2) levels, *P < 0.01 relative to air and #P < 0.05 relative to Cl2-alone group by 1-way ANOVA with Tukey's posttest. F: BALF and plasma keratinocyte chemokine (KC) levels, *P < 0.01 or #P < 0.05 relative to air, by 1-way ANOVA with Tukey's posttest. All data are means ± SE (n = 5–9).
Effects of neutropenia on Cl2 toxicity and nitrite therapy.
Figure 2 suggests an association between preventing PMN accumulation in the BAL and short-term survival benefit after Cl2 gas exposure. To directly test whether PMN are involved, mice were rendered neutropenic first, then exposed to Cl2 gas, and mortality followed over 12 h. Neutropenia was confirmed by no change in blood macrophage counts and >90% depletion of blood PMN 24 h after treatment with anti-Ly6 antibody compared with isotype control (not shown). Figure 3 shows that mortality was no different between mice exposed to Cl2 only vs. mice exposed to Cl2 after treatment with isotype control antibody. However, mortality was significantly lower in neutropenic mice. Moreover, nitrite therapy still afforded protection in mice treated with isotype control antibody but had no effect in neutropenic mice.
Fig. 3.
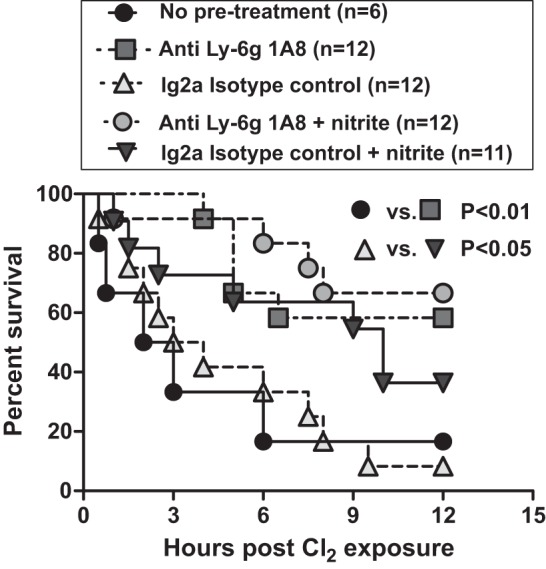
Effects of neutropenia on Cl2-induced mortality and nitrite therapy. Male mice were untreated (no pretreatment) or injected intraperitoneally with 200 μg of either anti Ly-6G or IgG2a isotype control antibody. 24 h later, all mice were exposed to Cl2 gas (600 ppm, 45 min) and then treated with saline or nitrite (10 mg/kg) 30 min after Cl2 exposure. Data show Kaplan-Meier survival curves. P < 0.01 between anti Ly-6G or IgG2a groups, P < 0.05 between IgG2a and IgG2a + nitrite groups, P = 0.6 between Ly-6G and Ly-6G + nitrite groups.
Effects of nitrite therapy on nitrite and nitrate concentrations.
Similar to our previous studies (11), circulating nitrite levels decreased in Cl2-exposed mice (Fig. 4A) with no significant changes in nitrate (Fig. 4B). We have also shown previously that IM injection of nitrite maximally increased circulating levels ∼5 min after administration and with a half-life of ∼20 min. (28). Consistent with this short plasma half-life, nitrite therapy had no effect on circulating nitrite at 6 h postadministration. Although not significant, trends toward increased plasma nitrate formation were evident, however, consistent with nitrite oxidation. More interestingly, trends (P between 0.08 and 0.1) toward increased lung tissue levels of nitrite and nitrate were observed in Cl2-exposed animals. Most striking, however, were the significant increases in nitrite and nitrate in the BALF in nitrite-therapy groups (Fig. 4, E and F).
Fig. 4.

Nitrite and nitrate levels after nitrite therapy. Male mice were exposed to Cl2 600 ppm, 45 min and then brought back to room air and 30 min thereafter received saline or nitrite 10 mg/kg by IM injection. Plasma nitrite (A) and nitrate (B), lung nitrite (C) and nitrate (D), and BALF nitrite (E) and nitrate (F) were measured after 6 h. Data shown are means ± SE, n = 5–9. *P < 0.05 relative to air by for A and *P < 0.05 or **P < 0.01 relative to air and Cl2 alone for E and F by 1-way ANOVA with Tukey's posttest.
Effects of sex on nitrite-dependent protection against Cl2 toxicity.
Recent studies have shown potential differences in the anti-platelet effects of therapeutic nitrate in males vs. females, nitrite efficacy being lost in females (32). To test the general applicability of nitrite therapy and assess whether sex is a modifying factor for Cl2-induced toxicity, matched male and female mice were compared. Figure 5 shows that female mice are more sensitive to Cl2-induced injury compared with males. Nitrite therapy afforded protection in males similar to that observed in Fig. 1. However, nitrite did not statistically improve survival in females although a trend (P = 0.09) was noted.
Fig. 5.
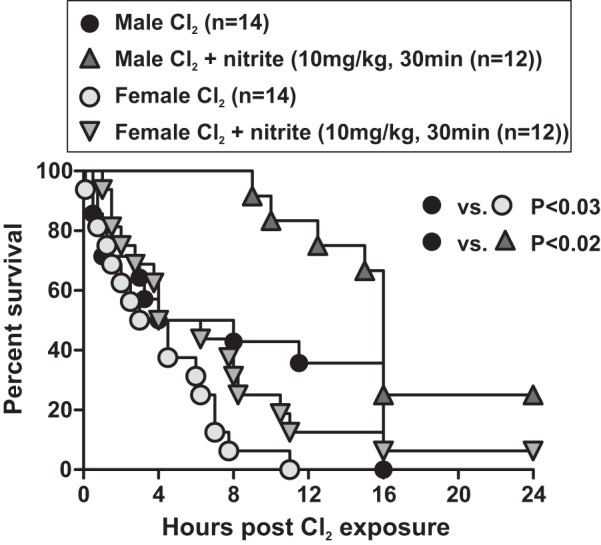
Effect of sex on chlorine toxicity and nitrite therapy. Male or female mice were exposed to Cl2 at 600 ppm, 45 min and then brought back to room air, and nitrite was administered as indicated in figure by IM injection 30 min postexposure. Data show Kaplan-Meier survival curves. P < 0.03 between male and female Cl2-alone groups; P < 0.02 for nitrite therapy in males and P = 0.09 for nitrite therapy in females.
DISCUSSION
In this study, we provide evidence that nitrite therapy improves 24-h survival in mice exposed to Cl2. Importantly, nitrite was efficacious when administered at 30 min or 60 min after Cl2 exposure, providing the first example, to our knowledge, of a postexposure IM injectable therapeutic that was effective in improving mortality in a model of Cl2 toxicity. Together with the potential ability of nitrite to be stockpiled, these data underscore the potential for nitrite therapy as a front line therapeutic for inhaled toxicants in mass-casualty situations.
The last decade has seen many studies document the potential for nitrite to replete NO signaling in acute and chronic diseases afflicting all major organ systems, which are characterized by loss of endogenous NO function (14, 18, 33). We chose to test nitrite therapy on the basis that endothelial NO synthase-dependent signaling is inhibited, and nitrite levels are decreased during the initial stages (hours) after Cl2 exposure (Fig. 4) (11). Also, nitrite afforded protection against acute lung injury in a model of sublethal Cl2 exposure, and similar results have been shown in a model of mechanical ventilator-induced injury (27, 28, 36). This suggests that the therapeutic mechanisms of action of nitrite may target aspects of lung injury common to distinct initiators of acute lung injury. Further studies testing nitrite therapy for lung injury induced by other infectious and noninfectious/mechanical stimuli are warranted.
Less clear is how nitrite improves survival after Cl2 exposure. We have not characterized precisely how mice were dying in these studies with hypoxemic stress and critical-end organ dysfunction likely key. Notably, nitrite was able to improve 24-h survival even when administered 1 h after Cl2 exposure, a time over which much damage to the lungs has already occurred and induction of inflammation likely. Our previous study using a sublethal Cl2 exposure protocol showed that nitrite protected against both increased permeability and inflammation components of Cl2 injury, which was associated with increased airway reepithelialization (28, 36). In the current study, we demonstrate a more prominent role for PMN, which may be attributable to a different model being used here (mice and lethal exposure protocol), testing of only one nitrite dose, and assessment of acute lung injury at one time point. Neutropenia alone improved survival and resulted in a loss of nitrite-dependent protection, suggesting that prevention of PMN-dependent injury is one mechanism by which nitrite elicits protective effects. A recent study also noted attenuated nitrogen mustard-induced skin injury in mice lacking the predominantly neutrophilic enzyme myeloperoxidase (13), suggesting a central role for PMN in mediating postexposure toxicities to diverse chemical threat agents. The anti-PMN effects of nitrite were not mediated by altering MIP-2 or KC, two principal chemokines responsible for PMN homing to the lungs in acute lung injury. In fact, nitrite therapy increased levels of these chemokines in the BALF and plasma. At first glance, such increases in MIP-2 and KC are expected to further stimulate PMN egress into the lungs; however, it is also possible that nitrite increased plasma chemokines more so than BALF levels, which would dissipate the chemokine gradient into the lung, lowering the driving force for PMN infiltration from the circulation into the pulmonary compartment. Other potential mechanisms by which nitrite could affect PMN infiltration include modulation of endothelial adhesion molecules and PMN activation, and future studies need to evaluate whether nitrite modulates PMN-dependent clearance of pathogens, which we have shown are inhibited after Cl2 exposure (10).
Other potential protective mechanisms of nitrite include improving blood flow, protecting against cell death, and allowing endogenous repair mechanisms to function. In the latter context, macrophage viability in the BAL was also improved by nitrite therapy. The dose of nitrite that was effective in the current study (10 mg/kg) was 10-fold higher compared with our previous studies that employed a sublethal Cl2-exposure protocol (28, 36). Thus the precise dose of nitrite required appears to depend in part on the exposure and the end point being assessed. In the current study, use of 10-fold higher or lower dose of nitrite resulted in a loss of protection. Similar “U”-shaped dependence for nitrite therapy has been observed previously for ischemia-reperfusion injury (7, 27). That said, even at the highest dose tested here, nitrite did not increase Cl2 toxicity (see Fig. 1), suggesting that a relatively large therapeutic dose and time window exist.
Interestingly, female mice were more sensitive to Cl2-induced mortality compared with males. Whether this is a consequence of estrogen remains to be tested. We used age-matched males and females, and therefore females were lighter compared with males; whether this is an effector for different responses also requires further investigation. Our primary goals were to test whether nitrite therapy was effective in female mice also. Recent studies have shown that nitrite-based anticoagulation therapy (via nitrate supplementation) may be less effective in female human volunteers (32). Similarly, our data show that nitrite was less effective at preventing Cl2-induced mortality in female mice compared with male mice. This could be a consequence for greater injury in females vs. males and/or less sensitivity for nitrite-based protection in females. That said, a trend toward significance was still noted, however, suggesting the general utility for nitrite therapy for males and females, although we note that this requires much further study.
We also observed that the degree of Cl2-induced toxicity can vary significantly depending on the exposure protocol. We utilized whole body exposure protocols, in contrast to nose-only exposure. The pros and cons of these approaches have been compared previously (6). Cl2 toxicity was significantly lower if five mice were exposed at the same time compared with two. We speculate that “huddling of mice” may have limited exposure of Cl2 to individual mice in the former setting as well as potentially introducing uncontrolled variables. For these reasons, we opted for a protocol with two animals per exposure. Moreover, variances in Cl2 toxicity responses were observed between different experiments. For example, for all data shown in this study, the time for 50% mortality in Cl2 gas-only groups varied from 3–12 h. The precise mechanisms for such variance is not clear but underscores the importance of ensuring that exposures are performed with a pairwise manner when comparing different experimental/therapeutic groups.
The purpose of this study was to test whether IM injection of nitrite may be an effective postexposure therapeutic in a lethal model of Cl2 exposure. The extent of Cl2-induced injury depends on the dose and exposure time. Although these variables can be controlled experimentally, testing of therapeutics across all potential Cl2 exposure scenarios that likely occur in an accident or military setting is not feasible. We therefore opted to use 12–24-h mortality as the end point, with the rationale being that a postexposure therapeutic that can be administered within 60 min of exposure, to model first-responder times, and which also improves short-term survival will be key in allowing victims to reach a primary care facility where targeted therapeutics can be initiated. A limitation of this study is the lack of testing of this concept and assessing mortality effects beyond 24 h. Future studies evaluating long-term outcomes after postexposure nitrite therapy are required.
GRANTS
This research was supported by the CounterACT Program, National Institutes of Health, Office of the Director, and the National Institute of Environmental Health Sciences, Grant Number 1U01ES023759 and 1R21ES024027-01
DISCLOSURES
R. Patel is a coinventor on a patent for use of nitrite salts for the treatment of cardiovascular conditions. No other conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.H., S.F.D., C.S., S.M., and R.P.P. conception and design of research; J.H., S.F.D., and J.-Y.O. performed experiments; J.H., J.-Y.O., and R.P.P. analyzed data; J.H., S.F.D., J.-Y.O., S.M., and R.P.P. edited and revised manuscript; J.H., S.F.D., J.-Y.O., C.S., S.M., and R.P.P. approved final version of manuscript; S.M. and R.P.P. interpreted results of experiments; R.P.P. prepared figures; R.P.P. drafted manuscript.
REFERENCES
- 1.Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, Yu Z, Sui A, Cheung M, Leishman E, Eidam HS, Ye G, Willette RN, Thorneloe KS, Bradshaw HB, Matalon S, Jordt SE. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 307: L158–L172, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bessac BF, Jordt SE. Sensory detection and responses to toxic gases: mechanisms, health effects, and countermeasures. Proc Am Thorac Soc 7: 269–277, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cevik Y, Onay M, Akmaz I, Sezigen S. Mass casualties from acute inhalation of chlorine gas. South Med J 102: 1209–1213, 2009. [DOI] [PubMed] [Google Scholar]
- 4.Chang W, Chen J, Schlueter CF, Rando RJ, Pathak YV, Hoyle GW. Inhibition of chlorine-induced lung injury by the type 4 phosphodiesterase inhibitor rolipram. Toxicol Appl Pharmacol 263: 251–258, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Mo Y, Schlueter CF, Hoyle GW. Inhibition of chlorine-induced pulmonary inflammation and edema by mometasone and budesonide. Toxicol Appl Pharmacol 272: 408–413, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng YS, Bowen L, Rando RJ, Postlethwait EM, Squadrito GL, Matalon S. Exposing animals to oxidant gases: Nose only vs. whole body. Proc Am Thorac Soc 7: 264–268, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet SF, Wang X, Kevil CG, Gladwin MT, Lefer DJ. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest 115: 1232–1240, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans RB. Chlorine: State of the art. Lung 183: 151–167, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Fanucchi MV, Bracher A, Doran SF, Squadrito GL, Fernandez S, Postlethwait EM, Bowen L, Matalon S. Post-exposure antioxidant treatment in rats decreases airway hyperplasia and hyperreactivity due to chlorine inhalation. Am J Respir Cell Mol Biol 46: 599–606, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gessner MA, Doran SF, Yu Z, Dunaway CW, Matalon S, Steele C. Chlorine gas exposure increases susceptibility to invasive lung fungal infection. Am J Physiol Lung Cell Mol Physiol 304: L765–L773, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Honavar J, Samal AA, Bradley KM, Brandon A, Balanay J, Squadrito GL, Mohankumar K, Maheshwari A, Postlethwait EM, Matalon S, Patel RP. Chlorine gas exposure causes systemic endothelial dysfunction by inhibiting eNOS-dependent signaling. Am J Respir Cell Mol Biol 45: 419–425, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoyle GW. Mitigation of chlorine lung injury by increasing cyclic AMP levels. Proc Am Thorac Soc 7: 284–289, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain AK, Tewari-Singh N, Inturi S, Orlicky DJ, White CW, Agarwal R. Myeloperoxidase deficiency attenuates nitrogen mustard-induced skin injuries. Toxicology 320: 25–33, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kevil CG, Kolluru GK, Pattillo CB, Giordano T. Inorganic nitrite therapy: historical perspective and future directions. Free Radic Biol Med 51: 576–593, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koohsari H, Tamaoka M, Campbell HR, Martin JG. The role of gamma delta T cells in airway epithelial injury and bronchial responsiveness after chlorine gas exposure in mice. Respir Res 8: 21, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leustik M, Doran S, Bracher A, Williams S, Squadrito GL, Schoeb TR, Postlethwait E, Matalon S. Mitigation of chlorine-induced lung injury by low-molecular-weight antioxidants. Am J Physiol Lung Cell Mol Physiol 295: L733–L743, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li C, Weng Z, Doran SF, Srivastava RK, Afaq F, Matalon S, Athar M. Chlorine induces the unfolded protein response in murine lungs and skin. Am J Respir Cell Mol Biol 49: 197–203, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lundberg JO, Gladwin MT, Ahluwalia A, Benjamin N, Bryan NS, Butler A, Cabrales P, Fago A, Feelisch M, Ford PC, Freeman BA, Frenneaux M, Friedman J, Kelm M, Kevil CG, Kim-Shapiro DB, Kozlov AV, Lancaster JR, Jr, Lefer DJ, McColl K, McCurry K, Patel RP, Petersson J, Rassaf T, Reutov VP, Richter-Addo GB, Schechter A, Shiva S, Tsuchiya K, van Faassen EE, Webb AJ, Zuckerbraun BS, Zweier JL, Weitzberg E. Nitrate and nitrite in biology, nutrition and therapeutics. Nat Chem Biol 5: 865–869, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov 7: 156–167, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Martin JG, Campbell HR, Iijima H, Gautrin D, Malo JL, Eidelman DH, Hamid Q, Maghni K. Chlorine-induced injury to the airways in mice. Am J Respir Crit Care Med 168: 568–574, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Matalon S, Maull EA. Understanding and treating chlorine-induced lung injury. Proc Am Thorac Soc 7: 253, 2010. [DOI] [PubMed] [Google Scholar]
- 22.McGovern T, Day BJ, White CW, Powell WS, Martin JG. AEOL10150: a novel therapeutic for rescue treatment after toxic gas lung injury. Free Radic Biol Med 50: 602–608, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGovern TK, Powell WS, Day BJ, White CW, Govindaraju K, Karmouty-Quintana H, Lavoie N, Tan JJ, Martin JG. Dimethylthiourea protects against chlorine induced changes in airway function in a murine model of irritant induced asthma. Respir Res 11: 138, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mo Y, Chen J, Schlueter CF, Hoyle GW. Differential susceptibility of inbred mouse strains to chlorine-induced airway fibrosis. Am J Physiol Lung Cell Mol Physiol 304: L92–L102, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Musah S, Chen J, Hoyle GW. Repair of tracheal epithelium by basal cells after chlorine-induced injury. Respir Res 13: 107, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Koren EG, Hogan BL, Gunn MD. Loss of basal cells precedes bronchiolitis obliterans-like pathological changes in a murine model of chlorine gas inhalation. Am J Respir Cell Mol Biol 49: 788–797, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pickerodt PA, Emery MJ, Zarndt R, Martin W, Francis RC, Boemke W, Swenson ER. Sodium nitrite mitigates ventilator-induced lung injury in rats. Anesthesiology 117: 592–601, 2012. [DOI] [PubMed] [Google Scholar]
- 28.Samal AA, Honavar J, Brandon A, Bradley KM, Doran S, Liu Y, Dunaway C, Steele C, Postlethwait EM, Squadrito GL, Fanucchi MV, Matalon S, Patel RP. Administration of nitrite after chlorine gas exposure prevents lung injury: effect of administration modality. Free Radic Biol Med 53: 1431–1439, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song W, Wei S, Liu G, Yu Z, Estell K, Yadav AK, Schwiebert LM, Matalon S. Postexposure administration of a β2-agonist decreases chlorine-induced airway hyperreactivity in mice. Am J Respir Cell Mol Biol 45: 88–94, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuck SA, Ramos-Barbon D, Campbell H, McGovern T, Karmouty-Quintana H, Martin JG. Time course of airway remodelling after an acute chlorine gas exposure in mice. Respir Res 9: 61, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Sickle D, Wenck MA, Belflower A, Drociuk D, Ferdinands J, Holguin F, Svendsen E, Bretous L, Jankelevich S, Gibson JJ, Garbe P, Moolenaar RL. Acute health effects after exposure to chlorine gas released after a train derailment. Am J Emerg Med 27: 1–7, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Velmurugan S, Kapil V, Ghosh SM, Davies S, McKnight A, Aboud Z, Khambata RS, Webb AJ, Poole A, Ahluwalia A. Antiplatelet effects of dietary nitrate in healthy volunteers: involvement of cGMP and influence of sex. Free Radic Biol Med 65: 1521–1532, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vitturi DA, Patel RP. Current perspectives and challenges in understanding the role of nitrite as an integral player in nitric oxide biology and therapy. Free Radic Biol Med 51: 805–812, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wenck MA, Van Sickle D, Drociuk D, Belflower A, Youngblood C, Whisnant MD, Taylor R, Rudnick V, Gibson JJ. Rapid assessment of exposure to chlorine released from a train derailment and resulting health impact. Public Health Rep 122: 784–792, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wozniak KL, Kolls JK, Wormley FL., Jr Depletion of neutrophils in a protective model of pulmonary cryptococcosis results in increased IL-17A production by gammadelta T cells. BMC Immunol 13: 65, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yadav AK, Doran SF, Samal AA, Sharma R, Vedagiri K, Postlethwait EM, Squadrito GL, Fanucchi MV, Roberts LJ, 2nd, Patel RP, Matalon S. Mitigation of chlorine gas lung injury in rats by postexposure administration of sodium nitrite. Am J Physiol Lung Cell Mol Physiol 300: L362–L369, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zarogiannis SG, Jurkuvenaite A, Fernandez S, Doran SF, Yadav AK, Squadrito GL, Postlethwait EM, Bowen L, Matalon S. Ascorbate and deferoxamine administration after chlorine exposure decrease mortality and lung injury in mice. Am J Respir Cell Mol Biol 45: 386–392, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]