

Abstract
Our recent publication showed that pharmacological blockade of arginases confers kidney protection in diabetic nephropathy via a nitric oxide (NO) synthase (NOS)3-dependent mechanism. Arginase competes with endothelial NOS (eNOS) for the common substrate l-arginine. Lack of l-arginine results in reduced NO production and eNOS uncoupling, which lead to endothelial dysfunction. Therefore, we hypothesized that l-arginine or l-citrulline supplementation would ameliorate diabetic nephropathy. DBA mice injected with multiple low doses of vehicle or streptozotocin (50 mg/kg ip for 5 days) were provided drinking water with or without l-arginine (1.5%, 6.05 g·kg−1·day−1) or l-citrulline (1.66%, 5.73 g·kg−1·day−1) for 9 wk. Nonsupplemented diabetic mice showed significant increases in albuminuria, blood urea nitrogen, glomerular histopathological changes, kidney macrophage recruitment, kidney TNF-α and fibronectin mRNA expression, kidney arginase activity, kidney arginase-2 protein expression, and urinary oxidative stress along with a significant reduction of nephrin and eNOS protein expression and kidney nitrite + nitrate compared with normal mice after 9 wk of diabetes. Surprisingly, l-arginine or l-citrulline supplementation in diabetic mice did not affect any of these parameters despite greatly increasing kidney and plasma arginine levels. These findings demonstrate that chronic l-arginine or l-citrulline supplementation does not prevent or reduce renal injury in a model of type 1 diabetes.
Keywords: l-arginine, l-citrulline, diabetic nephropathy, oral supplementation
each year, >100,000 Americans are diagnosed with kidney failure (30), with over 25% of Medicare spending ($42 billion) going to treat kidney disease. As of 2012, although nearly 30 million Americans (13% of the population) had the diagnosis of chronic kidney disease, most people were unaware (undiagnosed) that they had it (30). The most common cause of kidney failure is diabetes, which accounts for nearly 44% of new cases each year (30). Early alterations in diabetic kidneys include the development of glomerular hyperfiltration and hypertrophy followed by thickening of the glomerular basement membrane, mesangial matrix accumulation, increased urinary albumin excretion rate, and, ultimately, the progression to glomerular sclerosis and end-stage renal failure. Current therapies, including blood pressure and glucose control and other lifestyle changes, have been only modestly successful in delaying the progression of renal failure. Thus, it is important to identify pharmacotherapeutics that will prevent or slow down the development and progression of diabetic kidney disease.
Alterations in endothelial function are a common underlying event for vascular abnormalities observed in patients with diabetes. Endothelial dysfunction, characterized by reduced bioavailability of nitric oxide (NO) and increased oxidative stress, is a hallmark characteristic in diabetes (13) and diabetic nephropathy (DN) (15). NO is produced from l-arginine (l-Arg) by NO synthases (NOSs). Under conditions of low arginine levels or hyperglycemia, endothelial NOS (eNOS) is uncoupled, producing ROS and oxidative stress in lieu of NO (8, 34). Recently, low or lack of eNOS has been shown to exacerbate DN (32, 39). Therefore, elucidating the basis for vascular dysfunction in DN is critical (4).
Arginine is also the substrate of arginase, which produces ornithine (Orn) and urea. Thus, both NOS and arginase compete for l-arginine, raising the possibility that arginase may play a role in NO deficiency in diabetes. In fact, our recent publication (23) showed that pharmacological blockade or genetic deficiency of arginase-2 confers kidney protection in diabetic mouse models. We further showed that arginase inhibition mediates renal tissue protection in DN via an eNOS-dependent mechanism (37). These findings provided the rationale for the hypothesis that increasing arginine bioavailability may prevent or reduce the progression of DN.
Although oral supplementation with l-Arg can increase plasma l-Arg levels, oral supplementation with l-citrulline (l-Cit), a precursor for arginine biosynthesis, has been shown to be more efficient than oral l-Arg in increasing plasma l-Arg concentration (25), due to splanchnic catabolism of ingested l-Arg (10, 33). Some, but not all, reports have indicated that oral l-Arg or l-Cit supplementation may enhance NO-mediated cardiovascular function (3, 6). Moreover, several published reports have shown that increasing arginine availability in cultured cell models or by supplementation in vivo was able to overcome the effects of arginase and to enhance NO synthesis (5, 19, 29), suggesting that l-Arg or l-Cit supplementation may be beneficial in the case of DN. However, the efficacy of l-Arg or l-Cit supplementation in the progression of DN is not completely clear.
In this report, we show that oral supplementation with l-Arg or l-Cit did not prevent or reduce diabetic renal injury despite greatly increasing kidney and plasma arginine levels.
MATERIALS AND METHODS
Animal model.
As recommended by the NIH Animal Models of Diabetes Complication Consortium (AMDCC) (7, 9), 6-wk-old male DBA/2J mice (Jackson Laboratory, Bar Harbor, MN) were injected by streptozocin (STZ; Sigma, St. Louis, MO) via intraperiteneal injection at a dose of 50 mg/kg body wt for 5 consecutive days to induce diabetes. After 9 wk of diabetes, all mice were euthanized. Mouse plasma and 24-h urine were collected, and kidneys were removed for further study. All animal experiments were approved by the Institutional Animal Care and Use Committee of Pennsylvania State University College of Medicine.
Amino acid administration.
l-Arg (1.5%, pH 6.93, Sigma) or l-Cit (1.66%, pH 7.28, Sigma) in drinking water was used for oral supplementation for 9 wk. All mice had free access to drinking water.
Systolic blood pressure measurements.
The CODA blood pressure system (Kent Scientific, Torrington, CT) was used to measure systolic blood pressure as previously described (23, 37). Mice were allowed to rest quietly for 15 min at room temperature before blood pressure measurements. All measurements were performed at the same time for all groups to prevent any diurnal variations.
Histology and immunohistochemistry.
Mouse kidney tissues were fixed in 10% formalin and embedded in paraffin. Three-micrometer sections were cut. Periodic acid-Schiff (PAS) staining was performed in kidney sections as previously described (1, 23, 37). All glomeruli were examined at ×40 in a blinded manner. Images were taken with an Olympus BX51 microscope and DP71 digital camera using Microsuite Basic 2.6 image software. Images were obtained with a ×100 (oil) objective with a total magnification of ×1,000. Semiquantitative scores (0–4+) were assigned based on the masked readings. Mesangial matrix expansion or sclerosis was scored as previously described (23, 37). Immunohistochemistry was performed on paraffin-embedded sections with anti-mouse Mac-2 antibody (clone M3/38, Cedarlane, Burlington, NC) as previously described (23, 37).
Analytic methodology.
As previously described (23, 37), the urine albumin concentration was measured by ELISA using an Albuwell M kit (Exocell, Philadelphia, PA), urine creatinine was determined using a Creatinine Liquid Reagents Assay kit (Diazyme Laboratories, Poway, CA), blood urea nitrogen was determined using Vitros DT6011 chemistry slides (Ortho-Clinical Diagnostics, Rochester, NY), and body composition was measured using LF90 Minispec Time Domain Nuclear Magnetic Resonance Spectrometer (Burker Optics, Billerica, MA). Urinary thiobarbituric acid-reactive substance (TBARS) levels, which are recommended by the AMDCC (http://www.amdcc.org/shared/showFile.aspx?doctypeid=3&docid=33) as an index of oxidative stress in DN, were determined as previously described (20).
Kidney amino acid assay.
Mouse kidneys were homogenized in 300 μl of 3 M perchloric acid (Fisher Scientific, Fair Lawn, NJ) per 100 mg of tissue. After centrifugation at 4°C, supernatants were used for the amino acid assay (37).
Plasma amino acid assay.
Frozen plasma samples were thawed and vortexed, and 5 μl internal standard was spiked into 10 μl of each plasma sample. Subsequently, 30 μl methanol was added followed by centrifugation at 9,000 rpm for 10 min to precipitate proteins. The supernatant was further cleaned up by solid phase extraction using a 1-ml MCX cartridge as follows: the cartridge was preconditioned with methanol and water, and, after being loaded with the supernatant, the cartridge was washed with 2% formic acid and methanol and then eluted with 5% NH4OH in 50% methanol. The purified sample was evaporated to dryness and then reconstituted in 25 μl of 5 mM heptafluorobutyric acid. Reconstituted samples were then analyzed by a liquid chromotography-mass spectroscopy system (Waters, Milford, MA) in the MS core facility at Penn State College of Medicine.
Quantitative RT-PCR.
Total RNA was isolated using Tri reagent (Molecular Research Center, Cincinnati, OH) per the manufacturer's instructions. Single-strand cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) for two-step RT-PCR. Quantitative PCR was performed in an Eppendorf Mastercycler pro system using a Taqman gene expression assay (fibronectin: Mm01256744_m1 and TNF-α: Mm00443260_g1). GAPDH mRNA (GAPDH: Mm99999915_g1) was used as an internal reference. Data were analyzed using the 2−ΔΔCT equation, where CT is threshold cycle (1, 23, 36).
Western blot analysis.
Kidney tissues were homogenized in lysis solution (0.1% Triton X-100) supplemented with protease inhibitor cocktail tablets (Roche). A BCA protein assay (Thermo Fisher Scientific) was used to determine protein concentration. Fifteen micrograms of kidney lysates were separated on a NuPAGE 4∼12% Bis-Tris gel (Invitrogen), and separated proteins were transferred onto a polyvinylidene difluoride membrane (Invitrogen). All gels were run with a set of molecular weight markers (SeeBlue Plus2 Pre-Stained Standard, catalog no. LC5925, Life Technologies). Membranes were cut into half to allow independent probing for large proteins [nephrin, eNOS, inducible NOS (iNOS), or neuronal NOS (nNOS)] in the upper portion of the membrane and smaller proteins (arginase-2, β-actin, or GAPDH) in the lower portion of the membrane. After being blocked using 5% nonfat dry milk, membranes were incubated with primary antibody overnight at 4°C followed by incubation with appropriate secondary antibody for 1 h at room temperature. Membranes were probed with primary antibodies to arginase-2 (1:500, catalog no. sc-20151, Santa Cruz Biotechnology), eNOS (1:1,000, catalog no. ab76198, Abcam), iNOS (1:500, catalog no. ab3523, Abcam), nNOS-β (1:500, catalog no. sc-648, Santa Cruz Biotechnology), nephrin (1:500, catalog no. 20R-NP002, Fitzgerald Industries), β-actin (1:5,000, catalog no. A5441, Sigma-Aldrich), or GAPDH (1:1,000, catalog no. 5174S, Cell Signaling Technology). The secondary antibody used for the detection of arginase-2, iNOS, nNOS-β, and GAPDH was anti-rabbit horseradish peroxidase (HRP)-conjugated IgG (1:3,000, catalog no. 7074S, Cell Signaling Technology); for eNOS and β-actin, the secondary antibody was anti-mouse HRP-conjugated IgG (1:3,000, catalog no. 7076S, Cell Signaling Technology); and for nephrin, the secondary antibody was anti-guinea pig HRP-conjugated IgG (1:2,000, catalog no. 43R-1096, Fitzgerald Industries). In some cases, membranes were stripped and reprobed to allow the quantification of multiple proteins from the same blot. Membranes were developed using enhanced chemiluminescence solutions (Thermo Fisher Scientific) followed by exposure to X-ray film. Densitometry was performed using ImageJ (National Institutes of Health, http://rsbweb.nih.gov/ij/index.html) as previously described (23, 37).
Kidney arginase activity assay.
Kidney lysates were prepared in lysis solution (0.1% Triton X-100) supplemented with protease inhibitors. Arginase activity was measured as previously described (23, 37).
Kidney nitrate + nitrite assay.
Kidneys were homogenized in PBS (pH 7.4). After centrifugation at 4°C, supernatants were transferred to a clean tube and used for protein assays to measure protein concentration and nitrate + nitrite assays using a Nitrate/Nitrite Colorimetric Assay Kit (catalog no. 780001, Cayman Chemical, Ann Arbor, MI) according to the manufacturer's instructions.
Statistical analysis.
Comparisons between groups were examined using SPSS (version 19.0) software for Windows (SPSS, Chicago, IL). Data are expressed as means ± SE. One-way ANOVA was used when more than two groups were compared, and the significance of observed differences among the groups was evaluated with a least-significant-difference post hoc test. Statistical significance was identified at P < 0.05.
RESULTS
Effect of oral l-Arg or l-Cit supplementation on plasma and tissue levels of l-Arg and arginine metabolites in diabetic mice.
To assess the possible role of oral l-Arg or l-Cit supplementation on the pathogenesis of DN, male DBA/2J mice were used to generate type 1 diabetes. Diabetic mice were provided drinking water with or without l-Arg or l-Cit supplementation for 9 wk. First, the impact of oral l-Arg or l-Cit supplementation on levels of l-Arg and its metabolites was evaluated by measuring amino acid levels in the plasma and kidney after 9 wk of diabetes. Plasma l-Arg levels were significantly reduced in nonsupplemented diabetic mice compared with control nondiabetic mice (Fig. 1A). In contrast, plasma l-Cit and Orn levels significantly increased compared with normal mice (Fig. 1, B and C). Oral l-Arg supplementation greatly increased plasma l-Arg and Orn levels (Fig. 1, A and C) but had no effect on plasma l-Cit levels (Fig. 1B) compared with nonsupplemented diabetic mice. Oral l-Cit supplementation not only increased plasma l-Cit levels (Fig. 1B) but also plasma l-Arg (Fig. 1A) and Orn (Fig. 1C) levels in diabetic mice compared with nonsupplemented diabetic mice. As l-Arg and Orn compete for cellular uptake via the y+ transport system (12), the plasma l-Arg-to-Orn ratio provides an approximation of the relative efficiency of l-Arg uptake compared with that of l-Orn. The value of plasma l-Arg/Orn plummeted in diabetic mice but was modestly increased by supplementation with l-Arg and strongly enhanced by supplementation with l-Cit, to ∼60% of the value in nondiabetic mice (Fig. 1D).
Fig. 1.
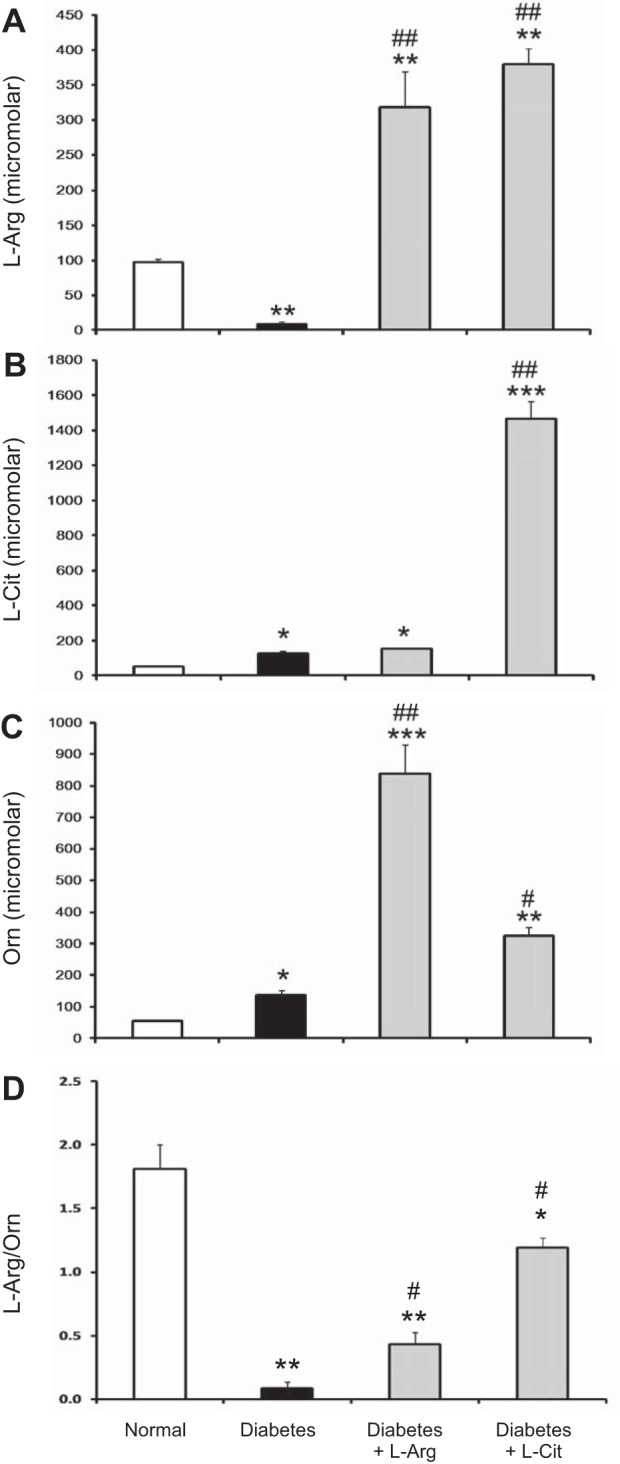
Effect of oral l-arginine (l-Arg) or l-citrulline (l-Cit) supplementation on plasma levels of l-Arg and arginine metabolites in diabetic mice. Streptozocin (STZ)-induced diabetic mice were fed with l-Arg or l-Cit as indicated in drinking water for 9 wk. Drinking water was used in the control (normal) group. Plasma l-Arg (A), l-Cit (B), ornithine (Orn; C), and l-Arg-to-Orn ratio (D) assays were performed using a liquid chromatography-mass spectrometry system. Results are presented as means ± SE. *P < 0.05, **P < 0.01, and ***P < 0.0001 compared with normal mice; #P < 0.01 and ##P < 0.001 compared with nonsupplemented diabetic mice.
In the kidney, levels of l-Arg, l-Cit, and Orn were not significantly altered in diabetic mice (Fig. 2, A–C). l-Arg and l-Cit supplementation were equally effective in greatly increasing l-Arg levels in the kidney above levels in both normal and nonsupplemented diabetic mice (Fig. 2A). Both l-Arg and l-Cit supplementation increased l-Orn levels in the kidney, but the increase was much less with l-Cit supplementation (Fig. 2C). Whereas l-Arg supplementation had no significant effect on kidney l-Cit levels, l-Cit supplementation resulted in large increases in kidney l-Cit levels (Fig. 2B).
Fig. 2.
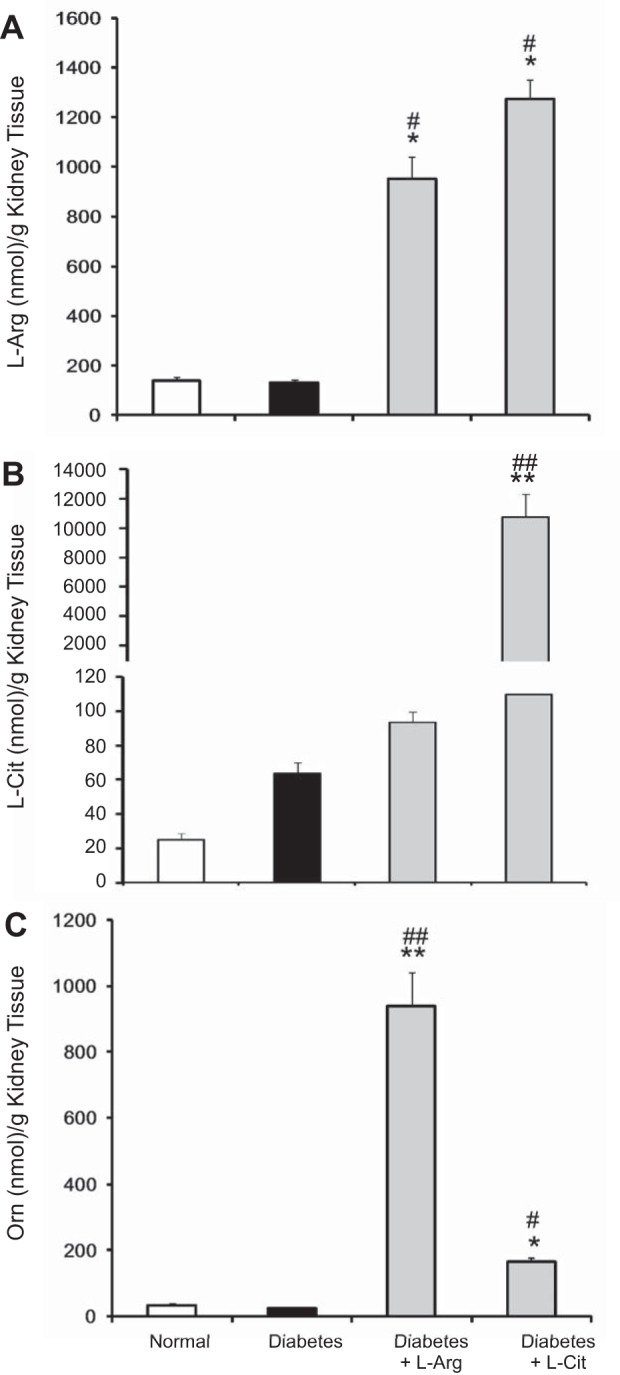
Effect of oral l-Arg or l-Cit supplementation on kidney levels of l-Arg and arginine metabolites in diabetic mice. Kidney l-Arg (A), l-Cit (B), and Orn (C) assays were performed in tissue lysates using a liquid chromatography-mass spectrometry system. Results are presented as means ± SE. *P < 0.01 and **P < 0.001 compared with normal mice; #P < 0.01 and ##P < 0.001 compared with nonsupplemented diabetic mice.
l-Arg or l-Cit supplementation did not affect characteristics of DN.
As shown in Table 1, nonsupplemented diabetic mice had increased blood glucose levels, increased water consumption, decreased body weight, decreased body fluid composition, and an increased kidney weight-to-body weight ratio compared with normal mice. l-Arg or l-Cit supplementation did not affect any of these parameters despite highly elevated l-Arg levels in the plasma and kidney (Figs. 1 and 2). Importantly, there were no changes in blood pressure between all groups.
Table 1.
Characteristics of mice in the diabetes study
| Normal | Diabetes | Diabetes + l-Arginine | Diabetes + l-Citrulline | |
|---|---|---|---|---|
| Number of mice/group | 8 | 8 | 7 | 6 |
| Body weight, g | 29 ± 1.05 | 18 ± 0.61‡ | 18 ± 0.42‡ | 15 ± 0.33‡ |
| Blood glucose, mg/dl | 149 ± 5.37 | 499 ± 0.98* | 495 ± 2.38* | 500 ± 0.5* |
| Water consumed, ml/day | 2.29 ± 0.37 | 6.00 ± 0.91* | 7.26 ± 0.48* | 5.18 ± 1.19* |
| Average amino acid consumed in drinking water, g·kg−1·day−1 | 0 | 0 | 6.05 | 5.73 |
| Average nitrogen consumed in drinking water, mmol·kg−1·day−1 | 0 | 0 | 138.9 | 98.1 |
| Systolic blood pressure, mmHg | 122 ± 2.44 | 112 ± 4.03 | 111 ± 4.09 | 109 ± 8.25 |
| Percent fluid | 7.5 ± 0.13 | 6.6 ± 0.11* | 5.5 ± 0.17* | 5.8 ± 0.09* |
| Kidney weight/body weight, mg/g | 0.92 ± 0.05 | 1.33 ± 0.02* | 1.54 ± 0.03* | 1.53 ± 0.05* |
| Plasma creatinine, mg/dl | 0.26 ± 0.08 | 0.62 ± 0.11* | 0.48 ± 0.04* | 0.94 ± 0.16* |
Data are means ± SE. Values were measured 9 wk after streptozotocin treatment.
P < 0.05 compared with the normal group;
P < 0.0001 compared with the normal group.
l-Arg or l-Cit supplementation did not prevent diabetic renal injury.
To determine whether l-Arg or l-Cit oral supplementation had any effect on diabetic kidney function, urine samples were collected to measure 24-h urinary albumin excretion rates and albumin-to-creatinine ratios as indicators of renal injury after 9 wk of diabetes. Nonsupplemented diabetic mice exhibited an increased urinary albumin excretion rate (Fig. 3A), albumin-to-creatinine ratio (Fig. 3B), and plasma creatinine (Table 1) compared with normal mice. l-Arg or l-Cit supplementation did not prevent diabetic renal injury as indicated by the lack of changes in any of these parameters and were similar to nonsupplemented diabetic mice.
Fig. 3.
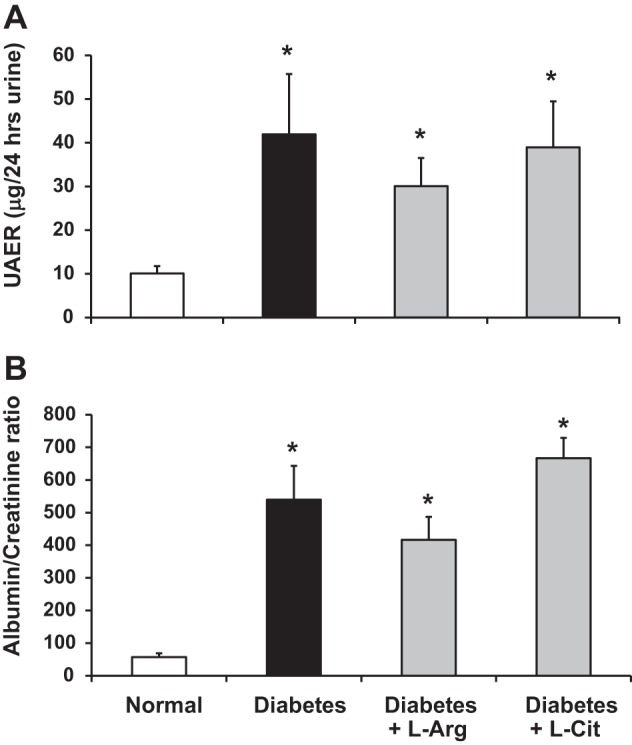
l-Arg or l-Cit supplementation did not attenuate diabetic kidney injury. Twenty-four-hour urine was collected for measurements of the urinary albumin excretion rate (UAER; A) and albumin-to-creatinine ratio (B). Data are presented as means ± SE. *P < 0.05 compared with the normal group.
l-Arg or l-Cit supplementation did not improve kidney histological changes in diabetic mice.
PAS staining of kidney sections showed increased glomerular cellularity and mesangial expansion in nonsupplemented diabetic mice (score: 0.98 ± 0.06, P < 0.01) compared with normal mice (score: 0.33 ± 0.06; Fig. 4). However, l-Arg or l-Cit supplementation did not reduce glomerular cellularity or mesangial expansion, as indicated by the absence of significant differences in PAS scores (score: 0.81 ± 0.06 and 0.70 ± 0.07) and were similar to nonsupplemented diabetic mice.
Fig. 4.
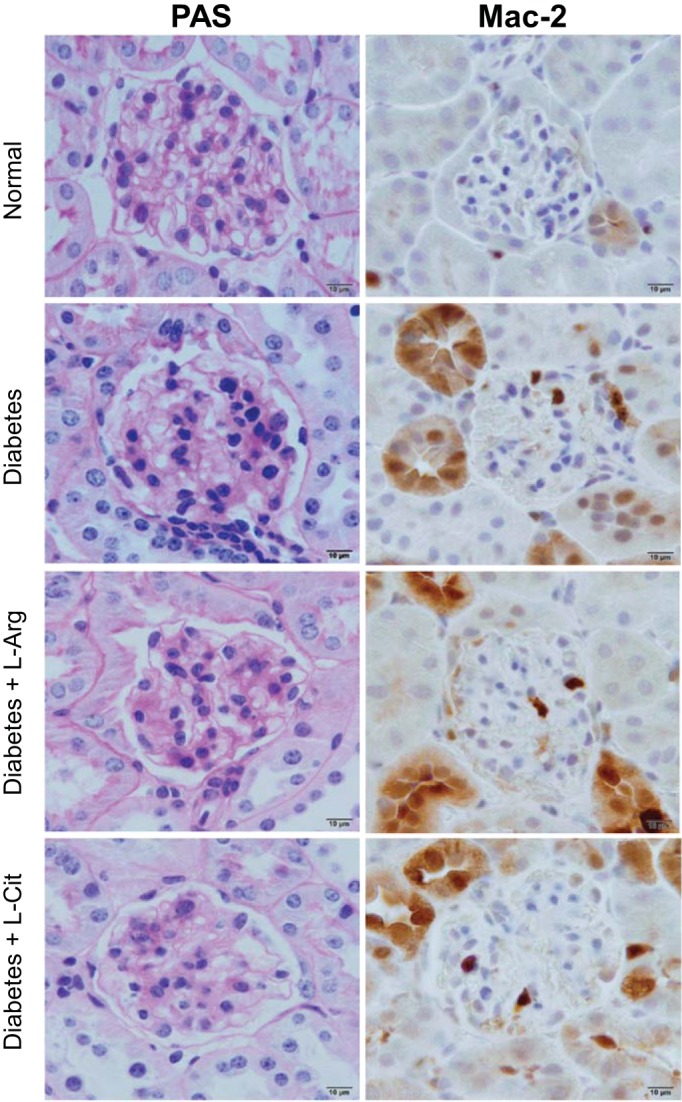
l-Arg or l-Cit supplementation did not improve renal histological changes or glomerular macrophage infiltration in diabetic mice. After 9 wk of diabetes with l-Arg or l-Cit supplementation as indicated, mice were euthanized and mouse kidneys were removed. Left: paraffin-embedded mouse kidney sections were performed with periodic acid-Schiff (PAS) staining, and images were taken with a ×100 (oil) objective with a total magnification of ×1,000. Semiquantitative scores (0∼4+) were assigned based on the masked readings, where 0 = no lesion and 1, 2, 3, and 4+ = mesangial matrix expansion or sclerosis involving ≤25%, 25∼50%, 50∼75%, or >75% of the glomerular tuft area, respectively. Right: glomerular macrophage recruitment was visualized by immunohistochemical staining using Mac-2 antibody at week 9 after STZ-induced diabetes.
l-Arg or l-Cit supplementation did not reduce increased glomerular macrophage infiltration in diabetic mice.
We (23) have previously shown that arginase is critical for macrophage infiltration in DN. Therefore, we tested whether supplementation with l-Arg or l-Cit would reduce kidney macrophage recruitment. Using immunohistochemistry (Mac-2-positive macrophages; Fig. 4), glomerular macrophage infiltration was significantly increased in nonsupplemented diabetic mice (0.73 ± 0.06 macrophages/glomerulus, P < 0.05) compared with normal mice (0.28 ± 0.04 macrophages/glomerulus). l-Arg or l-Cit supplementation did not significantly reduce glomerular macrophage recruitment (0.52 ± 0.04 macrophages/glomerulus and 0.84 ± 0.10 macrophages/glomerulus) compared with nonsupplemented diabetic mice.
l-Arg or l-Cit supplementation did not prevent increases in renal TNF-α or fibronectin expression in diabetic mice.
TNF-α plays an important role in inflammatory diseases and DN (2, 18, 37). Our data showed that renal TNF-α mRNA levels were significantly increased in diabetic mice (Fig. 5A). Both l-Arg and l-Cit supplementation failed to reduce renal TNF-α mRNA levels compared with nonsupplemented diabetic mice. Expression of fibronectin, a biomarker of tissue fibrosis, was also used to determine the role of l-Arg or l-Cit in DN. Similar to results with TNF-α mRNA, renal fibronectin mRNA levels significantly increased in nonsupplemented diabetic mice, and l-Arg or l-Cit supplementation also failed to reduce fibronectin mRNA levels in the diabetic mouse kidney (Fig. 5B).
Fig. 5.
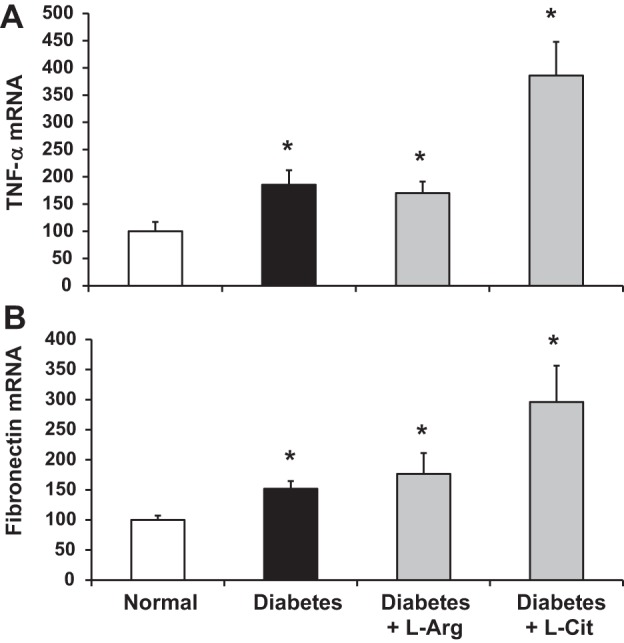
l-Arg or l-Cit supplementation did not reduce kidney TNF-α or fibronectin mRNA expression in diabetic mice. Quantitative RT-PCR was performed to detect TNF-α (A) or fibronectin (B) mRNA expression in mouse kidneys after 9 wk of STZ-induced diabetes. Levels of mRNA were normalized to GAPDH mRNA and expressed relative to average values for normal mice (arbitrarily set to 100). Data are presented as means ± SE. *P < 0.01 compared with the normal group.
l-Arg or l-Cit supplementation did not prevent increases in renal arginase activity or arginase-2 protein expression in diabetic mice.
Our previous data showed that renal arginase activity and arginase-2 protein expression are increased in diabetic mouse kidneys compared with normal mouse kidneys (37). In the present study, we determined whether l-Arg or l-Cit supplementation would affect arginase activity and/or arginase-2 protein expression in the diabetic kidney. Consistent with our previous study, renal arginase activity (Fig. 6A) and arginase-2 protein expression (Fig. 6B) in nonsupplemented diabetic mice were significantly elevated compared with normal mice. Interestingly, l-Arg or l-Cit supplementation had no significant effect on kidney arginase activity or arginase-2 protein expression compared with nonsupplemented diabetic mice.
Fig. 6.

l-Arg or l-Cit supplementation did not affect kidney arginase activity or arginase-2 protein expression in diabetic mice. Mouse kidney tissue lysates were prepared after 9 wk of STZ-induced diabetes with or without l-Arg or l-Cit supplementation as indicated. Arginase activity (A) was measured as described in materials and methods. Western blot analysis was performed to detect arginase-2 protein. The membrane was then stripped and reprobed to measure β-actin as a loading control. Quantification was performed by densitometry followed by normalization to β-actin (B). Data are presented as means ± SE; n = 4–8 mice/group. *P < 0.05 compared with the normal group. A representative Western blot is shown in this figure and in subsequent figures.
l-Arg or l-Cit supplementation did not prevent reductions in nephrin protein expression in diabetic mice.
Nephrin is a component protein in the slit diaphragm, which plays a pivotal role in maintaining kidney function, and decreased nephrin expression is associated with albuminuria. As shown in Fig. 7, nephrin protein expression was significantly reduced in nonsupplemented diabetic mice. l-Arg or l-Cit supplementation failed to prevent the reduction in nephrin protein expression.
Fig. 7.

l-Arg or l-Cit supplementation did not restore renal nephrin protein expression in diabetic mice. Kidney nephrin expression was detected using Western blot analysis at week 9 after STZ-induced diabetes. β-Actin was used as a loading control. Quantification was performed by densitometry followed by normalization to β-actin. Data are presented as means ± SE; n = 4–6 mice/group. *P < 0.05 compared with the normal group.
l-Arg or l-Cit supplementation did not prevent reductions in renal eNOS protein expression or NO production in diabetic mice.
Depletion or decreased expression of eNOS has been associated with exacerbation of diabetic kidney injury (37). It is also important to note that renal eNOS expression was elevated twofold above control values in diabetic mice treated with the arginase inhibitor S-(2-boronoethyl)-l-cysteine (37). Thus, the effect of increased renal arginase 2 in diabetes was not due merely to reducing substrate levels for eNOS but to reducing eNOS expression. As expected from previous studies, eNOS protein expression was significantly decreased in the nonsupplemented diabetic mouse kidney compared with the normal mouse kidney (Fig. 8). However, unlike the results obtained with arginase inhibition, l-Arg or l-Cit supplementation did not prevent the decrease in renal eNOS protein expression.
Fig. 8.

l-Arg or l-Cit supplementation did not restore renal endothelial nitric oxide synthase (eNOS) protein expression in diabetic mice. Kidney eNOS protein expression in tissue lysates was detected by Western blot analysis after 9 wk of STZ-induced diabetes. β-Actin was used as a loading control protein. Quantification was performed by densitometry followed by normalization to β-actin. Data are presented as means ± SE; n = 4–5 mice/group. *P < 0.05 compared with the normal group.
We also evaluated the impact of supplementation on renal expression of iNOS and nNOS (Fig. 9). Our data show that diabetes resulted in increased renal levels of iNOS, and the elevated levels were unaffected by supplementation with either l-Arg or l-Cit (Fig. 9A). Since nNOS has several splice variants expressed in the kidney, we used a COOH-terminal-specific antibody that can detect both nNOS-α and nNOS-β splice variants. The brain tissue lysate was used as a positive control for nNOS-α. While we could not detect nNOS-α in the kidney, expression of nNOS-β was not significantly different between all groups (Fig. 9B).
Fig. 9.

l-Arg or l-Cit supplementation did not restore renal inducible nitric oxide synthase (iNOS) or neuronal nitric oxide synthase (nNOS)-β protein expression in diabetic mice. Kidney iNOS (A) and nNOS-β (B) protein expression in tissue lysates was detected by Western blot analysis after 9 wk of STZ-induced diabetes. β-Actin or GAPDH was used as a loading control protein, and brain tissue lysate was used as a positive control for nNOS-α in B. Quantification was performed by densitometry followed by normalization to β-actin or GAPDH. Data are presented as means ± SE; n = 4–5 mice/group. *P < 0.05 compared with the normal group.
Reduced NO bioavailability plays an important role in diabetic renal injury (13, 15). To elucidate the role of l-Arg or l-Cit supplementation on NO bioavailability, kidney nitrate + nitrite was assayed as a measure of NO status. As shown in Fig. 10, the renal nitrate + nitrite concentration was significantly decreased in nonsupplemented diabetic mice. Surprisingly, oral supplementation of l-Arg or l-Cit failed to restore renal NO production in diabetic mice, as indicated by similar kidney nitrate + nitrite concentrations in supplemented and nonsupplemented diabetic mice.
Fig. 10.
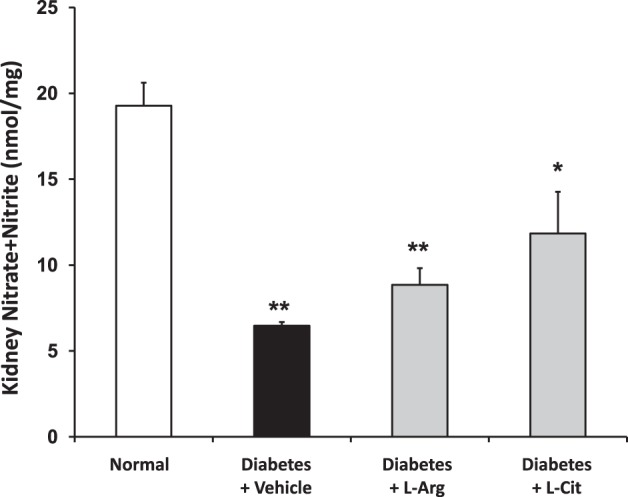
l-Arg or l-Cit supplementation did not restore renal nitrate + nitrite concentration in diabetic mice. Mouse kidney tissue lysates were used to measure the nitrate + nitrite concentration after 9 wk of diabetes. Data are presented as means ± SE. *P < 0.05 and **P < 0.001 compared with the normal group.
l-Arg or l-Cit supplementation did not prevent increases in an indicator of oxidative stress in diabetic mice.
Urinary TBARS was measured as an indicator of oxidative stress. As previously reported (37), levels of urinary TBARS in diabetic mice were more than double the levels in normal mice (Fig. 11). Surprisingly, these levels were not reduced by l-Arg or l-Cit supplementation but were further enhanced.
Fig. 11.
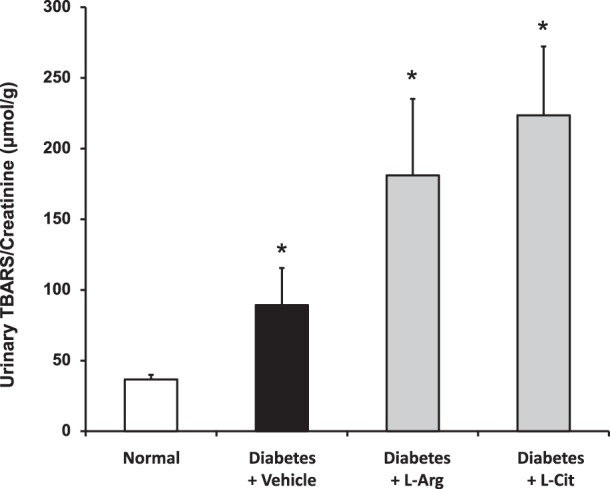
l-Arg or l-Cit supplementation did not restore urinary thiobarbituric acid-reactive substances (TBARS) in diabetic mice. Urine was collected for measurements of TBARS levels. Results are means ± SE. *P < 0.001 compared with the normal group.
DISCUSSION
Because NO plays a critical role in renal blood flow regulation, glomerular filtration rate, and renin and angiotensin generation and reabsorption (11), decreased NO bioavailability is considered to be an important factor in DN (15). Consistent with this view, decreased eNOS expression (32) or genetic depletion of eNOS (39) has been reported to exacerbate DN. Moreover, we have demonstrated that pharmacological blockade or genetic deficiency of arginase-2 confers kidney protection in diabetic mouse models (23) and that this effect is eNOS dependent (37). Since reduced l-Arg bioavailability has been associated with eNOS dysfunction, l-Arg or l-Cit supplementation are potential modalities to restore l-Arg levels and thus improve endothelial dysfunction. For example, in a rat model of chronic renal failure, l-Arg supplementation was beneficial, as evidenced by decreased blood pressure and decreased urinary protein excretion (35). However, the efficacy of l-Arg or l-Cit supplementation in a long-term type 1 diabetic mouse model is not completely clear.
In the present report, we examined the effect of oral supplementation of l-Arg or l-Cit in DN using STZ-induced type 1 diabetic mice. As expected, oral l-Arg or l-Cit supplementation greatly increased kidney and plasma l-Arg levels. To evaluate the effect of supplementation on diabetic renal function and markers of injury, multiple parameters were measured. In diabetic mice, albuminuria and blood urea nitrogen were significantly elevated compared with normal mice. These changes were associated with increased kidney macrophage recruitment, histological changes, and increased expression of TNF-α and fibronectin. Nephrin, a slit diaphragm component, is a very important protein in the pathophysiology of proteinuria (38). It has been previously reported that, compared with nondiabetic patients, nephrin protein expression was significantly reduced in diabetes (17). In the present study, we showed that nephrin protein expression was downregulated in the diabetic mouse kidney. Next, we examined the role of l-Arg or l-Cit supplementation in glomerular macrophage infiltration and kidney TNF-α mRNA expression. Macrophages are crucial in the progression of chronic inflammation through the secretion of multiple cytokines, such as TNF-α (27), IL-1 (28), and monocyte chemotactic protein-1 (14, 26, 31), and we have previously shown that glomerular macrophage infiltration is increased in diabetic kidneys (36). TNF-α is mostly secreted by macrophages and is elevated in the diabetic kidney. Despite greatly elevating l-Arg levels in the plasma and kidneys, oral l-Arg or l-Cit oral supplementation failed to prevent or ameliorate any of these markers of diabetes-induced kidney injury.
Endothelial dysfunction associated with reduced NO bioavailability has been strongly indicated as a critical factor in DN (13, 15), and we have previously established the importance of renal arginase-2 and eNOS in DN (23, 37). Because l-Arg supplementation increased kidney eNOS protein expression in chronic renal failure (15), we evaluated the impact of l-Arg supplementation on renal arginase, eNOS, and NO production (as measured as levels of nitrate + nitrite in the kidney). We confirmed previous results showing increased renal arginase activity and arginase-2 expression in diabetes as well as reduced eNOS expression. In the present study, we also showed that diabetes resulted in increased renal expression of iNOS but not nNOS-β and that the expression levels seen in diabetes were unaffected by supplementation with l-Arg or l-Cit. nNOS has several splice variants expressed in the kidney, especially in the collecting duct and macula densa (16, 21). Hyndman et al. (16) have shown that nNOS-β in the collecting duct is critically important for blood pressure regulation, and Lu et al. (21) have shown the importance of nNOS-β in the macula densa. Similar to Hyndman and colleagues (16), we could not detect nNOS-α in the kidney, whereas it was highly expressed in brain tissue lysate as a positive control.
We also showed decreased NO bioavailability in the diabetic kidney as measured by nitrate + nitrite. However, l-Arg or l-Cit supplementation failed to have any beneficial effect on any of these parameters of arginine/NO metabolism, in striking contrast to the beneficial results obtained with inhibition of arginase (23, 37). In this regard, it has been noted elsewhere that supplementation to increase l-Arg availability does not always enhance NO-mediated effects (3, 6).
In summary, our data demonstrate that oral l-Arg or l-Cit supplementation did not prevent or reduce any markers of renal injury in STZ-induced type 1 diabetes, despite successfully increasing l-Arg levels in the plasma and kidney. There are several possible explanations for the failure of oral supplementation of l-Arg and l-Cit in the treatment of DN. For example, our results may indicate that cellular uptake of l-Arg in affected renal cells, particularly those expressing eNOS, may be impaired. The plasma l-Arg concentration in normal mice is ∼100 μM, which is in the Km range of 100–160 μM for cellular uptake of l-Arg via the y+ and y+L transport systems (12). As l-Arg and Orn compete for cellular uptake with equivalent efficiency, significant reductions in the plasma l-Arg-to-Orn ratio would be expected to result in a reduction in relative efficiency of cellular arginine uptake. In this regard, it is important to note that l-Cit supplementation restored the l-Arg-to-Orn ratio to a significant fraction of the normal value. Also, large absolute increases in plasma l-Arg concentration well above the Km of the transport system, even with reductions in the plasma l-Arg-to-Orn ratio, would still be expected to enhance cellular uptake. In any case, supplementation with either l-Arg or l-Cit enhanced the plasma l-Arg-to-Orn ratio above the level in diabetic mice, particularly in the case of l-Cit supplementation, and increased uptake was indicated by the increased levels of l-Arg in kidney tissue after supplementation with l-Arg or l-Cit. However, we cannot exclude the possibility that l-Arg uptake was impaired in a subpopulation of cells expressing eNOS, which would not be apparent from measurements of l-Arg in bulk kidney tissue. Alternatively, diabetes may lead to altered subcellular compartmentation of enzymes or other proteins in the arginine/NO pathway that result in a lack of response to increases in extracellular l-Arg. Finally, it is important to consider the possibility that the diabetic renal complications may not be associated with depletion of arginine but with arginase-dependent production of some downstream metabolite(s), such as polyamines (22). This would be consistent with the differences in results obtained with arginase inhibition versus supplementation with l-Arg or l-Cit. Moreover, if the production of downstream metabolites rather than arginine depletion is important in diabetic renal pathology, this raises a concern that supplementation with l-Arg or l-Cit could be detrimental rather than beneficial. To evaluate the possibility that NO-independent mechanisms may be involved in the renal complications, future studies should determine the effects of supplementation with l-Arg or l-Cit on renal polyamine metabolism in diabetes.
In contrast with the present results, a recent report (24) concluded that oral l-Cit supplementation is beneficial in STZ-induced DN in a different mouse strain (C57BL/6). It should be noted, however, that C57BL/6 mice are nephropathy resistant and that l-Cit supplementation in that report was nearly twice as long as in the present study (16 vs. 9 wk) and that amino acid levels in the plasma or kidney were not determined. Nevertheless, the different results are perplexing and remain unresolved. Whether the different results obtained with supplementation in these studies reflect differences in the rodent models used or in some other element(s) of the experimental systems remains to be determined.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-094930 and DK-094930S1.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: H.Y., T.G., T.K.C., and A.S.A. performed experiments; H.Y. and A.S.A. analyzed data; H.Y., S.M.M., and A.S.A. interpreted results of experiments; H.Y., T.K.C., and A.S.A. prepared figures; H.Y. and A.S.A. drafted manuscript; H.Y., T.G., T.K.C., S.M.M., and A.S.A. approved final version of manuscript; T.K.C., S.M.M., and A.S.A. edited and revised manuscript; S.M.M. and A.S.A. conception and design of research.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the MS Core Facility at Penn State College of Medicine.
REFERENCES
- 1.Awad AS, Gao T, Gvritishvili A, You H, Liu Y, Cooper TK, Reeves WB, Tombran-Tink J. Protective role of small pigment epithelium-derived factor (PEDF) peptide in diabetic renal injury. Am J Physiol Renal Physiol 305: F891–F900, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Awad AS, Huang L, Ye H, Duong ET, Bolton WK, Linden J, Okusa MD. Adenosine A2A receptor activation attenuates inflammation and injury in diabetic nephropathy. Am J Physiol Renal Physiol 290: F828–F837, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Bai Y, Sun L, Yang T, Sun K, Chen J, Hui R. Increase in fasting vascular endothelial function after short-term oral l-arginine is effective when baseline flow-mediated dilation is low: a meta-analysis of randomized controlled trials. Am J Clin Nutr 89: 77–84, 2009. [DOI] [PubMed] [Google Scholar]
- 4.Balakumar P, Chakkarwar VA, Krishan P, Singh M. Vascular endothelial dysfunction: a tug of war in diabetic nephropathy? Biomed Pharmacother 63: 171–179, 2009. [DOI] [PubMed] [Google Scholar]
- 5.Beleznai T, Feher A, Spielvogel D, Lansman SL, Bagi Z. Arginase 1 contributes to diminished coronary arteriolar dilation in patients with diabetes. Am J Physiol Heart Circ Physiol 300: H777–H783, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boger RH. l-Arginine therapy in cardiovascular pathologies: beneficial or dangerous? Curr Opin Clin Nutr Metab Care 11: 55–61, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Breyer MD, Bottinger E, Brosius FC, 3rd, Coffman TM, Harris RC, Heilig CW, Sharma K. Mouse models of diabetic nephropathy. J Am Soc Nephrol 16: 27–45, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Brodsky SV, Gao S, Li H, Goligorsky MS. Hyperglycemic switch from mitochondrial nitric oxide to superoxide production in endothelial cells. Am J Physiol Heart Circ Physiol 283: H2130–H2139, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Brosius FC, 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T. Mouse models of diabetic nephropathy. J Am Soc Nephrol 20: 2503–2512, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castillo L, Chapman TE, Yu YM, Ajami A, Burke JF, Young VR. Dietary arginine uptake by the splanchnic region in adult humans. Am J Physiol Endocrinol Metab 265: E532–E539, 1993. [DOI] [PubMed] [Google Scholar]
- 11.Chen C, Mitchell KD, Navar LG. Role of endothelium-derived nitric oxide in the renal hemodynamic response to amino acid infusion. Am J Physiol Regul Integr Comp Physiol 263: R510–R516, 1992. [DOI] [PubMed] [Google Scholar]
- 12.Closs EI, Simon A, Vekony N, Rotmann A. Plasma membrane transporters for arginine. J Nutr 134: 2752S–2759S and 2765S–2767S, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Creager MA, Luscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Circulation 108: 1527–1532, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Giunti S, Barutta F, Perin PC, Gruden G. Targeting the MCP-1/CCR2 system in diabetic kidney disease. Curr Vasc Pharmacol 8: 849–860, 2010. [DOI] [PubMed] [Google Scholar]
- 15.Goligorsky MS, Chen J, Brodsky S. Workshop: endothelial cell dysfunction leading to diabetic nephropathy: focus on nitric oxide. Hypertension 37: 744–748, 2001. [DOI] [PubMed] [Google Scholar]
- 16.Hyndman KA, Boesen EI, Elmarakby AA, Brands MW, Huang P, Kohan DE, Pollock DM, Pollock JS. Renal collecting duct NOS1 maintains fluid-electrolyte homeostasis and blood pressure. Hypertension 62: 91–98, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jim B, Ghanta M, Qipo A, Fan Y, Chuang PY, Cohen HW, Abadi M, Thomas DB, He JC. Dysregulated nephrin in diabetic nephropathy of type 2 diabetes: a cross sectional study. PLOS ONE 7: e36041, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalantarinia K, Awad AS, Siragy HM. Urinary and renal interstitial concentrations of TNF-α increase prior to the rise in albuminuria in diabetic rats. Kidney Int 64: 1208–1213, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Kohli R, Meininger CJ, Haynes TE, Yan W, Self JT, Wu G. Dietary l-arginine supplementation enhances endothelial nitric oxide synthesis in streptozotocin-induced diabetic rats. J Nutr 134: 600–608, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Lapenna D, Ciofani G, Pierdomenico SD, Giamberardino MA, Cuccurullo F. Reaction conditions affecting the relationship between thiobarbituric acid reactivity and lipid peroxides in human plasma. Free Radic Biol Med 31: 331–335, 2001. [DOI] [PubMed] [Google Scholar]
- 21.Lu D, Fu Y, Lopez-Ruiz A, Zhang R, Juncos R, Liu H, Manning RD, Jr, Juncos LA, Liu R. Salt-sensitive splice variant of nNOS expressed in the macula densa cells. Am J Physiol Renal Physiol 298: F1465–F1471, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morris SM., Jr Arginine metabolism: boundaries of our knowledge. J Nutr 137: 1602S–1609S, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Morris SM, Jr, Gao T, Cooper TK, Kepka-Lenhart D, Awad AS. Arginase-2 mediates diabetic renal injury. Diabetes 60: 3015–3022, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romero MJ, Yao L, Sridhar S, Bhatta A, Dou H, Ramesh G, Brands MW, Pollock DM, Caldwell RB, Cederbaum SD, Head CA, Bagi Z, Lucas R, Caldwell RW. l-Citrulline protects from kidney damage in type 1 diabetic mice. Front Immunol 4: 480, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwedhelm E, Maas R, Freese R, Jung D, Lukacs Z, Jambrecina A, Spickler W, Schulze F, Boger RH. Pharmacokinetic and pharmacodynamic properties of oral l-citrulline and l-arginine: impact on nitric oxide metabolism. Br J Clin Pharmacol 65: 51–59, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang WW, Qi M, Warren JS. Monocyte chemoattractant protein 1 mediates glomerular macrophage infiltration in anti-GBM Ab GN. Kidney Int 50: 665–671, 1996. [DOI] [PubMed] [Google Scholar]
- 27.Tipping PG, Leong TW, Holdsworth SR. Tumor necrosis factor production by glomerular macrophages in anti-glomerular basement membrane glomerulonephritis in rabbits. Lab Invest 65: 272–279, 1991. [PubMed] [Google Scholar]
- 28.Tipping PG, Lowe MG, Holdsworth SR. Glomerular interleukin 1 production is dependent on macrophage infiltration in anti-GBM glomerulonephritis. Kidney Int 39: 103–110, 1991. [DOI] [PubMed] [Google Scholar]
- 29.Topal G, Brunet A, Walch L, Boucher JL, David-Dufilho M. Mitochondrial arginase II modulates nitric-oxide synthesis through nonfreely exchangeable l-arginine pools in human endothelial cells. J Pharmacol Exp Ther 318: 1368–1374, 2006. [DOI] [PubMed] [Google Scholar]
- 30.United States Renal Data System. Annual Data Report (online). http://www.usrds.org/atlas07.aspx [20 October 2014]. [Google Scholar]
- 31.Wada T, Furuichi K, Sakai N, Iwata Y, Yoshimoto K, Shimizu M, Takeda SI, Takasawa K, Yoshimura M, Kida H, Kobayashi KI, Mukaida N, Naito T, Matsushima K, Yokoyama H. Up-regulation of monocyte chemoattractant protein-1 in tubulointerstitial lesions of human diabetic nephropathy. Kidney Int 58: 1492–1499, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Wang CH, Li F, Hiller S, Kim HS, Maeda N, Smithies O, Takahashi N. A modest decrease in endothelial NOS in mice comparable to that associated with human NOS3 variants exacerbates diabetic nephropathy. Proc Natl Acad Sci USA 108: 2070–2075, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Windmueller HG, Spaeth AE. Metabolism of absorbed aspartate, asparagine, and arginine by rat small intestine in vivo. Arch Biochem Biophys 175: 670–676, 1976. [DOI] [PubMed] [Google Scholar]
- 34.Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci USA 93: 6770–6774, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamizu K, Shinozaki K, Ayajiki K, Gemba M, Okamura T. Oral administration of both tetrahydrobiopterin and l-arginine prevents endothelial dysfunction in rats with chronic renal failure. J Cardiovasc Pharmacol 49: 131–139, 2007. [DOI] [PubMed] [Google Scholar]
- 36.You H, Gao T, Cooper TK, Brian Reeves W, Awad AS. Macrophages directly mediate diabetic renal injury. Am J Physiol Renal Physiol 305: F1719–F1727, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.You H, Gao T, Cooper TK, Morris SM, Jr, Awad AS. Arginase inhibition mediates renal tissue protection in diabetic nephropathy by a nitric oxide synthase 3-dependent mechanism. Kidney Int 84: 1189–1197, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang A, Huang S. Progress in pathogenesis of proteinuria. Int J Nephrol 2012: 314251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao HJ, Wang S, Cheng H, Zhang MZ, Takahashi T, Fogo AB, Breyer MD, Harris RC. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol 17: 2664–2669, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]