

Abstract
Endothelial nitric oxide (NO) is the primary mediator of flow-mediated dilation (FMD) in human adipose microvessels. Impaired NO-mediated vasodilation occurs after acute and chronic hypertension, possibly due to excess generation of reactive oxygen species (ROS). The direct role of pressure elevation in this impairment of human arteriolar dilation is not known. We tested the hypothesis that elevation in pressure is sufficient to impair FMD. Arterioles were isolated from human adipose tissue and cannulated, and vasodilation to graded flow gradients was measured before and after exposure to increased intraluminal pressure (IILP; 150 mmHg, 30 min). The mediator of FMD was determined using pharmacological agents to reduce NO [NG-nitro-l-arginine methyl ester (l-NAME), 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (c-PTIO)], or H2O2 [polyethylene glycol (PEG)-catalase], and mitochondrial (mt) ROS was quantified using fluorescence microscopy. Exposure to IILP decreased overall FMD (max %dilation: 82.7 ± 4.9 vs. 62 ± 5.6; P < 0.05). This dilation was abolished by treatment with l-NAME prepressure and PEG-catalase after IILP (max %dilation: l-NAME: 23.8 ± 6.1 vs. 74.8 ± 8.6; PEG-catalase: 71.8 ± 5.9 vs. 24.6 ± 10.6). To examine if this change was mediated by mtROS, FMD responses were measured in the presence of the complex I inhibitor rotenone or the mitochondrial antioxidant mitoTempol. Before IILP, FMD was unaffected by either compound; however, both inhibited dilation after IILP. The fluorescence intensity of mitochondria peroxy yellow 1 (MitoPY1), a mitochondria-specific fluorescent probe for H2O2, increased during flow after IILP (%change from static: 12.3 ± 14.5 vs. 127.9 ± 57.7). These results demonstrate a novel compensatory dilator mechanism in humans that is triggered by IILP, inducing a change in the mediator of FMD from NO to mitochondria-derived H2O2.
Keywords: flow-mediated dilation, mitochondria, reactive oxygen species, vascular endothelium
endothelial dysfunction is a well-established predictor of future cardiovascular events (16, 17, 43) and is often identified by impaired flow-mediated dilation (FMD), a key physiological response to shear stress that is typically mediated by release of nitric oxide (NO) from the endothelium. Hypertension, a major risk factor for stroke and myocardial infarction, elevates vascular reactive oxygen species (ROS), which can reduce NO bioavailability (4, 26). With chronic loss of NO in disease, compensatory mechanisms can preserve FMD. For example, mice with a genetic deletion of endothelial (e)NOS maintain dilation through compensatory pathways via PGI2, cytochrome P450-derived metabolites of arachidonic acid, or H2O2 (3, 15). Furthermore, it has previously been established that H2O2 is the vasoactive mediator released from the endothelium in response to increased shear stress in the coronary and adipose microcirculations of patients with coronary artery disease (CAD).
It is unknown how ubiquitous the plasticity in the FMD response is, i.e., whether compensatory mediators of dilation are only induced in response to chronic disease or if similar mechanisms can be induced after acute physiological stress (e.g., exercise). Furthermore, even though acute and chronic hypertension can modulate endothelium-dependent dilation (36), it is not known whether the elevation in pressure per se is sufficient for the change in endothelial function. We hypothesized that an acute increase in intraluminal pressure in isolated adipose microvessels from healthy individuals would shift the mechanism of FMD from NO to H2O2. Using an in vivo approach, we have previously published that exercise-induced increases in systolic pressure to levels >170 mmHg led to sustained impairment in FMD (19, 36). We postulated these increases in systolic pressure translated to intraluminal pressures of 120–150 mmHg in the microvasculature. Our more recent work indicated a loss of agonist-induced dilation under these conditions [increased intraluminal pressure (IILP) of 150 mmHg for 30 min], with a marked increase in endothelial superoxide production (10).
MATERIALS AND METHODS
Tissue acquisition and general protocol.
All protocols were approved by the Institutional Review Board of the Medical College of Wisconsin. Discarded human omentum, subcutaneous or pericardial adipose was obtained at the time of surgery and placed in cold 4°C HEPES buffer solution. Arterioles were cleaned of fat and connective tissue and were prepared for continuous measurements of diameter as described previously (31). Tissue from patients with hypertension, CAD, and diabetes were excluded from this study.
Materials.
Endothelin-1 (ET-1) was obtained from Peninsula Laboratories. Other chemicals were obtained from Sigma-Aldrich. ET-1 was prepared in saline with 1% bovine serum albumin. Rotenone and MitoPY1 (Tocris Bioscience) were prepared in DMSO. Other agents were prepared in distilled water or physiological saline solution. All concentrations represent the final steady-state concentrations in the tissue bath.
Cannulated artery preparation.
The vasodilator response to flow in microvessels was evaluated as previously described (28). In an organ chamber, both ends of the vessel (50–200 μm) were cannulated with glass micropipettes filled with Krebs buffer and pressurized (60 mmHg) for video microscopy of diameter change. Flow gradients were generated as described by Kuo et al. (23, 24) using pipettes of identical impedance. Data are reported as percentage of maximal diameter at a given flow rate or pressure gradient. Pressure gradients of 5–100 cmH2O (correlating to flow rates of ∼5–20 μl/min) were generated, and steady-state diameter was assessed at 5 min after each change. Two flow-response curves were generated for each vessel, one before and one after the 30-min IILP. When possible, two vessels from the same tissue were used, with and without pharmacological intervention [100 μM NG-nitro-l-arginine methyl ester (l-NAME); 500 U/ml polyethylene glycol (PEG)-catalase; 1 μM rotenone; 10 μM mitoTempol; 100 μM 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (c-PTIO); and 10 μM sepiapterin].
Increased intraluminal pressure.
To increase intraluminal pressure in the cannulated microvessels, reservoirs connected to both sides of the vessel were simultaneously raised to a height creating a pressure of 150 mmHg for 30 min, followed by a 30-min recovery period (60 mmHg) before the second dose-response curve. For fluorescence studies (MitoPY1), this procedure was performed before the first dose-response curve and compared with a separate vessel from the same tissue without pressure treatment (time control).
Assessment of mitochondrial ROS.
The fluorescent probe MitoPY1 (Tocris Bioscience; 10 μM) (5, 35) was used to assess mitochondrial H2O2. Vessels were prepared in the same manner as vasodilator studies, except that HEPES buffer (275 mM NaCl, 7.99 mM KCl, 4.9 mM MgSO4, 3.2 mM CaCl2, 2.35 mM KH2PO4, 0.07 mM EDTA, 12 mM glucose, and 20 mM HEPES acid) was used. The pH was stabilized at 7.4 at 37°C. Vessels were cannulated with MitoPY1 added to the buffer perfusing the lumen of the vessel and maintained at 37°C for the duration of the experiment. After sufficient time for cellular integration of the probe (30–45 min), IILP was increased to 150 mmHg for 30 min. After lowering the pressure to 60 mmHg, the buffer in the organ chamber was changed and fluorescence of the vessel was detected before and after inducing a flow gradient of 100 cmH20 for 5 min. All vessels exposed to IILP were compared with a separate vessel from the same tissue without pressure treatment (time control).
MitoPY1 fluorescence was measured with a Nikon eclipse TE200 microscope equipped with a ×20 objective and Hamamatsu C4742–95 camera using an excitation wavelength of 489 nm with emission detection at 540 nm. Fluorescence was quantified by free-hand selecting the vessel and subtracting the fluorescent intensity of the background using MetaMorph (Version NX). Values are expressed as relative increase from baseline (0 vs. 100 cmH2O pressure gradient).
Statistical methods.
Data are presented as mean value ± SE. For all flow-response curves, differences between groups at each flow gradient were determined using a two-way, repeated-measures ANOVA. A post hoc Tukey test was used for comparison of more than two means following ANOVA. A probability value of P < 0.05 was considered to be statistically significant. For fluorescence studies, a one-way ANOVA was used with post hoc Tukey test.
RESULTS
Discarded human adipose tissue was collected at the time of surgery from 42 patients undergoing a variety of surgical procedures (e.g., bowel resection, reconstructive surgery). Only 2 of 42 had any cardiovascular risk factors. None of the subjects had clinically diagnosed CAD. Patient demographics are show in Table 1.
Table 1.
Patient demographics
| Total (n = 42) | |
|---|---|
| Characteristics | |
| Sex, M/F | 14/28 |
| Age, yr (average ± SE) | 48.0 ± 2.4 |
| Fat depot, sub Q/visceral | 14/28 |
| Underlying diseases/risk factors | |
| Coronary artery disease | 0 |
| Hypertension | 0 |
| Hypercholesterolemia | 2 |
| Diabetes mellitus | 0 |
| Congestive heart failure | 0 |
| None of the above | 40 |
Effect of IILP on FMD.
FMD in microvessels from individuals without cardiovascular disease was abrogated by l-NAME (Fig. 1, A and B). Following IILP, peak FMD was reduced by 20.5% (Fig. 1, A and B) and was further reduced by PEG-catalase (Fig. 1B), indicating a primary role for H2O2 in maintaining vasodilation. IILP had no effect on endothelium-independent dilation to papaverine (Fig. 1C). In the presence of both l-NAME and PEG-catalase, FMD was eliminated after exposure to IILP (max %dilation: pre-IILP −0.2 ± 0.2 vs. post IILP 3.2 ± 2.1; n = 4).
Fig. 1.
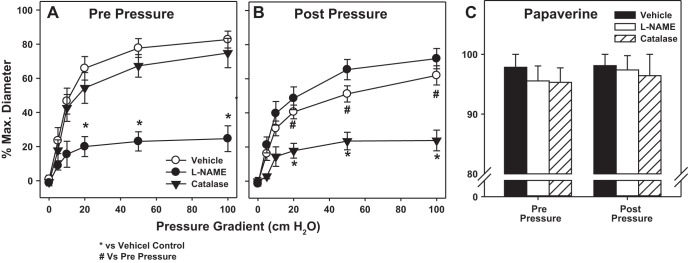
Increased intraluminal pressure (IILP) in microvessels from healthy individuals decreases flow-mediated dilation (FMD). The mechanism of FMD changed from nitric oxide (NO) [inhibited by NG-nitro-l-arginine methyl ester (l-NAME) prepressure; A] to hydrogen peroxide (inhibited by catalase postpressure; B). IILP had no effect on endothelial-independent dilation to papaverine (C). *P < 0.05 vs. vehicle control; #P < 0.05 vs. prepressure by two-way ANOVA repeated measures (RM).
Source of hydrogen peroxide mediating FMD.
To determine the source of the H2O2 responsible for FMD, we combined functional studies and fluorescence imaging. Rotenone, an inhibitor of mitochondrial electron transport chain complex I, had no effect on FMD in vessels before exposure to IILP but after IILP rotenone reduced FMD (%max diameter: 21.2 ± 3.5; P < 0.05 vs. before; Table 2). In separate experiments, the mitochondrial targeted free radical scavenger mitoTempol inhibited FMD similarly after IILP, with no effect under control conditions (%max diameter: pre-IILP = 87.9 ± 4.8; post-IIP = 22.3 ± 4.2; P < 0.05). These findings indicate that mitochondrial free radicals contribute in a prominent mechanistic way to FMD after exposure to IILP.
Table 2.
Vessel diameter measurements
| Untreated |
||||
|---|---|---|---|---|
| Max ∅ | Starting ∅ | Pre-IILP ∅ | Post-IILP ∅ | |
| Vehicle (8) | 233 ± 31 | 182 ± 26 | 217 ± 29 | 218 ± 39 |
| l-NAME (8) | 212 ± 15 | 141 ± 8 | 195 ± 14 | 194 ± 17 |
| PEG-catalase (6) | 191 ± 14 | 153 ± 9 | 183 ± 13 | 171 ± 14 |
| Rotenone (5) | 180 ± 23 | 135 ± 18 | 161 ± 21 | 156 ± 24 |
| c-PTIO (4) | 175 ± 17 | 100 ± 13 | 175 ± 19 | 157 ± 13 |
Values are means ± SE; nos. parentheses are no. of measurements. l-NAME, NG-nitro-l-arginine methyl ester; c-PTIO, 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide; PEG, polyethylene glycol; ∅, diameter; IILP, increased intraluminal pressure.
MitoPY1, a fluorescent probe specific for mitochondrial H2O2 (5, 7), was used to confirm H2O2 production in response to increased intraluminal flow. Following IILP, MitoPY1 fluorescence increased during flow compared with vessels studied in parallel that were not exposed to pressure (Fig. 2).
Fig. 2.

IILP increases mitochondrial free radical production in response to intraluminal flow. A: mitochondrial H2O2 generation in the presence or absence of flow was measured using mitochondria peroxy yellow 1 (MitoPY1), a cell-permeable fluorescent dye that is avidly taken up by the mitochondria. B: quantification of MitoPY1 signal in response to flow compared with baseline (0→100 cmH2O). C and D: FMD in the presence of complex 1 inhibitor rotenone or mitochondrial free radical scavenger mitoTempol. *P < 0.05 vs. 0 flow control t-test (B); #P < 0.05 vs. prepressure two-way ANOVA RM (C and D).
Role of NOS.
Before IILP, the NO scavenger c-PTIO reduced maximal percent FMD to 18.0 ± 2.3, similar to the effect of l-NAME, suggesting that NOS-derived NO is the principal mediator of FMD under normal conditions. Similar to l-NAME, c-PTIO had no effect on vasodilation after IILP (Fig. 3).
Fig. 3.
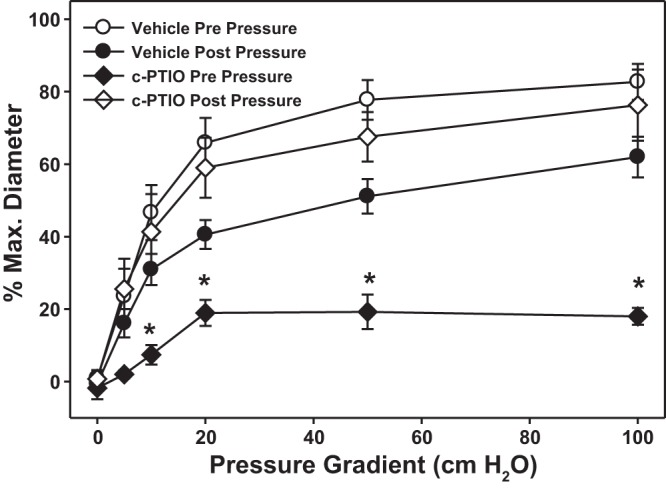
In vessels from healthy subjects NO is the mediator of FMD. The NO-scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (c-PTIO) was used to differentiate involvement of NO or the dependency on nitric oxide synthase (NOS). *P < 0.05 vs. prepressure two-way ANOVA RM.
Sepiapterin prevents transition from NO to H2O2 as mediator of FMD in microvessels.
Under normal conditions, eNOS generates NO. However, the absence of the cofactor tetrahydrobiopterin (BH4), uncouples eNOS resulting in generation of superoxide and H2O2 rather than NO (1). To test the contribution of uncoupled NOS to ROS generation following IILP, we treated isolated vessels with sepiapterin (10 μM), a stable, cell permeable precursor of BH4 synthesis. Sepiapterin prevented the decrease in dilation after IILP. Furthermore, the mechanism of FMD following IILP remained l-NAME-inhibitable, indicating the response was NOS mediated (Fig. 4, A and B). To confirm NO as the mediator of FMD, c-PTIO was added to the tissue bath. In the presence of sepiapterin, c-PTIO decreased the FMD response before and after exposure to IILP in a similar manner to l-NAME (Fig. 4, C and D). In the presence of sepiapterin, scavenging H2O2 with catalase had no effect on FMD either before or after IILP (Fig. 4, E and F).
Fig. 4.

Sepiapterin preserves NO as the mechanism of FMD after exposure to IILP. In the presence of sepiapterin, FMD is similarly abrogated before and after IILP by both the NOS inhibitor l-NAME (A and B) and the NO-scavenger c-PTIO (C and D). Polyethylene glycol (PEG)-catalase had no effect on FMD in vessels treated with sepiapterin (E and F). Sepiapterin control curves are the same in pre- and postpressure. *P < 0.05 vs. prepressure two-way ANOVA RM. #P < 0.05, EC50 vs. treated.
DISCUSSION
The current study documents a transition in the mechanism of FMD in human arterioles following a brief exposure to IILP (Fig. 5). We confirm that under normal conditions the mechanism of FMD in the microcirculation of individuals without cardiovascular disease involves eNOS-derived NO (with H2O2 providing the remaining smaller component). The major new findings of this study are threefold. First, in individuals without cardiovascular disease or its risk factors, the magnitude of FMD (mainly the NO component) is reduced following a transient increase in intraluminal pressure. Second, the dilation after IILP is mediated by a compensatory increase in H2O2, derived in part from the mitochondrial electron transport chain. Third, sepiapterin prevents the pressure-induced loss of FMD and also prevents the IILP-induced shift in vasoactive mediator from NO to mitochondrial-derived H2O2. Together, these results suggest that formation of H2O2 after IILP is mediated by the mitochondria with levels of NO bioavailability playing a crucial role for regulation of mitochondrial ROS production.
Fig. 5.
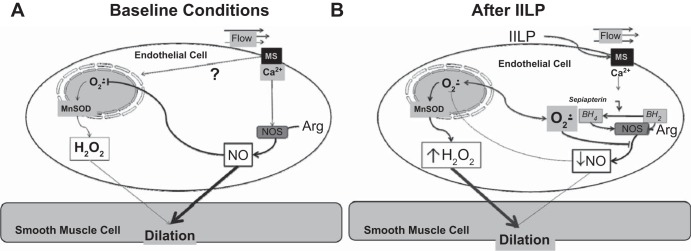
Mechanism of FMD. Mechanical shear (flow)-induced dilation in human microvessels under baseline (A) and after exposure to acute increase intraluminal pressure (B).
Mechanism of FMD in health and disease.
In subjects without CAD, NO is the primary mediator of FMD in resistance (36) and conduit (18, 33) vessels. It is well established in both animal models and humans that external stressors or disease reduces NO bioavailability in the vasculature. One of the major risk factors for cardiovascular disease is hypertension. Here we show that even brief elevations in pressure within the vessel are sufficient to produce a significant impairment in endothelial function by suppressing NO-mediated dilation. Furthermore, after exposure to increased pressure, the vasodilator response to the flow stimulus is mediated by a compensatory rise in mitochondrial-derived H2O2, which serves as the primary dilator mechanism.
Hypertension elevates vascular ROS, which scavenge NO (41, 42). Previous studies have shown that compensatory mechanisms can preserve FMD in the presence of chronic disease in humans (19, 28, 38) and in animal models of cardiovascular disease (3, 20, 21). For example, mouse models with genetic deletion of eNOS have compensatory dilator pathways that involve metabolites of arachidonic acid or H2O2, (3, 15). Our data indicate that such compensatory dilator mechanisms can be invoked in a relatively short period of time (30 min). In human vessels, this unique ability to transition mechanisms of FMD has been described by us and others (9, 37, 39) but does not occur in all situations. In pig coronary arterioles, FMD is mediated by NO. However, acute inhibition of NOS (24) or chronic hypercholesterolemia (25) eliminates FMD in the porcine heart with no compensatory dilation. The data presented here demonstrate an adaptive response to IILP that results in a switch in mediators with preservation of the FMD response.
Coupled vs. uncoupled NOS in disease.
One of the major risk factors for cardiovascular disease is increased arterial pressure, and uncoupling of eNOS has been documented to contribute to the progression of hypertension (13). The ability of eNOS to produce superoxide during stress conditions (e.g., absence of cofactors or substrate) is frequently associated with cardiovascular disease states. That sepiapterin, a precursor of the eNOS cofactor BH4, prevents uncoupling of eNOS and blocked the switch from NO to H2O2 as the mechanism of FMD further underlies the importance of NO bioavailability for the maintenance of normal vascular function. These results also suggest a possible role of uncoupled eNOS as one of the signaling enzymes involved in provoking mitochondrial ROS generation under stress conditions. As our fluorescence data indicate, IILP upregulates mitochondrial-derived H2O2, which we have previously shown to be the mediator of FMD in arterioles from subjects with CAD (27). As l-NAME only affected dilation before IILP, the role of eNOS-derived ROS as the main source of vasoactive H2O2 appears minor. However, it must be considered that l-NAME does not block NOS-production of ROS in all circumstances (2).
These data suggest a key role of NO in the signaling pathway of FMD under control situations since treatment of vessels with c-PTIO (scavenger of NO) abrogated FMD in controls but had no effect after IILP. This parallels the response seen to l-NAME, which blocks NOS activity, typically preventing either NO or O2·− formation by NOS. Addition of sepiapterin restores NOS coupling and the NO mechanism of FMD (l-NAME inhibitable pre- and postpressure in presence of sepiapterin). In the presence of sepiapterin, H2O2 did not contribute to FMD.
Mitochondrial ROS as signaling molecules.
The mechanism by which IILP provokes the switch in vasodilator mechanism from NO to mitochondrial H2O2 remains unknown. Multiple ion channels (22), receptors (e.g., AT1R) and organelles (e.g., cilia; Ref. 34) have been shown to be involved in the mechanochemical signal transduction of shear. A number of shear sensors are also redox sensitive (30), which may contribute to shifting the mechanism of dilation, as IILP increases vascular superoxide levels (10).
NO suppresses mitochondrial ROS production through direct quenching of superoxide and inhibition of mitochondrial electron transport chain components (12). This could potentially explain how a loss of NO shifts dilation to mitochondrial generated H2O2, and how restoration of NOS production of NO reverts the mechanism of dilation back to NO. Alternatively, superoxide release from uncoupled eNOS may invoke a ROS-induced ROS release mechanism (44) where signaling levels of H2O2 released from NOS induce the release of vasoactive levels of H2O2 from the mitochondria to elicit dilation. Recent data from our laboratory support a critical role for the involvement of ceramide in the switch from NO to H2O2 in subjects with coronary disease (14). A similar role in pressure-induced changes in endothelial function has not been explored.
Study limitations.
There are several potential limitations to our study. Adipose tissue was collected from diverse depots, including visceral and subcutaneous locations. However, we observed no site-specific differences in FMD or its mechanisms (40). Also, the tissue was obtained from discarded specimens. To maintain required confidentiality, it was not possible to collect comprehensive clinical data (medication lists, quantitative laboratory data). However, the resulting heterogeneity would make any differences we observed more robust. One cannot exclude the presence of residual effects of patients' prescribed medications on isolated microvessels; however, such influences on vascular tone are likely minimal due to washout since vessels are superfused with >200 ml of drug-free buffer solution for more than an hour before initial diameter measurements are made (31). Consistent with this expectation, the effects of pharmacological dilator agents added during our protocols are reversed by washing the vessels with fresh buffer.
Nonspecificity of fluorescence tracers is a general concern. MitoPY1 is highly specific for mitochondrial H2O2 (6, 7), but we cannot exclude that other oxidative metabolites are also be detected. This concern is reduced by the observation that PEG-catalase, which is highly specific for H2O2, eliminates the MitoPY1 signal, indicating specificity of this novel florescent probe. Similarly, rotenone eliminated FMD after IILP. While at higher doses rotenone has been reported to reverse the electron transport chain and result in ROS generation (8), under the conditions of our experiments rotenone blocks electron transport chain production of ROS (14, 28). Together with supportive data using the mitochondrial ROS scavenger mitoTempol, we interpret our findings to indicate a necessary role for mitochondrial H2O2 in FMD after exposure to IILP.
Novelty.
This study is the first to identify a signaling change in the vasoactive mediator released from the endothelium of human microvessels after a short increase in intraluminal pressure. This observed transition occurs in a matter of minutes, which may have implications for exercise, labile hypertension, and stress-induced increases in mean arterial blood pressure that occur throughout daily life.
GRANTS
This work was supported by National Institutes of Health Grants R01-HL-113612 (to D. D. Gutterman), R21-OD-018306 (to A. M. Beyer), and T32-HL-007792 (to M. J. Durand) and American Heart Association Grant 14POST18780022 (to M. J. Durand).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.M.B., S.A.P., and D.D.G. conception and design of research; A.M.B., M.J.D., and J.H. performed experiments; A.M.B. and M.J.D. analyzed data; A.M.B. and D.D.G. interpreted results of experiments; A.M.B. prepared figures; A.M.B. drafted manuscript; M.J.D., J.H., T.C.G., and D.D.G. approved final version of manuscript; T.C.G. and D.D.G. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank the surgeons and nurses at Froedtert Memorial Lutheran Hospital, the Division of Cardiothoracic Surgery at the Medical College of Wisconsin, the Cardiothoracic Surgery Division at the Zablocki Veterans Affairs Medical Center in Milwaukee, the Aurora Medical Group Cardiovascular and Thoracic Surgery, and the Cardiothoracic Surgery Group of Milwaukee for providing tissue.
REFERENCES
- 1.Channon K. Tetrahydrobiopterin: regulator of endothelial nitric oxide synthase in vascular disease. Trends Cardiovasc Med 14: 323–327, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MH, Chen YR, Druhan LJ, Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468: 1115–1118, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chlopicki S, Kozlovski VI, Lorkowska B, Drelicharz L, Gebska A. Compensation of endothelium-dependent responses in coronary circulation of eNOS-deficient mice. J Cardiovasc Pharmacol 46: 115–123, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Cooke M, John P, Dzau M, Victor J. Nitric oxide synthase: role in the genesis of vascular disease. Annu Rev Med 48: 489–509, 1997. [DOI] [PubMed] [Google Scholar]
- 5.Dickinson BC, Chang CJ. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J Am Chem Soc 130: 9638–9639, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickinson BC, Lin VS, Chang CJ. Preparation and use of MitoPY1 for imaging hydrogen peroxide in mitochondria of live cells. Nat Protoc 8: 1249–1259, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dikalov SI, Kirilyuk IA, Voinov M, Grigor'ev IA. EPR detection of cellular and mitochondrial superoxide using cyclic hydroxylamines. Free Radic Res 45: 417–430, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Durand MJ, Gutterman DD. Diversity in mechanisms of endothelium-dependent vasodilation in health and disease. Microcirculation 20: 239–247, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durand MJ, Phillips SA, Widlansky ME, Otterson MF, Gutterman DD. The vascular renin angiotensin system contributes to blunted vasodilation induced by transient high pressure in human adipose microvessels. Am J Physiol Heart Circ Physiol 307: H25–H32, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erusalimsky JD, Moncada S. Nitric oxide and mitochondrial signaling from physiology to pathophysiology. Arterioscler Thromb Vasc Biol 27: 2524–2531, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease from marvel to menace. Circulation 113: 1708–1714, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Freed JK, Beyer AM, LoGiudice JA, Hockenberry JC, Gutterman DD. Ceramide changes the mediator of flow-induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circ Res 115: 525–532, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gödecke A, Schrader J. Adaptive mechanisms of the cardiovascular system in transgenic mice-lessons from eNOS and myoglobin knockout mice. Basic Res Cardiol 95: 492–498, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Gokce N, Keaney JF, Jr., Hunter LM, Watkins MT, Menzoian JO, Vita JA. Risk stratification for postoperative cardiovascular events via noninvasive assessment of endothelial function: a prospective study. Circulation 105: 1567–1572, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Harrison D, Freiman P, Armstrong M, Marcus M, Heistad D. Alterations of vascular reactivity in atherosclerosis. Circ Res 61: II74–180, 1987. [PubMed] [Google Scholar]
- 18.Joannides R, Haefeli WE, Linder L, Richard V, Bakkali EH, Thuillez C, Lüscher TF. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation 91: 1314–1319, 1995. [DOI] [PubMed] [Google Scholar]
- 19.Jurva JW, Phillips SA, Syed AQ, Syed AY, Pitt S, Weaver A, Gutterman DD. The effect of exertional hypertension evoked by weight lifting on vascular endothelial function. J Am Coll Cardiol 48: 588–589, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Kang KT, Sullivan JC, Sasser JM, Imig JD, Pollock JS. Novel nitric oxide synthase-dependent mechanism of vasorelaxation in small arteries from hypertensive rats. Hypertension 49: 893–901, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Kang KT, Sullivan JC, Spradley FT, d'Uscio LV, Katusic ZS, Pollock JS. Antihypertensive therapy increases tetrahydrobiopterin levels and NO/cGMP signaling in small arteries of angiotensin II-infused hypertensive rats. Am J Physiol Heart Circ Physiol 300: H718–H724, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Köttgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol 182: 437–447, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuo L, Arko F, Chilian WM, Davis MJ. Coronary venular responses to flow and pressure. Circ Res 72: 607–615, 1993. [DOI] [PubMed] [Google Scholar]
- 24.Kuo L, Chilian WM, Davis MJ. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am J Physiol Heart Circ Physiol 261: H1706–H1715, 1991. [DOI] [PubMed] [Google Scholar]
- 25.Kuo L, Davis MJ, Cannon MS, Chilian WM. Pathophysiological consequences of atherosclerosis extend into the coronary microcirculation. Restoration of endothelium-dependent responses by l-arginine. Circ Res 70: 465–476, 1992. [DOI] [PubMed] [Google Scholar]
- 26.Lind L. Endothelium-dependent vasodilation in hypertension: a review. Blood Press 9: 4–15, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res 108: 566–573, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res 93: 573–580, 2003. [DOI] [PubMed] [Google Scholar]
- 30.Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, Suzuki M, Zhang DX. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am J Physiol Heart Circ Physiol 298: H466–H476, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miura H, Liu Y, Gutterman DD. Human coronary arteriolar dilation to bradykinin depends on membrane hyperpolarization contribution of nitric oxide and Ca2+-activated K+ channels. Circulation 99: 3132–3138, 1999. [DOI] [PubMed] [Google Scholar]
- 33.Mullen MJ, Kharbanda RK, Cross J, Donald AE, Taylor M, Vallance P, Deanfield JE, MacAllister RJ. Heterogenous nature of flow-mediated dilatation in human conduit arteries in vivo relevance to endothelial dysfunction in hypercholesterolemia. Circ Res 88: 145–151, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Nauli SM, Jin X, Hierck BP. The mechanosensory role of primary cilia in vascular hypertension. Int J Vasc Med 2011: 376281, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohsaki Y, O'Connor P, Mori T, Ryan RP, Dickinson BC, Chang CJ, Lu Y, Ito S, Cowley AW. Increase of sodium delivery stimulates the mitochondrial respiratory chain H2O2 production in rat renal medullary thick ascending limb. Am J Physiol Renal Physiol 302: F95–F102, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paniagua OA, Bryant MB, Panza JA. Role of endothelial nitric oxide in shear stress-induced vasodilation of human microvasculature diminished activity in hypertensive and hypercholesterolemic patients. Circulation 103: 1752–1758, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Phillips SA, Das E, Wang J, Pritchard K, Gutterman DD. Resistance and aerobic exercise protects against acute endothelial impairment induced by a single exposure to hypertension during exertion. J Appl Physiol 110: 1013–1020, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phillips SA, Hatoum O, Gutterman DD. The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. Am J Physiol Heart Circ Physiol 292: H93–H100, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Phillips SA, Jurva JW, Syed AQ, Kulinski JP, Pleuss J, Hoffmann RG, Gutterman DD. Benefit of low-fat over low-carbohydrate diet on endothelial health in obesity. Hypertension 51: 376–382, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sato A, Sakuma I, Gutterman DD. Mechanism of dilation to reactive oxygen species in human coronary arterioles. Am J Physiol Heart Circ Physiol 285: H2345–H2354, 2003. [DOI] [PubMed] [Google Scholar]
- 41.Touyz R, Schiffrin E. Reactive oxygen species in vascular biology: implications in hypertension. Histochem Cell Biol 122: 339–352, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension what is the clinical significance? Hypertension 44: 248–252, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Yoshida T, Kawano H, Miyamoto S, Motoyama T, Fukushima H, Hirai N, Ogawa H. Prognostic value of flow-mediated dilation of the brachial artery in patients with cardiovascular disease. Intern Med 45: 575–579, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Zinkevich NS, Gutterman DD. ROS-induced ROS release in vascular biology: redox-redox signaling. Am J Physiol Heart Circ Physiol 301: H647–H653, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]