

Abstract
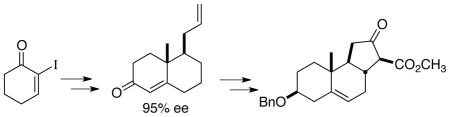
Organocatalyzed enantioselective allylation of 2-iodocyclohexenone followed by methylation and oxy-Cope rearrangement delivered enantiomerically-enriched 2-methyl 3-allyl cyclohexanone, that engaged in acid-catalyzed Robinson annulation to give the bicyclic enone. Subsequent elaboration of the pendant allyl group into an α-diazo β-keto ester set the stage for Rh-mediated cyclization to deliver the tricyclic A-B-C core of the Veratrum alkaloids.
Introduction
The Veratrum alkaloids (Fig. 1) contain a 6-6-5-6 carbocyclic scaffold connected to either a piperidine system or a 5–6 tetrahydrofuran/piperidine system.1 Members of this family of natural products include veratramine 1, cyclopamine 2, jervine 3 and cycloposine 4. In particular, cyclopamine has been investigated for its activity against certain cancers, especially basal cell carcinoma, pancreatic cancer, medulloblastoma and small cell lung cancer.2 These cancers propagate via the Hedgehog pathway, of which cyclopamine 2 is a known inhibitor.
Figure 1.

Members of the Veratrum family of alkaloids.
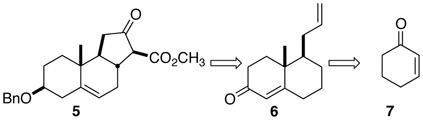
To date, no direct synthetic route to the 6-6-5 tricyclic core system of the Veratrum alkaloids has been described. We envisoned (Eq.1) an approach to the β-keto ester 5 by way of the key enone 6.
 |
(1) |
Results and Discussion
The Wieland-Miescher ketone 8 (Fig. 2), both in racemic3 and enantiomerically-pure4 form, has been a workhorse for polyalicyclic synthesis. We hypothesized that the enantiomerically-pure enone 6 and the deconjugated ether 9 could be similarly versatile chirons.
Figure 2.

Chirons for polyalicyclic synthesis.
Preparation of the enone 6
The preparation (Scheme 1) of the enone 6 began with the asymmetric allylation proceeded smoothly crystalline α-iodo enone 10.5 As we have reported,6 using the Schaus7 protocol, allyl borane 11 in the presence of catalytic R-Br2BINOL, to deliver the alcohol 12 in high yield and 95% ee. For this conversion to be practical, the catalyst ($200/gram) needed to be recycled. We were pleased to find that an aqueous extraction using 5% NaOH enabled 90–95% catalyst separation and recovery.
Scheme 1.

A Kumada coupling8 was employed to convert 12 to 13. The oxy-Cope rearrangement of 13 in toluene using KH in paraffin9 initially presented some difficulty, due to the low molecular weight of 14. Extractive workup with low boiling diethyl ether reduced the amount of volatile product lost during the purification. Direct filtration of the concentrated toluene extract through silica gel then delivered the pure ketone 14.
Rounding out the synthesis was a Robinson annulation with methyl vinyl ketone to give the enantiomerically pure enone 6. The acid-mediated Robinson annulation proceeded to ~35% conversion before decomposition set in. Attempts to increase conversion through additional equivalents of methyl vinyl ketone were not successful. However, the unreacted ketone 14 was readily recovered in the course of the purification of 6.
Preparation of the Ether 9
Many polyalicyclic natural products, including 1–4, have an equatorial hydroxyl group in the A ring with the alkene beginning at C-5. We therefore applied (Scheme 2) the classic deconjugating protocol10 to the enone 6. The crude enol acetate 15 was reduced11 with NaBH4 to give the alcohol 16 as a 4:1 mixture of diastereomers. A survey of protecting groups led to the crystalline 3-bromobenzoate 17, the structure, including absolute configuration, of which was confirmed by X-ray crystallography (Supporting Information).
Scheme 2.

Exposure of the recrystallized 17 to CH3OH/K2CO3 liberated the pure equatorial alcohol 18, which was carried on to the benzyl ether 9. The protection of 18 with BnBr and NaH gave incomplete conversion, even at elevated temperatures. Alternatively, with KH in paraffin,12 the alcohol was converted to the benzyl ether 9 smoothly at room temperature.
Preparation of a Tricyclic β-Keto Ester
The alkaloids 1–4 each have a trans-fused five-membered C-ring. Given the literature precedent,13 it seemed likely that the α-diazo β-keto ester 20 (Scheme 3) would, under Rh catalysis, cyclize to 5.
Scheme 3.

In a preliminary investigation, the terminal alkene of racemic 9 was selectively converted to the methyl ketone 19 using the modified Wacker oxidation developed by Sigman,14 t-butyl hydroperoxide in the presence of a Pd(quinox)Cl2complex. Diazo transfer15 on the derived β-keto ester delivered the desired α-diazo β-keto ester 20.
In the event, cyclization proceeded smoothly, to give the crystalline tricyclic ketone 5 as a single diastereomer. The assignment of the relative configuration of 5 was supported by the ring methines at δ 62.3, 48.5, and 37.7 in the 13C NMR, as well as by the 12.0 Hz coupling constant of the β-keto ester methine in the 1H NMR.
Conclusion
We have developed a scalable, enantioselective route to the bicyclic chirons 6 and 9, and to the tricyclic β-keto ester 5. We believe that these now readily-available chirons will have wide applicability in target-directed synthesis.16
Experimental
General Procedures
1H-NMR and 13C-NMR measured at 400 MHz for 1H, 90 MHz and 100 MHz for 13C in CDCl3. 13C multiplicities were determined with the aid of a JVERT pulse sequence, differentiating the signals for methyl and methine carbons as “d”, from methylene and quaternary carbon as “u”. The infrared (IR) spectra were determined as neat oil. High resolution mass spectra (HRMS) were obtained by electron impact (EI) or chemical ionization (CI). Rf values indicated refer to thin layer chromatography (TLC) on 2.5 × 10 cm, 250 micron analytical plates coated with silica gel GF, developed in the solvent system indicated. Flash chromatography was performed using silica gel 60 (40–60 μm). Solvents are referred as volume/volume mixtures. All air and moisture sensitive reactions were performed in oven dried glassware under a positive pressure of nitrogen. All solvents were purified immediately prior to use. Tetrahydrofuran (THF) and diethyl ether (Et2O) were distilled from sodium metal/benzophenone under nitrogen. Dichloromethane, toluene and acetonitrile (MeCN) were distilled from CaH2 under nitrogen. All reaction mixtures were stirred magnetically under nitrogen atmosphere. MTBE is t-butyl methyl ether, PE is 30–60 petroleum ether, DME is dimethoxyethane, and MeOH is methanol. HRMS were recorded in EI mode on a double-focusing instrument.
2-Iodocyclohex-2-enone 10
Following the reported procedure,5 2-cyclohexenone (35.0 g, 0.365 mol) was dissolved in a 1:1 mixture of THF:H2O and stirred at 0 °C for 5 min. Next, K2CO3 (60.375 g, 0.438 mol) was added, followed by DMAP (8.893 g, 7.29 mmol). The reaction mixture turned a series of colors before finally turning brown. The solution was stirred for 10 min. I2 (111.1 g, 0.437 mol) was then added slowly to avoid forming a solid mass at the bottom of the flask. The reaction turned black and was stirred overnight open to the atmosphere at room temperature. The mixture was then partitioned between EtOAc and, sequentially, 0.1 M aqueous HCl, saturated aqueous Na2S2O3, and brine. The combined organic layers were dried (Na2SO4) and concentrated. The residue was filtered through a silica plug to give enone 10 (72.89 g, 90% yield) as a yellow crystalline solid. The spectral data matched that of the previously synthesized material.5b TLC: Rf (MTBE/PE, 1:4) = 0.40; 1H-NMR (CDCl3) δ 7.77 (t, J = 4.4 Hz, 1H), 2.67 (t, J = 6.8 Hz, 2H), 2.44 (m, 2H), 2.09 (m, 2H); 13C-NMR δ u: 192.2, 103.9, 37.3, 30.0, 22.9; d: 159.5.
B-Allyl-1,3,2-dioxaborinane 11
Following the reported procedure,17 a solution of trimethyl borate (22.76 mL, 200 mmol) was dissolved in 200 mL Et2O at −78 °C and allylmagnesium bromide (1.0 M in Et2O, 200 mL, 200 mmol) was added. The resulting white suspension was stirred at this temperature for 2 h before the dry ice bath was removed and the solution allowed to warm to −20 °C. 3 M aqueous HCl (200 mL) was added, and the mixture turned clear as the solids dissolved. The solution was stirred for 20 min at room temperature. The layers were separated and the aqueous layer was extracted x3 with Et2O. The organic layers were dried and concentrated to about 200 mL on a rotovap with the bath set no higher than 30 °C. Then 100 mL of dry Et2O was added along with 1,3-propanediol (15.2 g, 200 mmol) and activated 4Å molecular sieves (36 g). The reaction mixture was stirred at room temperature for 20 h. The reaction mixture was filtered through glass wool and the molecular sieves were washed x2 with Et2O. The reaction mixture was concentrated without having the water bath exceed 30 °C. The resulting clear solution was dissolved in 200 mL pentane and the excess 1,3-propanediol (bottom layer) was removed via pipette. The solution was filtered through celite and concentrated under reduced pressure to give compound 11 (17.1 g, 68% yield) as a clear oil. The spectra matched that of the previously synthesized compound.7 1H-NMR (CDCl3) δ 5.86 (m, 1H), 4.92 (m, 2H), 3.99 (t, J = 5.6 Hz, 4H), 1.95 (m, 2H), 1.64 (d, J = 7.2 Hz, 2H); 13C-NMR δ u: 113.9, 61.8, 27.3; d: 135.5.
(R)-1-Allyl-2-iodocyclohex-2-enol 12
Following the reported procedure,7 to a round bottom flask was added (R)-BINOL-Br2 (0.750 g, 1.689 mmol), t-BuOH (4.65 g, 62.83 mmol) and the allylborane 11 (6.861 g, 54.46 mmol). This mixture was stirred for 10 min at room temperature, then 2-iodocyclohex-2-enone 10 (9.3 g, 41.89 mmol) was added all at once, and the mixture was stirred for 24 h at room temperature. The mixture was then diluted with PE and extracted with 5% aqueous NaOH solution x3. The aqueous phase was then back extracted once with diethyl ether and the combined organics were dried (Na2SO4) and concentrated. The residue was passed through a small plug of silica gel to give alcohol 1218 (10.6 g, 96% yield) as a light yellow oil. [α]20D = –34.6° (c = 1.00, CH2Cl2), 95% ee; TLC: Rf (MTBE/PE, 1:4) = 0.54; 1H-NMR (CDCl3) δ 6.57 (m, 1H), 5.78 (m, 1H), 5.19 (d, J = 6.0 Hz, 1H), 5.15 (s, 1H), 2.44 (d, J = 7.2 Hz, 2H), 2.04 (m, 4H), 1.89 (m, 1H), 1.72 (m, 2H); 13C-NMR δ u: 119.0, 111.8, 73.0, 47.2, 34.2, 29.7, 18.9; d: 141.9, 132.9; IR: 3435, 1638, 1436, 1329, 1171, 1081, 981, 918, 764; HRMS calcd for C9H12I (M – OH): 246.9984, obsd: 246.9992. Recovery of catalyst. The basic aqueous phase was acidified with conc. aqueous HCl until the pH was ~1. The aqueous layer was extracted x3 with Et2O and the combined organics were dried (Na2SO4), rotovaped to silica gel and purified through a silica plug to give recovered (R)-BINOL-Br2 (0.690 g, 92% recovered). Determination of enantiomeric excess. An HPLC was used, along with a UV/Vis detector and a Chiracel OD column (150 × 20 mm). 20 μL of a 1 mg/mL sample of alcohol 12 in PE was injected and a linear run (1 mL/min) was employed ranging from 0.2% isopropanol in hexane at 0 min to 0.3% isopropanol in hexane at 70 min. The detector was set at a wavelength of 254 nm. The retention time of the major enantiomer was 19.012 min and of the minor enantiomer was 21.062 min.
(R)-1-Allyl-2-methylcyclohex-2-enol 136
To a slurry of NiCl2·dppp (23 mg, 0.5 mol%) in dry Et2O (40 mL) was added MeMgBr (3.0 M in Et2O, 6.7 mL, 20.04 mmol) via syringe and the mixture was then stirred at room temperature for 10 min under N2. Iodide 12 (2.30 g, 8.71 mmol) was slowly added in 10 mL of Et2O. The reaction was stirred overnight at room temperature. Once the reaction was complete by TLC, Na2SO3 (5 g) was added and the mixture stirred vigorously for 5 min. The atmosphere was purged completely with N2 and the solution was sparged with N2. The solution was then rapidly quenched with saturated aqueous Na2S2O3 under N2 atmosphere. PE was then immediately added to dilute the solution. The layers were separated and the aqueous layer was extracted x3 with Et2O. The solution was then dried (Na2SO4 and Na2SO3) and concentrated. The solution was filtered through a small plug of silica gel to give alcohol 13 (1.196 g, 90% yield) as a light yellow oil. [α]20D = –34.9° (c = 1.00, CH2Cl2); TLC: Rf (MTBE/PE, 1:4) = 0.46; 1H-NMR δ 5.78 (m, 1H), 5.64 (m, J = 1.6 Hz, 1H), 5.15 (m, 1H), 5.11 (t, J = 1.2 Hz, 1H), 2.40 (dt, J = 7.2, 1.2 Hz, 2H), 1.98 (m, 2H), 1.78 (m, 4H), 1.64 (m, 4H); 13C-NMR δ u: 137.0, 118.2, 71.6, 43.6, 35.7, 25.6, 19.1; d: 134.1, 126.6, 17.8; IR: 1639, 1440, 1174, 973, 912 cm−1; HRMS calcd for C10H15(M – OH): 135.1174, obsd: 135.1173.
(R)-3-Allyl-2-methylcyclohexanone 14
Following the literature procedure,6,19 18-crown-6 (4.113 g, 15.58 mmol) was dissolved in dry toluene (80 mL) and t-BuOK (3.489 g, 31.16 mmol) was added and the mixture stirred under N2 for 5 min at room temperature. Alcohol 13 (3.383 g, 22.26 mmol) in 15 mL of dry toluene was added dropwise over 5 min. The mixture was then heated to 85 °C (bath temperature) for 1 h, cooled to room temperature and quenched with saturated aqueous NH4Cl. The aqueous layer was extracted x3 with PE and the combined organic layers were dried (Na2SO4). The toluene/PE solution was then diluted with more PE and placed directly on a column for purification by silica gel chromatography to afford ketone 14 (2.162 g, 64% yield, mixture of trans/cis diastereomers) as a light yellow oil. [α]20D = +10.0° (c = 1.00, CH2Cl2, 2:1 mixture of trans:cis diastereomers); TLC: Rf (MTBE/PE, 1:4) = 0.67; 1H-NMR δ 5.85-5.73 (m, 1H, trans diastereomer), 5.75-5.63 (m, 1H, cis diastereomer), 5.11-5.03 (m, 2H, trans diastereomer), 5.03-4.98 (m, 2H, cis diastereomer), 2.66-2.00 (m, 6H), 1.93-1.41 (m, 4H), 1.05 (d, J = 12.8 Hz, 3H, trans diastereomer), 1.02 (d, J = 12.8 Hz, 3H, cis diastereomer); 13C-NMR (trans diastereomer) δ u: 213.4, 117.1, 41.5, 38.2, 30.3, 25.7; d: 135.5, 49.4, 45.2, 11.9; (cis diastereomer) δ u: 214.5, 116.3, 39.8, 33.7, 26.7, 23.7; d: 136.5, 48.7, 41.9, 11.5; IR: 1711, 1640, 1447, 1221, 999, 914 cm−1; HRMS calcd for C10H17O (M + H): 153.1279, obsd: 153.1283.
(4aR,5R)-5-Allyl-4a-methyl-4,4a,5,6,7,8-hexahydronaphthalen-2(3H)-one 6
To a solution of ketone 14 (1.360 g, 8.95 mmol) in dry toluene (30 mL) was added p-toluenesulfonic acid monohydrate (0.170 g, 0.89 mmol) and a small amount of methylene blue dye as a free radical inhibitor. The mixture was stirred for 5 min at room temperature, then methyl vinyl ketone (1.565 g, 22.37 mmol) was added and the mixture was heated to reflux overnight. The reaction was then cooled to room temperature and quenched with saturated aqueous NaHCO3. The layers were separated and the aqueous layer was extracted x3 with Et2O. The combined organic layers were then dried (Na2SO4) and the ether was removed (setting the rotovap bath no higher than 30 °C). The toluene solution was diluted with PE and placed directly on a column for purification by silica gel chromatography to afford enone 6 (417 mg, 66% brsm, 23% overall) as an orange oil along with 887 mg of ketone 14. [α]20D = +69.8° (c = 1.00, CH2Cl2); TLC: Rf (MTBE/PE, 1:4) = 0.35; 1H-NMR δ 5.86-5.66 (m, 2H), 5.08-4.98 (m, 2H), 2.50-2.08 (m, 6H), 1.92-1.71 (m, 4H), 1.43-1.25 (m, 3H), 1.13 (s, 3H); 13C-NMR δ u: 199.5, 171.8, 116.2, 39.2, 35.4, 34.1, 33.9, 33.5, 26.8, 26.3; d: 137.7, 124.2, 48.4, 16.8; IR: 1673, 1615, 1441, 995, 912 cm−1; HRMS calcd for C14H21O (M + H): 205.1592, obsd: 205.1588.
(4aR,5R)-5-Allyl-4a-methyl-1,2,3,4,4a,5,6,7-octahydronaphthalen-2-ol 16
The enone 6 (0.400 g, 1.96 mmol) was dissolved in acetic anhydride (5 mL) and acetyl chloride (0.769 g, 9.80 mmol) was then added. The mixture was heated to 65 °C for 4 h. The acetic anhydride and acetyl chloride were removed via bulb to bulb distillation under vacuum (heating no higher than 40 °C). The residue was then dissolved in CH2Cl2, rotovaped to silica gel and purified by column chromatography to afford (4aR,5R)-5-allyl-4a-methyl-3,4,4a,5,6,7-hexahydronaphthalen-2-yl acetate 15 along with unidentified contaminants (0.320 g total weight). This crude mixture (which included acetate 15) was dissolved in 13 mL of a 1:1 mixture of THF:t-BuOH. The solution was cooled to 0 °C and stirred for 5 min. NaBH4 (0.395 g, 10.41 mmol) in a 1:1 mixture of H2O:THF (3 mL) was then added dropwise at 0 °C. The reaction mixture was placed in the freezer (-20 °C) for 48 h, then stirred at room temperature for 48 h. The solvent was removed under reduced pressure and 0.5 M aqueous HCl was added to the residue. The aqueous layer was extracted x3 with Et2O. The combined organic layers were dried (Na2SO4) and concentrated. The residue was purified via column chromatography to afford alcohol 16 (206 mg, 50% yield from 6) as a 4:1 mixture of diastereomers (clear oils). [α]20D = –71.0° (c = 1.00, CH2Cl2, 4:1 mixture of diastereomers); TLC: Rf (MTBE/PE, 1:4) = 0.28; 1H-NMR (CDCl3) δ 5.77 (m, 1H), 5.40 (m, 1H), 5.01 (m, 2H), 3.54 (m, 1H), 2.35-1.07 (m, 14H), 0.95 (s, 3H); 13C-NMR δ u: 140.7, 115.5, 42.2, 36.9, 34.6, 31.6, 25,9, 23.0; d: 138.7, 122.8, 71.9, 46.1, 18.2; IR: 1641, 1439, 1357, 1049, 906 cm−1; HRMS calcd for C14H21(M – OH): 189.1643, obsd: 189.1640.
(4aR,5R)-5-Allyl-4a-methyl-1,2,3,4,4a,5,6,7-octahydronaphthalen-2-yl 3-bromobenzoate 17
Alcohol 16 (0.227 g, 1.10 mmol), m-Br benzoic acid (0.244 g, 1.21 mmol), and DMAP (34 mg, 0.275 mmol) were combined in a round bottom flask along with 5 mL CH2Cl2. The solution was cooled to 0 °C and stirred for 5 min. DCC in 2 mL CH2Cl2 was added slowly. The ice bath was removed and the reaction was stirred overnight at room temperature. The solids were filtered out and washed with CH2Cl2. The filtrate was concentrated and the residue rotovaped to silica gel and chromatographed to give ester 17 (353 mg, 94% yield) as a clear oil (4:1 mixture of diastereomers). [α]20D = –39.2° (c = 1.00, CH2Cl2, after crystallization, 14:1 mixture of diastereomers); TLC: Rf (MTBE/PE, 5:95) = 0.69; 1H-NMR (CDCl3) δ 8.19 (t, J = 1.6 Hz, 1H), 8.00 (dt, J = 7.6, 1.2 Hz, 1H), 7.70 (dt, J = 8.0, 1.2 Hz, 1H), 7.34 (t, J = 8.0 Hz, 1H), 5.80 (m, 1H), 5.50 (s, 1H), 5.05 (m, 2H), 4.88 (m, 1H), 2.47 (m, 2H), 2.34 (dd, J = 12.4, 1.2 Hz, 1H), 2.02 (m, 4H), 1.76 (m, 3H), 1.26 (m, 3H), 1.03 (s, 3H); 13C-NMR δ u: 164.7, 139.4, 132.7, 122.4, 115.6, 38.0, 37.0, 36.6, 34.5, 27.8, 26.0, 23.0; d: 138.6, 135.7, 132.6, 129.9, 128.2, 124.1, 75.1, 46.0, 18.2; IR: 1719, 1285, 1255, 1123, 987, 747 cm−1.
Enantioenriched alcohol 18
Ester 17 was recrystallized by dissolving in a minimal amount of boiling MeOH and allowing the solution to stand and cool until crystals formed. The diastereomeric ratio of the isolated crystals ranged from 9:1 to 20:1. [α]20D = –39.2° (14:1 ratio), mp = 77–79 °C. After recrystallization, ester 17 (103 mg, 0.264 mmol) was stirred in 4 mL of MeOH. Then K2CO3 (146 mg, 1.059 mmol) was added and the suspension was stirred at room temperature for 2.5 h. The MeOH was removed under reduced pressure and the residue was rotovaped to silica gel and chromatographed to give enantioenriched alcohol 18 (50 mg, 92% yield) [α]20D = –92.8° (c = 1.00, CH2Cl2, 20:1 diastereomeric ratio) as a clear oil. The spectra matched those of alcohol 16.
(1R,6S,8aR)-1-Allyl-6-(benzyloxy)-8a-methyl-1,2,3,5,6,7,8,8a-octahydronaphthalene 9
The alcohol 18 (90 mg, 0.44 mmol) was dissolved in 5 mL THF and KH(P) (70 mg @ 50% w/w in paraffin, 0.874 mmol) was added. The mixture was stirred at room temperature for 30 min and then tetrabutylammonium iodide (16 mg, 0.044 mmol) was added, followed by BnBr (75 mg, 0.437 mmol). The reaction mixture was stirred for 1 h at room temperature and then rotovaped to silica gel and chromatographed to give benzyl ether 9 (110 mg, 85% yield) as a clear oil. TLC: Rf (MTBE/PE, 1:4) = 0.82; 1H-NMR δ 7.39-7.22 (m, 5H), 5.76 (m, 1H), 5.38 (m, 1H), 5.00 (m, 2H), 4.56 (s, 2H), 3.29 (m, 1H), 2.44 (m, 1H), 2.35-1.00 (m, 12H), 0.95 (s, 3H); 13C-NMR δ u: 140.9, 139.0, 115.6, 70.0, 39.1, 37.3, 36.8, 34.6, 28.4, 26.0, 23.0; d: 138.8, 128.4, 127.6, 127.5, 122.7, 78.6, 46.1, 18.2; IR: 1451, 1357, 1094, 909, 735 cm−1; HRMS calcd for C14H21 (M – OBn): 189.1643, obsd: 189.1647.
1-((1R,6S,8aR)-6-(Benzyloxy)-8a-methyl-1,2,3,5,6,7,8,8a-octahydronaphthalen-1-yl)propan-2-one 19
Pd(quinox)Cl214 (14 mg, 8.5 mol%) and AgSbF6 (40 mg, 24 mol%) were stirred with 3 mL CH2Cl2 for 10 min at room temperature in the dark. t-Butyl hydroperoxide (70% in H2O, 677 mg, 5.27 mmol) was added at room temperature and the mixture stirred for 10 min. The solution was then cooled to 0 °C and stirred for 5 min. Benzyl ether 9 (130 mg, 0.439 mmol) in 1 mL CH2Cl2 was then added slowly over 5 min. The reaction mixture was stirred until the starting material was consumed, as monitored by TLC. The excess TBHP was reduced by adding saturated aqueous Na2S2O3 and stirring for 30 min. The aqueous layer was extracted x3 with CH2Cl2. The combined organics were washed with brine, dried (Na2SO4) and concentrated. The residue was chromatographed to afford ketone 19 (130 mg, 61% yield) as a clear oil. TLC: Rf (MTBE/PE, 1:4) = 0.42; 1H-NMR δ 7.39-7.25 (m, 5H), 5.42 (s, 1H), 4.59 (s, 2H), 3.36-3.25 (m, 1H), 2.59-2.40 (m, 2H), 2.30-2.20 (m, 2H), 2.17 (s, 3H), 2.10-1.09 (m, 9H), 0.98 (s, 3H); 13C-NMR δ u: 208.2, 139.2, 137.9, 69.0, 43.7, 38.0, 35.7, 35.6, 27.2, 24.6, 23.5; d: 127.4, 126.6, 126.4, 121.7, 77.4, 40.6, 29.6, 17.5; IR: 1714, 1453, 1359, 1274, 1094, 738 cm−1; HRMS calcd for C21H29O2 (M+H): 313.2168, obsd: 313.2176.
Methyl 2-diazo-4-((1R,6S,8aR)-6-(benzyloxy)-8a-methyl-1,2,3,5,6,7,8,8a-octahydronaphthalen-1-yl)-3-oxobutanoate 20
NaH (60% in mineral oil, 50 mg, 1.25 mmol) and dimethyl carbonate (187 mg, 2.08 mmol) were stirred in 3 mL of dry DME. The solution was heated to reflux (bath temperature = 95 °C, condenser attached) and 2 drops MeOH were added. Ketone 19 (65 mg, 0.208 mmol) in 2.5 mL DME was added and the mixture was heated to reflux for 12 h. The solution was cooled and 10% aqueous HCl was added. The aqueous layer was extracted x3 with Et2O. The combined organics were dried (Na2SO4) and concentrated. The residue was chromatographed to give methyl 4-((1R,6S,8aR)-6-(benzyloxy)-8a-methyl-1,2,3,5,6,7,8,8a-octahydronaphthalen-1-yl)-3-oxobutanoate (59 mg) as a clear oil. TLC: Rf (MTBE/PE, 1:4) = 0.39; 1H-NMR δ 7.31 (m, 5H), 5.40 (m, 1H), 4.56 (s, 2H), 3.74 (s, 3H), 3.46 (s, 2H), 3.30 (m, 1H), 2.65 (dd, J = 16.8, 2.4 Hz, 1H), 2.45 (m, 1H), 2.33 (dd, J = 16.8, 10.4 Hz, 1H), 2.24 (m, 1H), 2.10-1.06 (m, 9H), 0.96 (s, 3H); 13C-NMR δ u: 202.7, 167.6, 140.1, 138.9, 70.1, 49.5, 44.2, 39.0, 36.8, 36.6, 28.3, 25.6, 24.5; d: 128.4, 127.6, 127.5, 122.7, 78.4, 52.4, 41.4, 18.5; IR: 1742, 1712, 1442, 1314, 1241, 1079 cm−1; HRMS calcd for C23H30O4Na: 393.2042 (M + Na), obsd: 393.2037.
The β-keto ester (55 mg, 0.149 mmol) was dissolved in 3 mL dry MeCN and MsN315(36 mg, 0.297 mmol) was added. The solution was stirred for 5 min at 0 °C and NEt3 (45 mg, 0.445 mmol) was added in 1 mL of MeCN. The solution was stirred overnight at room temperature. The solution was evaporated directly onto silica gel and chromatographed to give diazo ester 20 (50 mg, 65% yield from 19) as a light yellow oil. TLC: Rf (MTBE/PE, 1:4) = 0.44; 1H-NMR δ 7.34 (m, 5H), 5.42 (m, 1H), 4.59 (s, 2H), 3.86 (s, 3H), 3.38-3.26 (m, 1H), 2.97 (dd, J = 16.0, 2.4 Hz, 1H), 2.75 (dd, J = 16.0, 10.8 Hz, 1H), 2.51-1.12 (m, 11H), 1.04 (s, 3H); 13C-NMR δ u: 193.0, 161.8, 140.4, 139.0, 74.2, 70.0, 41.0, 39.0, 37.0, 36.7, 28.3, 25.7, 24.3; d: 128.4, 127.6, 127.5, 122.6, 78.5, 52.2, 42.2, 18.6; IR: 2061, 1748, 1718, 1447, 1319, 1247, 1088 cm−1; HRMS calcd for C23H28O4N2Na (M + Na): 419.1947, obsd: 419.1942.
(3aR,7S,9aR,9bS)-Ethyl 7-(benzyloxy)-9a-methyl-2-oxo-2,3,3a,4,6,7,8,9,9a,9b-decahydro-1H-cyclopenta[a]naphthalene-3-carboxylate 5
Rhodium(II) octanoate (2 mg) was stirred in 2.5 mL CH2Cl2 that had been filtered through anhydrous K2CO3. Diazo ester 20 (50 mg, 0.126 mmol) was dissolved in 3 mL CH2Cl2 (filtered through anhydrous K2CO3) and added dropwise under N2. The solution was stirred for 1 h at room temperature. The reaction mixture was evaporated onto silica gel and chromatographed to afford tricyclic ketone 5 (32 mg, 70% yield) as an off white solid. TLC: Rf (MTBE/PE, 1:4) = 0.20; mp = 107-110 ¼C (recrystallized from MeOH); 1H-NMR δ 7.37-7.29 (m, 5H), 5.43 (m, 1H), 4.59 (s, 2H), 3.77 (s, 3H), 3.33 (m, 1H), 2.91 (d, J = 12.0 Hz, 1H), 2.58-2.18 (m, 6H), 2.05-1.17 (m, 6H), 1.10 (s, 3H); 13C-NMR δ u: 210.1, 169.3, 141.2, 138.7, 70.1, 38.9, 38.7, 38.2, 36.5, 31.4, 27.9; d: 128.4, 127.6, 127.6, 120.8, 78.1, 62.3, 52.5, 48.5, 37.7, 18.4; IR: 1756, 1728, 1438, 1264, 1128, 1094, 736 cm−1; HRMS calcd for C23H29O4 (M + H): 369.2066, obsd: 369.2060.
Supplementary Material
Acknowledgments
We thank John Dykins for high resolution mass spectrometry under the financial support of NSF 054117, Dr. Shi Bai for NMR spectra financially supported by NSF CRIF:MU, CHE 080401, Glenn Yap for X-ray crystallography and the National Institutes of Health (GM060287) for financial support of this work.
Footnotes
Supporting Information: 1H NMR and 13C NMR spectra for all new compounds as well as 50% probability figures and .cif files for 17 are available free of charge via the internet at http://pubs.acs.org.
References
- 1.Keeler RF. Phytochemistry. 1968;7:303–306. [Google Scholar]
- 2.For a review on the topic of cyclopamine, including its chemistry, biology and medicinal relevance, see Heretsch P, Tzagkaroulaki L, Giannis A. Angew Chem Int Ed. 2010;49:3418–3427. doi: 10.1002/anie.200906967. and references within.
- 3.For the initial preparation of the Wieland-Miescher ketone, see: Wieland P, Miescher K. Helv Chim Acta. 1950;33:2215–2228.Hajos ZG, Parrish DR. 2102623. German Patent DE. 1971 Jul 29;3,975,440. USP. 1976 Aug 17; Example 21.Hajos ZG, Parrish DR. Org Synth. 1990Coll. 7:368–372.Bui T, Barbas CF. Tetrahedron Lett. 2000;41:6951–6954.
- 4.For recent preparations of the Wieland-Miescher ketone, see Bradshaw B, Extebarria-Jardi G, Bonjoch J, Viozquez SF, Guillena G, Najera C. Adv Synth Catal. 2009;351:2482–2490.and references within. Guillena G, Najera C, Viozquez SF. Synlett. 2008:3031–30.
- 5.(a) Krafft ME, Cran JW. Synlett. 2005:1263–1266. [Google Scholar]; (b) Pandey G, Balakrishnan M. J Org Chem. 2008;73:8128–8131. doi: 10.1021/jo801761f. [DOI] [PubMed] [Google Scholar]
- 6.Taber DF, Gerstenhaber DA, Berry JF. J Org Chem. 2011;76:7614–7617. doi: 10.1021/jo2013753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Schaus SE, Lou S, Moquist PN. J Am Chem Soc. 2006;128:12660–12661. doi: 10.1021/ja0651308. [DOI] [PubMed] [Google Scholar]; (b) Barnett DS, Moquist PN, Schaus SE. Angew Chem Int Ed. 2009;48:8679–8682. doi: 10.1002/anie.200904715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiso Y, Yamamoto K, Tamao K, Kumada M. J Am Chem Soc. 1972;94:4374–4376. [Google Scholar]
- 9.Taber DF, Nelson CG. J Org Chem. 2006;71:8973–8974. doi: 10.1021/jo061420v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Mase T, Ichita J, Marino JP, Koreeda M. Tetrahedron Lett. 1989;30:2075–2078. [Google Scholar]; (b) Moon SS, Stuhmiller LM, McMorris JC. J Org Chem. 1989;54:26–28. [Google Scholar]; (c) Moon SS, Stuhmiller LM, Chadha RK, McMorris JC. Tetrahedron. 1990;46:2287–2306. [Google Scholar]
- 11.Hua Z, Carache DA, Tian Y, Li YM, Danishefsky SJ. J Org Chem. 2005;70:9849–9856. doi: 10.1021/jo051556d. [DOI] [PubMed] [Google Scholar]
- 12.Huang H, Nelson CG, Taber DF. Tetrahedron Lett. 2010;51:3545–3546. [Google Scholar]
- 13.Taber DF, Ruckle RE. J Am Chem Soc. 1986;108:7686–7693. doi: 10.1021/ja00284a037. [DOI] [PubMed] [Google Scholar]
- 14.(a) Michel BW, Carmelio AM, Cornell CN, Sigman MS. J Am Chem Soc. 2009;131:6076–6077. doi: 10.1021/ja901212h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cornell CN, Sigman MS. J Am Chem Soc. 2005;127:2796–2797. doi: 10.1021/ja043203m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taber DF, Ruckle Robert E, Jr, Hennessy Michael J. J Org Chem. 1986;51:4077–4078. [Google Scholar]
- 16.For a complementary preparation of an angularly-substituted trans-anti-trans 6-6-5 chiron, see Taber DF, Taluskie KV. J Org Chem. 2006;71:2797–2801. doi: 10.1021/jo052656m.Taber DF, Joerger JM. J Org Chem. 2008;73:4155–4159. doi: 10.1021/jo800454w.
- 17.Brown HC, Rachela PJ, Pellechia J. J Org Chem. 1990;55:1868–1874. [Google Scholar]
- 18.Wadamoto M, Yamamoto H. J Am Chem Soc. 2005;127:14556–14557. doi: 10.1021/ja0553351. [DOI] [PubMed] [Google Scholar]
- 19.For rate acceleration of the oxy-Cope rearrangement using 18-crown-6, see Evans D, Golob AM. J Am Chem Soc. 1975;97:4765–4766.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.