

Abstract
Alzheimer’s disease (AD) is clinically characterized with progressive memory loss and cognitive decline. Synaptic dysfunction is an early pathological feature that occurs prior to neurodegeneration and memory dysfunction. Mounting evidence suggests that aggregation of amyloid-β (Aβ) and hyperphosphorylated tau leads to synaptic deficits and neurodegeneration, thereby to memory loss. Among the established genetic risk factors for AD, the ɛ4 allele of apolipoprotein E (APOE) is the strongest genetic risk factor. We and others previously demonstrated that apoE regulates Aβ aggregation and clearance in an isoform-dependent manner. While the effect of apoE on Aβ may explain how apoE isoforms differentially affect AD pathogenesis, there are also other underexplored pathogenic mechanisms. They include differential effects of apoE on cerebral energy metabolism, neuroinflammation, neurovascular function, neurogenesis, and synaptic plasticity. ApoE is a major carrier of cholesterols that are required for neuronal activity and injury repair in the brain. Although there are a few conflicting findings and the underlying mechanism is still unclear, several lines of studies demonstrated that apoE4 leads to synaptic deficits and impairment in long-term potentiation, memory and cognition. In this review, we summarize current understanding of apoE function in the brain, with a particular emphasis on its role in synaptic plasticity and the underlying cellular and molecular mechanisms, involving low-density lipoprotein receptor-related protein 1 (LRP1), syndecan, and LRP8/ApoER2.
Keywords: Alzheimer’s disease, ApoER2, Apolipoprotein E, HSPG, LRP1, synaptic plasticity
ALZHEIMER’S DISEASE
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly. Two neuropathological hallmarks of AD are extracellular Aβ aggregation and intraneuronal neurofibrillary tangles, mainly composed of hyperphosphorylated tau (Golde et al., 2010). Although the exact pathogenic mechanism of AD is still debated, accumulating evidence supports that abnormal aggregation of Aβ in the brain is an early event that triggers pathogenic cascades. Aβ species, including the most abundant Aβ40 and more toxic Aβ42, are produced by two sequential proteolytic cleavages of amyloid precursor protein (APP) by β- and γ-secretases (Kim et al., 2007; McGowan et al., 2005). Genetic mutations responsible for dominant early-onset familial AD are found in APP and presenilin 1 and 2, essential components of γ-secretase complex. In general, such mutations increase Aβ production or increase Aβ42 to Aβ40 ratio. However, only less than 1–3% of AD cases account for familial AD carrying these genetic mutations. Most cases of AD are late-onset and most individuals with late-onset sporadic AD (LOAD) do not have mutations that enhance Aβ production. Therefore, defective Aβ clearance may underlie the increase of Aβ level and AD pathogenesis. Although non-genetic environmental factors, such as age and low education, affect the risk of sporadic AD, the strongest genetic risk factor of LOAD is the APOE ɛ4 allele (Kim et al., 2009a). Mounting evidence suggests that the influence of apoE on AD may be mainly via its effect on clearance and aggregation of Aβ, even though cellular mechanisms for both processes are still unclear.
SYNAPTIC DYSFUNCTION IN ALZHEIMER’S DISEASE
Major clinical characteristic of AD patients is progressive decline in memory with an initial impairment of short-term declarative memory, followed by losses of multiple cognitive functions. Given the fact that synaptic dysfunction is directly related to memory loss, it is important to realize that synaptic loss is an early pathological feature of AD (Selkoe, 2002). In various efforts to find semiquantitative correlations between the progressive cognitive impairment and pathological alterations, such as severity of plaque or neurofibrillary tangles, the best statistical correlation was found between the loss of synaptic density and the degree of cognitive impairment in AD (Terry et al., 1991). Several studies support that loss of synaptic number and density precedes the demise of the neuron (Walsh and Selkoe, 2004). An increasing line of evidence suggests that accumulation of soluble Aβ may be a key factor of synaptic loss and cognitive impairment (Haass and Selkoe, 2007). In addition, synaptic deficit tends to correlate well with soluble Aβ level in various APP transgenic mouse models. Therefore, it appears that lowering soluble Aβ level in the brain is a promising therapeutic strategy to restore synaptic function and prevent cognitive decline.
APOE IN ALZHEIMER’S DISEASE
ApoE4, found in 65–80% of cases of sporadic and familial Alzheimer's disease, increases the occurrence of AD by ∼12 fold in two ɛ4 allele carriers, compared with those with no ɛ4 allele (Farrer et al., 1997). Rare apoE2 allele has a protective effect against LOAD compared to other alleles (Corder et al., 1993). Numerous studies demonstrated the differential effect of apoE isoforms on the clearance and aggregation of Aβ (Kim et al., 2009a). Although the mechanisms underlying the Aβ-mediated pathological effects of apoE are still not clear, mounting evidence suggests that the physical and genetic interactions between apoE and Aβ may have critical roles in AD pathogenesis. Interestingly, apoE colocalizes with amyloid plaques (Namba et al., 1991). ApoE gene deletion in APP transgenic mice leads to dramatically decreased amyloid plaques without affecting Aβ production (Bales et al., 1997). ATP-binding cassette transporter A1 (ABCA1) overexpression in APP transgenic mice also showed a similar phenotype (Wahrle et al., 2008). ABCA1-mediated lipidation of apoE may play a key role in facilitating Aβ degradation by Aβ-degrading enzymes (Jiang et al., 2008). Furthermore, overexpression of apoE receptor, low-density lipoprotein receptor (LDLR), dramatically decreased Aβ deposition in vivo (Kim et al., 2009b). These genetic approaches provide strong support for apoE-targeting therapeutic strategy. Indeed, several groups tested whether targeting apoE with a more practical approach would have functional effects on Aβ and other pathological process in vivo. For example, an Aβ 12–28 synthetic peptide, which contains the apoE-binding site, reduced cerebral Aβ accumulation in animal models of Aβ amyloidosis (Kuszczyk et al., 2013; Pankiewicz et al., 2014; Sadowski et al., 2004; 2006). In addition, apoE mimetic peptide affected APP trafficking and inhibited Aβ production (Minami et al., 2010). Because apoE is tightly related to the regulation of Aβ level and AD pathogenesis, several modes of apoE modulation, such as altering its expression level or status of its lipidation, are pursed as promising therapeutic targets (Bien-Ly et al., 2012; Castellano et al., 2011; Cramer et al., 2012; Kim et al., 2009b; 2011; 2012a; 2012b; Liao et al., 2014; Wahrle et al., 2008;).
GENERAL FUNCTION AND STRUCTURE OF APOE
ApoE transports cholesterol and other lipids in the plasma and brain through cell surface apoE binding receptors (Lane-Donovan et al., 2014). In brain, apoE is the predominant apolipoprotein of high-density lipoprotein (HDL)-like lipoprotein particle, whereas ApoA-1 is major form of plasma HDL. Following the receptor-mediated endocytosis of lipoprotein particles, apoE is either degraded or recycled back to cell surface. ApoE is highly expressed in liver and brain. Within brain, apoE is mainly synthesized by astrocytes and to a limited extent by microglia. Cholesterol is a major component of neuronal and glial membranes and myelin sheath and is produced by glia and neuron. However, mature neurons appear not to produce sufficient cholesterol for membrane synthesis and repair. Therefore, lipid transports may be critical to maintaining neuronal activity (Dietschy and Turley, 2004).
Human ApoE, composed of 299 amino acids, has two separate N-terminal and C-terminal domains joined by a flexible hinge region. The N-terminal domain and C-terminal domain have the receptor-binding and the lipid-binding region, respectively. There are three major isoforms (apoE2, apoE3, and apoE4) in humans, whereas mice express only one type of apoE protein (Kim et al., 2009a). ApoE3 is the most predominant isoform, accounting almost 80% of alleles in general population, while two other forms are minor. Isoforms differ at one or two positions in their primary sequence. ApoE3 has cysteine-112 and arginine-158, whereas apoE4 and apoE2 have arginine and cysteine, respectively at both positions. The differences of one or two amino acids among isoforms significantly alter the apoE’s folding structure and change its ability to bind lipid and receptor (Mahley and Huang, 2012). ApoE3 and apoE4 have a much higher affinity to LDLR, compared with apoE2. So far, more than ten receptors of LDLR family have been identified and most members of the LDLR family bind to apoE. In addition to the lipoprotein endocytic function, some apoE receptors also mediate cellular signaling by binding to other extracellular ligands and intracellular adaptors. For example, reelin-mediated signaling through LRP8/APOER2 and very-low-density-lipoprotein receptor is crucial to synaptic plasticity (Lane-Donovan et al., 2014).
ROLE OF APOE IN SYNAPTIC PLASTICITY AND COGNITION
Differential effects of ApoE isoforms on neurite outgrowth in vitro
Although the differential effects of apoE isoforms on Aβ aggregation and clearance have been extensively studied (Kim et al., 2009a), Aβ-independent pathogenic mechanisms have not been thoroughly investigated. In addition to their effects on Aβ clearance, apoE isoforms affect synaptic plasticity in an isoform-dependent manner. Neuronal injuries are known to induce apoE expression and such increase in apoE level may help to repair the nervous system by delivering cholesterols and lipids to neurons (Mahley and Huang, 2012). Among the processes associated with synaptic plasticity, the effects of apoE isoforms on a neurite outgrowth have been widely investigated. Neurite is defined as any protruding projection from the neuronal cell body, either axon or dendrite. Most studies have demonstrated that apoE3 isoform promotes neurite outgrowth in primary embryonic and adult cortical neurons, as well as neuronal cell lines (Nathan et al., 1994; 2002; Sun et al., 1998). Furthermore, apoE3 was shown to augment neuronal sprouting in a developing organotypic hippocampal slice system (Teter et al., 1999). However, the effect of apoE4 on neurite outgrowth phenotype was not consistent between studies. While some studies reported that apoE4 had detrimental effects on neurite outgrowth (Teter et al., 2002), others found no effects (DeMattos et al., 1998) or even stimulating effects (Puttfarcken et al., 1997).
ApoE isoform dependent synaptic phenotypes in mouse models
Findings from in vitro cell culture studies appear to be consistent with most results obtained with apoE mouse models (Table 1). For example, compensatory sprouting after entorhinal cortex lesion was impaired in transgenic mice expressing apoE4 under the control of human promoter, compared with apoE3 transgenic mice (White et al., 2001). Such synaptic structural differences observed in neurons might explain findings from cognitive performance tests with apoE isoform mice. In transgenic mouse models with neuronal apoE over-expression, apoE4 mice, primarily females, had impairments in learning a water maze test and in vertical exploratory activity, compared with apoE3 mice (Raber et al., 2000). Interestingly, there was no difference in soluble Aβ levels between apoE3/APP and apoE4/APP transgenic mice, suggesting that the differential effect of apoE isoform on cognition may be due to its ability to modulate functional neuronal deficits triggered by Aβ or APP. Using another transgenic mouse model where the apoE isoform is expressed in astrocytes, Hartman et al. also demonstrated that apoE4 mice were more severely impaired in working memory in a radial arm maze test (Hartman et al., 2001). Other previous studies also reported an association between the apoE4 and spine deficits in vivo (Basak and Kim, 2010; Dumanis et al., 2009; Ji et al., 2003; Wang et al., 2005).
Table1.
Summary of APOE transgenic and knock-in mouse models
| Transgene | Promoter | Phenotypes | Age (months) | Note | REF |
|---|---|---|---|---|---|
| Human apoE3,E4 | Human apoE | Impaired neuronal plasticity in apoE4 TG compared to apoE3 TG after entorhinal cortex lesion | 3 | Measured inner molecular layer and immunoreactivity of synaptophysin and GAP-43 | (White et al., 2001) |
| Human apoE3,E4 | NSE | Impairment of WM task and vertical exploratory behavior in apoE4 TG compared to WT and apoE3 TG | 5–6 | More susceptible in female and aged mice | (Raber et al., 1998) |
| Human apoE3,E4 TG crossed with human APP | NSE | ApoE3 TG is less susceptible to the detrimental effect of APP/Aβ on WM performance than apoE4 TG | 6 | More susceptible in female and aged mice | (Raber et al., 2000) |
| Human apoE3,E4 and apoE KO | GFAP | ApoE3,E4 TG and KO were emotionally more reactive than WT. ApoE4 TG showed impaired performance in RAM | 5–14 | Performed other multiple cognitive tests | (Hartman et al., 2001) |
| Human apoE2,E3,E4 and apoE KO | Human apoE | ApoE4 TG had synaptic loss accompanied by an increase in synapse size during aging, compared to the other genotypes | 6–24 | Measured synapse per neuron ratio and synapse size in dentate gyrus | (Cambon et al., 2000) |
| apoE KO | N/A | Infusion of apoE3 and E4 into apoE KO mice improved WM performance, compared to apoE KO | 8 | Infused recombinant apoE3 and E4 | (Masliah et al., 1997) |
| Human apoE3,E4 and apoE KO | Human apoE | ApoE3 expression preserved neuronal integrity in aged mice better than apoE4 expression or apoE KO did | 12 | Performed WM and measured immunoreactivity of synaptophysin and GAP-43 | (Veinbergs et al., 1999) |
| Human apoE2 TG crossed with Tg2576 or PDAPP | Human transferrin | Spine density loss was ameliorated by apoE2 overexpression both in Tg2576 and PDAPP | 2,5,8 (PDAPP) 4.5,11 (Tg2576) | (Lanz et al., 2003) | |
| Human apoE3,E4 | NSE | ApoE3, but not apoE4, expression protected against neuronal damage and the age-dependent neurodegeneration compared to apoE KO | 3–4, 7–9 | (Buttini et al., 1999) | |
| Human apoE2,E3,E4 KI crossed with R1.40 APP | endogenous | All human apoE genotypes had increased brain cholesterol and Aβ levels | 28 days | Differential effect of apoE on cholesterol metabolism and Aβ levels in periphery relative to CNS | (Mann et al., 2004) |
| Human apoE3,E4 KI and Arg-61 apoE | endogenous | The levels of apoE4 and Arg-61 apoE in brain were lower than the that of apoE3 and WT, respectively | 5, 12, 24 | Domain interaction was introduced in Arg-61 apoE mice by gene targeting | (Ramaswamy et al., 2005) |
| Human apoE3/3, E4/4, E3/0 KI | endogenous | Inflammatory response; apoE4/4 > apoE3/0 > apoE3/3 | Inhibition of inflammation depends upon the dose of available apoE3 protein | (Vitek et al., 2007) | |
| Human apoE2/2,E3/3,E 4/4 KI | endogenous | Genotype dependent apoE level; apoE2/2 > apoE3/3 > apoE4/4 | 3–5 | Preferential degradation of apoE4 in astrocyte may lead to reduced secretion and reduced brain apoE4 level | (Riddell et al., 2008) |
| Human apoE3/3,E4/4 KI | endogenous | Reduced excitatory synaptic transmission and dentritic arborization in the amygdala of apoE4 KI compared to E3 KI | 30 weeks | No significant gliosis, amyloid deposition or neurofibrillary tangles in apoE4 KI mice | (Wang et al., 2005) |
| Human apoE2,E3, E4 KI and apoE KO | endogenous | LTP magnitude; WT and apoE3 > apoE KO and apoE2 > apoE4 |
2–4 | (Trommer et al., 2004) | |
| Human apoE3,E4 KI | endogenous | Hippocampal LTP was enhanced at a younger age in apoE 4 KI but not in apoE3 KI | 8 weeks, 6–7 | Showed age dependent effect | (Kitamura et al., 2004) |
| Human apoE3,E4 KI and apoE KO | endogenous | ApoE4 KI, predominantly in female, showed deficit in spatial memory compared to apoE3 KI | 4–5 | (Grootendorst et al., 2005) | |
| Human apoE3, E4 KI and apoE KO | endogenous | ApoE4 KI showed impaired performance in WM and fear conditioning task | 15–18 | Female apoE4 KI was more deteriorated | (Bour et al., 2008) |
| Human apoE2,E3,E4 KI | endogenous | ApoE4 KI showed reduced cortical dendritic spine density and complexity in cortex compared to other KI mice | 4weeks, 3, 12 | Showed age dependent effect | (Dumanis et al., 2009) |
| Human apoE2,E3,E4 KI and apoE KO | endogenous | LTP was inhibited by oligomeric aβ 1–42 with susceptibility of; apoE4 > apoE3 = apoE KO > apoE2 | 2–4 | apoE4: gain of negative function apoE2: loss of protective function | (Trommer et al., 2005) |
| Human apoE3,E4 KI | endogenous | ApoE4 KI exhibited deficits in RAM, which was improved after exercise. | 10–12 | (Nichol et al., 2009) | |
| Human apoE3,E4 KI crossed with human APP | endogenous | apoE4 KI showed reduction in locomotor activity and cognitive impairment compared to apoE3 KI | 6–7 | (Kornecook et al., 2010) | |
| apoE KO | N/A | Impaired memory in apoE KO mice Decreased cholinergic activity in hippocampus and frontal cortex of apoE KO | 6 | (Gordon et al., 1995) | |
| Human apoE2,E3,E4 KI | Endogenous or GFAP | Cognitive performance decreased with age in all isoform mice. ApoE4 KI had better cognitive performance and higher anxiety than apoE2 and apoE3 KI | 6–8, 10–13, 14–22 | Used young, middle-aged and old females | (Siegel et al., 2012) |
REF, reference; NSE, neuron-specific enolases; GFAP, glial fibrillary acidic protein; KO, knock-out; KI, knock-in; TG, transgenic mouse; WM, water maze test; RAM, radial arm maze test; LTP, long term potentiation
Although most studies reported that apoE3 increased synaptic plasticity and had neuroprotective effects to a greater extent, relative to apoE4, the effects of apoE4 were not consistent between studies. Both negative effects and minor beneficial effects on neurites and synaptic functions have been associated with apoE4 (Cambon et al., 2000; Masliah et al., 1997; Veinbergs et al., 1999). In one sense, such conflicting data from in vivo studies reflect the inconsistent results obtained from in vitro experiments (DeMattos et al., 1998; Puttfarcken et al., 1997; Teter et al., 2002). Unlike other two isoforms, apoE2 has not been studies extensively but consistent data appear to support its neuroprotective function (Hudry et al., 2013). In one study, dendritic spine loss in the hippocampus of young APP transgenic mice was ameliorated by apoE2 over-expression with a transferrin promoter, although an age-dependent reduction in spine density was not prevented by apoE2 expression (Lanz et al., 2003).
It has been generally assumed that matching apoE protein levels between isoform mice will circumvent any potential artifacts due to the difference in apoE expression levels. Indeed, since most apoE transgenic mice were generated with promoters from extraneous genes, such as neuron-specific enolase, glial fibrillary acidic protein, thymocyte differentiation antigen 1, or platelet-derived growth factor, transgenic founder lines with equivalent apoE expression levels between isoforms were often selected for further experiments (Table 1). However, later studies strongly suggest that there are considerable differences in apoE protein levels between APOE isoform knock-in mice where apoE transcription is driven by an endogenous APOE promoter (Kim et al., 2009a). Given significant differences in apoE levels under normal physiological conditions, the relevance of findings from transgenic mice with equivalent apoE protein levels becomes less straightforward.
However, some phenotypes observed in apoE transgenic mouse models have been reproduced in apoE knock-in mouse models. For example, apoE4 knock-in mice showed significantly reduced excitatory synaptic transmission and dendritic arborization in the absence of noticeable neuropathological changes (Wang et al., 2005). Consistent with such synaptic deficits, long-term potentiation (LTP) was reduced in apoE4 knock-in mice, compared with apoE3 mice (Trommer et al., 2004), although another study showed young apoE4 mice had enhanced LTP (Kitamura et al., 2004). Differences in experimental conditions, such as high-frequency stimulation versus tetanic stimulation, might contribute to the apparently conflicting results. Surprisingly, LTP in apoE2 knock-in mice was significantly less than apoE3 mice and comparable with apoE4 mice (Trommer et al., 2004). Although it was hypothesized that the weaker binding affinity of apoE2 to apoE receptors may underlie the LTP reduction in apoE2 mice, further studies are needed to clarify the underlying mechanism. Nonetheless, subsequent studies demonstrate that apoE4 knock-in mice, particularly females, have deficits in spatial memory performance tests, compared to apoE3 mice (Bour et al., 2008; Grootendorst et al., 2005). As discussed here, apoE isoforms have differential effects on synaptic function in mouse models. However, whether these findings from mouse models are directly relevant to human AD still remains unknown.
Apparently normal cognition in a human subject without functional APOE gene
Recently, one study provided a unique insight into a longstanding debate in the AD field (Mak et al., 2014). An apparently simple question of whether we should increase or decrease apoE levels for AD therapy has been a controversial topic in the field (Osherovich, 2009). Some groups and pharmaceutical companies are trying to “increase” the levels of apoE, even apoE4, to boost apoE’s ability to transport lipid and clear/degrade Aβ levels, whereas others are developing strategies to “decrease” apoE levels or block apoE4’s toxic effects. These entirely opposite approaches are based on completely different assumption of disease mechanism. Does apoE4 affect AD risk by the gain of detrimental functions or the loss of protective functions? While apoE does have lipid transporter functions under normal physiological conditions, whether its physiological function is directly relevant to its role under pathological AD conditions is still unknown. One of the strongest arguments against the apoE reduction approach was the impairment of cholesterol and other lipid transport in the brain, leading to severe cognitive impairment. The study by Mak et al. (2014) provides one of the first answers to this critical question. The authors identified a patient with no detectable apoE level due to a frameshift mutation in APOE gene. As expected from apoE’s role in lipid transport, this patient had high levels of cholesterol and triglycerides in plasma, leading to familial dysbetalipoproteinemia. However, he did not have any severe abnormality in his cognition and other neurological function. In addition, the levels of Aβ42, total tau, and phospho-tau in his cerebrospinal fluid were normal. These findings suggest that therapeutics aiming to reduce apoE levels in the brain will unlikely cause adversary cognitive effect. While this study apparently provides support for apoE reduction approach, the results do not necessarily provide direct insight into the role of apoE in AD pathogenesis. The elegant study by Mak et al., addressed the normal physiological function of apoE in the brain. Further follow-up study with this patient during his aging and the lipoprotein profiles from his cerebrospinal fluid will dramatically enhance our understanding of apoE in AD pathogenesis.
POTENTIAL CELLULAR AND MOLECULAR MECHANISMS
Regulation of neurite outgrowth by LRP1
Membrane trafficking and cargo delivery along the actin and microtubule cytoskeleton are essential for dendritic and axonal outgrowth and guidance. Neurites are packed with actin, microtubule bundles, and microtubule-binding proteins, such as tau protein. Numerous studies demonstrated that apoE isoforms affect neurite outgrowth in an isoform-dependent manner. The stimulation of neurite outgrowth by apoE is, in part, mediated by LRP1 (Fagan et al., 1996; Holtzman et al., 1995; Narita et al., 1997; Nathan et al., 2002). Attenuation of apoE’s effect on neurite outgrowth by anti-LRP1 antibody or receptor associated protein (RAP) provides strong evidence for the involvement of LRP1. In addition, Shi et al. (2009) demonstrated that activation of LRP1 by other ligands, such as tissue-type plasminogen activator (tPA) or α2-macroglobulin, activates Src family kinase and TrK receptor in neurons (Fig. 1B). Phosphorylation of Trk receptors was necessary for neurite outgrowth. However, it is unknown whether apoE can also activate the same signal transduction pathway triggered by other apoE receptor ligands. If the LRP1-mediated effect is the major mechanism, it is unclear how apoE isoforms would exert the differential effects on neurite sprouting, given the fact that LRP1 has little isoform-specific difference in its binding affinity to apoE3 and apoE4 (Kowal et al., 1990; Ruiz et al., 2005).
Fig. 1.

Signal transduction pathways to regulate neurite outgrowth. (A) Syndecan family member proteins may mediate apoE-dependent neurite outgrowth by interacting with actin filament and Src family kinase (SFK). (B) Activation of LRP1, triggered by ligands such as tissue-type plasminogen activator (tPA) or α-2-macroglobulin, activates SFK and TrK receptor, leading to neurite outgrowth.
Regulation of neurite outgrowth by syndecan family member proteins
Other apoE receptors could also mediate apoE isoform-specific effect on neurite outgrowth by its differential biding affinity to each apoE isoform. For example, apoE is known to bind to heparan sulfate proteoglycan (HSPG) through its N-terminal domain (142–147 amino acid domain) (Libeu et al., 2001). HSPGs consist of a core protein and one or more covalently attached heparin sulfate polysaccharides. They are localized at the basal lamina and in the dendrites. HSPG can take up extra-cellular apoE-containing lipoproteins, independent of LDLR and LRP1. Higher intracellular levels of apoE3, compared to apoE4, in neurons are associated with isoform-specific effects of apoE on neurite outgrowth phenotype. Interestingly, this differential retention of apoE isoforms in neurons is mediated by HSPG (Ji et al., 1998).
Among many HSPGs, syndecan family member proteins appear to be promising candidates that might mediate apoE-isoform dependent neurite outgrowth and dendritic spine maturation. The members of syndecan family are transmembrane HSPGs and their core proteins share a highly conserved cytoplasmic domain. Cytoplasmic domain interacts with actin filaments and tyrosine kinases of the Src family (Fig. 1A). For example, N-syndecan (syndecan-3) acts as a receptor for heparin-binding growth-associated molecule (HB-GAM) and promotes neurite outgrowth (Kinnunen et al., 1996). Upon HB-GAM binding, N-syndecan facilitates neurite outgrowth through a signal transduction system involving tyrosine kinases of the Src family (Kinnunen et al., 1998). In addition, syndecan-2 also induces the maturation of dendritic spines. Deletion of the C-terminal region of syndecan-2 abrogates the effect of syndecan-2 on spine formation (Ethell and Yamaguchi, 1999). Activation of EphB2 receptor tyrosine kinase and the subsequent interaction of syndecan-2 with PDZ domain proteins, such as syntenin and calcium/calmodulin-dependent serine protein kinase, appear to relay the extracellular signals to the intracellular cytoskeleton/signaling complex (Ethell et al., 2001). Interestingly, syndecan-1 is known to bind and internalize apoE-containing lipoprotein particles, independent of LRP1 in certain cell types (Stanford et al., 2009; Wilsie et al., 2006). Presumably, the interaction of apoE with syndecan occurs through electrostatic interactions between positively charged helix 4 domains of apoE and negatively charged sulfate groups in syndecan HSPG (Libeu et al., 2001). Binding of apoE to syndecan may induce conformational changes in syndecan, leading to recruitment of kinases, PDZ-domain proteins or cytoskeletal proteins. The recruitment of cytoplasmic proteins in turn may trigger a signal cascade that regulates actin assembly at the neurites (Fig. 1A).
Regulation of actin dynamics by LRP8/ApoER2
Dendritic spines are small protrusions along the dendrite. They are the primary sites of excitatory input on most neurons. Dendritic spine dysfunction and degeneration are thought to be one of the earliest pathogenic events in AD and correlate well with cognitive deficits in AD patients (Selkoe, 2002). Long-term changes in synaptic activity are associated with alterations in spine number, size, and shape. Based on their shapes, spines are classified as thin, stubby, or mushroom spines. Mushroom spines are considered as relatively more stable connections, while thin spines are dynamic and can respond to synaptic activity (Bourne and Harris, 2008). Synaptic enhancement leads to an enlargement of thin spines into mature mushroom spines. Spines with large heads are generally stable and express large numbers of AMPARs, and contribute to strong synaptic connections. In contrast, spines with smaller heads are less stable and represent weak synaptic connections.
Dendritic spine morphogenesis is dynamically regulated primarily by the polymerization and depolymerization of actin filaments within spines. Dynamic rearrangements of actin are regulated by Rho-like small GTPases, Rho-guanine-nucleotide exchange factors (GEFs) and Rho-GTPase activating proteins (GAPs) (Cingolani and Goda, 2008; Penzes and Jones, 2008). For example, stimulation of NMDA receptors during the LTP induction activates Rho GTPases signaling pathway that regulate myosin II motors (Rex et al., 2010). The destabilization of the actin cytoskeleton by myosin II motor allows the synapse to be rebuilt in order to store new information.
Currently, there is no strong evidence that directly links apoE with the modulation of actin dynamics. However, one study demonstrated that activation of apoE receptor family member, LRP8/ApoER2, inhibits the function of n-cofilin and this subsequently leads to stabilization of actin filaments (Chai et al., 2009) (Fig. 2). Non-muscle cofilin (N-cofilin) is found in the neurons and functions as an actin-depolymerizing protein. Modulation of cofilin activity has been directly linked with alteration in dendritic spine morphogenesis, synaptic transmission and plasticity (Kramar et al., 2009; Shankar et al., 2007). Binding of reelin to ApoER2 induces activation of cytosolic adapter protein Disabled-1 (Dab1) and Src family kinases (Fig. 2). Through a series of phosphorylation events, n-cofilin becomes inactivated and F-actin cannot be depolymerized in the leading processes of migrating neurons. Stabilization of the actin cytoskeleton by the reelin-ApoER2 signaling pathway might occur at the dendritic spine as well. If so, modulation of actin dynamics at spine will directly affect dendritic spine morphogenesis. Interestingly, apoE is known to interfere with reelin binding to ApoER2 (D’Arcangelo et al., 1999). It is tempting to speculate that apoE may mediate its effect on dendritic spine morphogenesis by indirectly affecting reelin signaling (Niu et al., 2008). However, it is unknown whether cofilin and other small GTPase actin regulators directly mediate differential effects of apoE isoforms on spine growth and elimination via apoE receptors. Alternative mechanisms include the differential effect of apoE isoform on glutamate receptor and apoER2 recycling (Chen et al., 2010). Further in vivo study is needed to determine the direct relevance of these findings on AD pathogenesis.
Fig. 2.
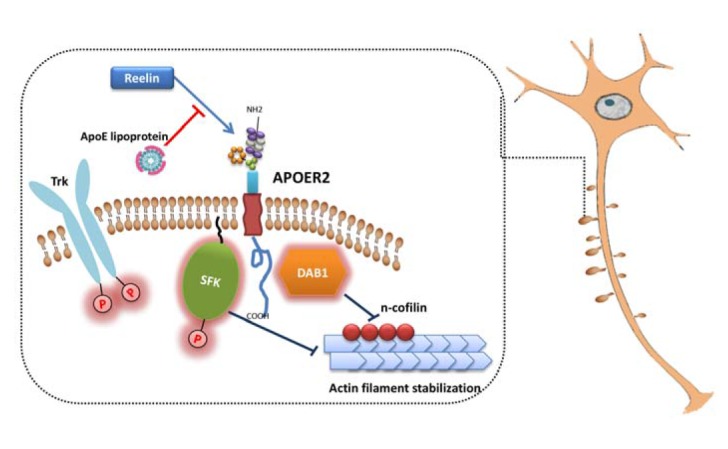
Role of ApoER2 in dendritic spine morphogenesis. Binding of reelin to ApoER2 induces activation of Diabled-1 and SFK. This subsequently inhibits function of n-cofilin, which leads to filament stabilization.
New emerging role of microtubules in dendritic spine morphogenesis
It has been generally believed that microtubules are stable and they are distributed only along the dendritic shaft but not in dendritic spines. Therefore, most studies have investigated the dominant roles of actin in dendritic spine morphogenesis and have ignored the potential function of microtubules in dendritic spine. However, emerging data suggests that neuronal microtubules are dynamic and can transiently move from the dendritic shaft to dendritic spines (Gu et al., 2008; Hu et al., 2008). Neuronal activity-dependent transient translocation of microtubules into dendritic spines may be mechanistically linked with the formation of spine head protrusions and rapid spine dynamics (Jaworski et al., 2009). The direct link between microtubule dynamics and actin dynamics warrant further investigation.
Given the fact that tau, the major component of neurofibrillary tangle, is a microtubule stabilizing protein, there have been several studies aimed to investigate the functional link among apoE, tau, and microtubules. The effect of apoE on microtubule organization may be mediated by alteration in microtubule-associated protein tau. Interestingly, apoE3 binds avidly to tau through the direct interaction between the N-terminal domain of apoE and the microtubule-binding repeat regions of tau, whereas apoE4 does not interact strongly with tau (Strittmatter et al., 1994). However, there is no strong evidence demonstrating apoE’s localization to the neuronal cytosol under normal physiological conditions. Limited data suggest that a particular fragment of apoE (1–272 amino acids) can translocate to cytosol and interacts with cytoskeletal proteins (Chang et al., 2005). The physiological relevance of the interaction between apoE fragment and tau warrants further mechanistic studies. However, apoE could affect tau and microtubule by indirectly modulating the signal transduction pathways that regulate tau kinases’ activity. For example, the addition of apoE2 caused the strong decrease in phosphorylation of GSK 3β and tau, apoE4 the least (Hoe et al., 2006). In addition, RAP treatment abolished the effect of apoE on tau phosphorylation. This finding strongly suggests that the alteration of tau phosphorylation by apoE is mediated by its extracellular interactions with apoE receptors. Although in vitro studies have provided some insights, in vivo studies providing more definitive mechanisms behind the apoE isoform-dependent effects on tau are still lacking. If there is a causal relationship between apoE and tau, it will be critical to understand whether this is due to a direct or indirect interaction.
CONCLUSION
Although there have been numerous reports demonstrating differential roles of apoE isoforms in various aspects of AD pathogenesis, the underlying mechanism for the increased risk of AD by apoE4 is unclear yet. Our studies suggest that the main effect of apoE on risk for AD is via its effect on Aβ clearance and aggregation (Castellano et al., 2011; Kim et al., 2009b; 2011; 2012a; Liao et al., 2014). However, apoE modulates not only Aβ clearance but also other neuropathological changes observed in AD, such as cerebral energy metabolism, neuroinflammation, neurovascular function, neurogenesis, and synaptic plasticity. Given the large overlap between Aβ- and ApoE-mediated pathologic effects, it is difficult to dissect out the exact pathological mechanism mediated by ApoE4 from the purely Aβ-mediated effects in human AD cases. Although the presence of APOE ɛ4 allele does not necessarily cause AD, apoE may cooperate with Aβ to accelerate AD development and progression in an isoform-dependent manner. With a series of failures in Aβ-targeting clinical trials, development of alternative therapeutics for AD is urgently needed. In this regard, targeting apoE may be an attractive therapeutic strategy for AD, either by targeting apoE alone or targeting apoE and Aβ together.
As overviewed in this review, apoE regulates synaptic function in an isoform-dependent manner. In general, apoE4 is associated with detrimental effects on synaptic plasticity, compared with other isoforms. However, there are a significant amount of conflicting findings in isoform-dependent roles of apoE in synaptic plasticity. We cannot exclude the possibility that the absence of more neuroprotective (or less neurotoxic) apoE2 and apoE3, rather than the presence of more neurotoxic apoE4, increases the risk for AD. Clarifying conflicting findings and understanding whether the apoE4-mediated effect on synapse and cognition is due to a gain of detrimental functions or loss of protective functions will have critical implications for the development of apoE-targeting treatments. Therefore, further studies are warranted to determine how apoE affects synaptic plasticity and other AD pathologies.
Acknowledgments
This work was supported by grants from BrightFocus Foundation, Alzheimer’s Association, NIH AG016574, and GHR Foundation to JK.
REFERENCES
- Bales K.R., Verina T., Dodel R.C., Du Y., Altstiel L., Bender M., Hyslop P., Johnstone E.M., Little S.P., Cummins D.J., et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat. Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- Basak J.M., Kim J. Differential effects of ApoE isoforms on dendritic spines in vivo: linking an Alzheimer’s disease risk factor with synaptic alterations. J. Neurosci. 2010;30:4526–4527. doi: 10.1523/JNEUROSCI.0505-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien-Ly N., Gillespie A.K., Walker D., Yoon S.Y., Huang Y. Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J. Neurosci. 2012;32:4803–4811. doi: 10.1523/JNEUROSCI.0033-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bour A., Grootendorst J., Vogel E., Kelche C., Dodart J.C., Bales K., Moreau P.H., Sullivan P.M., Mathis C. Middle-aged human apoE4 targeted-replacement mice show retention deficits on a wide range of spatial memory tasks. Behav. Brain Res. 2008;193:174–182. doi: 10.1016/j.bbr.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Bourne J.N., Harris K.M. Balancing structure and function at hippocampal dendritic spines. Annu. Rev. Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M., Orth M., Bellosta S., Akeefe H., Pitas R.E., Wyss-Coray T., Mucke L., Mahley R.W. Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: isoform-specific effects on neurodegeneration. J. Neurosci. 1999;19:4867–4880. doi: 10.1523/JNEUROSCI.19-12-04867.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambon K., Davies H.A., Stewart M.G. Synaptic loss is accompanied by an increase in synaptic area in the dentate gyrus of aged human apolipoprotein E4 transgenic mice. Neuroscience. 2000;97:685–692. doi: 10.1016/s0306-4522(00)00065-8. [DOI] [PubMed] [Google Scholar]
- Castellano J.M., Kim J., Stewart F.R., Jiang H., DeMattos R.B., Patterson B.W., Fagan A.M., Morris J.C., Mawuenyega K.G., Cruchaga C., et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai X., Forster E., Zhao S., Bock H.H., Frotscher M. Reelin stabilizes the actin cytoskeleton of neuronal processes by inducing n-cofilin phosphorylation at serine3. J. Neurosci. 2009;29:288–299. doi: 10.1523/JNEUROSCI.2934-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S., ran Ma T., Miranda R.D., Balestra M.E., Mahley R.W., Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA. 2005;102:18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Durakoglugil M.S., Xian X., Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc. Natl. Acad. Sci. USA. 2010;107:12011–12016. doi: 10.1073/pnas.0914984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani L.A., Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Corder E.H., Saunders A.M., Strittmatter W.J., Schmechel D.E., Gaskell P.C., Small G.W., Roses A.D., Haines J.L., Pericak-Vance M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Cramer P.E., Cirrito J.R., Wesson D.W., Lee C.Y., Karlo J.C., Zinn A.E., Casali B.T., Restivo J.L., Goebel W.D., James M.J., et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Arcangelo G., Homayouni R., Keshvara L., Rice D.S., Sheldon M., Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–479. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- DeMattos R.B., Curtiss L.K., Williams D.L. A minimally lipidated form of cell-derived apolipoprotein E exhibits isoform-specific stimulation of neurite outgrowth in the absence of exogenous lipids or lipoproteins. J. Biol. Chem. 1998;273:4206–4212. doi: 10.1074/jbc.273.7.4206. [DOI] [PubMed] [Google Scholar]
- Dietschy J.M., Turley S.D. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004;45:1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- Dumanis S.B., Tesoriero J.A., Babus L.W., Nguyen M.T., Trotter J.H., Ladu M.J., Weeber E.J., Turner R.S., Xu B., Rebeck G.W., et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J. Neurosci. 2009;29:15317–15322. doi: 10.1523/JNEUROSCI.4026-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethell I.M., Yamaguchi Y. Cell surface heparan sulfate proteoglycan syndecan-2 induces the maturation of dendritic spines in rat hippocampal neurons. J. Cell Biol. 1999;144:575–586. doi: 10.1083/jcb.144.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethell I.M., Irie F., Kalo M.S., Couchman J.R., Pasquale E.B., Yamaguchi Y. EphB/syndecan-2 signaling in dendritic spine morphogenesis. Neuron. 2001;31:1001–1013. doi: 10.1016/s0896-6273(01)00440-8. [DOI] [PubMed] [Google Scholar]
- Fagan A.M., Bu G., Sun Y., Daugherty A., Holtzman D.M. Apolipoprotein E-containing high density lipoprotein promotes neurite outgrowth and is a ligand for the low density lipoprotein receptor-related protein. J. Biol. Chem. 1996;271:30121–30125. doi: 10.1074/jbc.271.47.30121. [DOI] [PubMed] [Google Scholar]
- Farrer L.A., Cupples L.A., Haines J.L., Hyman B., Kukull W.A., Mayeux R., Myers R.H., Pericak-Vance M.A., Risch N., van Duijn C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349–1356. [PubMed] [Google Scholar]
- Golde T.E., Petrucelli L., Lewis J. Targeting Abeta and tau in Alzheimer’s disease, an early interim report. Exp. Neurol. 2010;223:252–266. doi: 10.1016/j.expneurol.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon I., Grauer E., Genis I., Sehayek E., Michaelson D.M. Memory deficits and cholinergic impairments in apolipoprotein E-deficient mice. Neurosci. Lett. 1995;199:1–4. doi: 10.1016/0304-3940(95)12006-p. [DOI] [PubMed] [Google Scholar]
- Grootendorst J., Bour A., Vogel E., Kelche C., Sullivan P.M., Dodart J.-C., Bales K., Mathis C. Human apoE targeted replacement mouse lines: h-apoE4 and h-apoE3 mice differ on spatial memory performance and avoidance behavior. Behav. Brain Res. 2005;159:1–14. doi: 10.1016/j.bbr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Gu J., Firestein B.L., Zheng J.Q. Microtubules in dendritic spine development. J. Neurosci. 2008;28:12120–12124. doi: 10.1523/JNEUROSCI.2509-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hartman R.E., Wozniak D.F., Nardi A., Olney J.W., Sartorius L., Holtzman D.M. Behavioral phenotyping of GFAP-ApoE3 and -ApoE4 transgenic mice: ApoE4 mice show profound working memory impairments in the absence of Alzheimer’s-like neuropathology. Exp. Neurol. 2001;170:326–344. doi: 10.1006/exnr.2001.7715. [DOI] [PubMed] [Google Scholar]
- Hoe H.S., Freeman J., Rebeck G.W. Apolipoprotein E decreases tau kinases and phospho-tau levels in primary neurons. Mol. Neurodegener. 2006;1:18. doi: 10.1186/1750-1326-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman D.M., Pitas R.E., Kilbridge J., Nathan B., Mahley R.W., Bu G., Schwartz A.L. Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc. Natl. Acad. Sci. USA. 1995;92:9480–9484. doi: 10.1073/pnas.92.21.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Viesselmann C., Nam S., Merriam E., Dent E.W. Activity-dependent dynamic microtubule invasion of dendritic spines. J. Neurosci. 2008;28:13094–13105. doi: 10.1523/JNEUROSCI.3074-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E., Dashkoff J., Roe A.D., Takeda S., Koffie R.M., Hashimoto T., Scheel M., Spires-Jones T., Arbel-Ornath M., Betensky R., et al. Gene transfer of human apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl. Med. 2013;5:212ra161. doi: 10.1126/scitranslmed.3007000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski J., Kapitein L.C., Gouveia S.M., Dortland B.R., Wulf P.S., Grigoriev I., Camera P., Spangler S.A., Di Stefano P., Demmers J., et al. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron. 2009;61:85–100. doi: 10.1016/j.neuron.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Ji Z.S., Pitas R.E., Mahley R.W. Differential cellular accumulation/retention of apolipoprotein E mediated by cell surface heparan sulfate proteoglycans. Apolipoproteins E3 and E2 greater than e4. J. Biol. Chem. 1998;273:13452–13460. doi: 10.1074/jbc.273.22.13452. [DOI] [PubMed] [Google Scholar]
- Ji Y., Gong Y., Gan W., Beach T., Holtzman D.M., Wisniewski T. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer’s disease patients. Neuro Sci. 2003;122:305–315. doi: 10.1016/j.neuroscience.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Jiang Q., Lee C.Y., Mandrekar S., Wilkinson B., Cramer P., Zelcer N., Mann K., Lamb B., Willson T.M., Collins J.L., et al. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Onstead L., Randle S., Price R., Smithson L., Zwizinski C., Dickson D.W., Golde T., McGowan E. Abeta40 inhibits amyloid deposition in vivo. J. Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Basak J.M., Holtzman D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009a;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Castellano J.M., Jiang H., Basak J.M., Parsadanian M., Pham V., Mason S.M., Paul S.M., Holtzman D.M. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A[beta] clearance. Neuron. 2009b;64:632–644. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Jiang H., Park S., Eltorai A., Stewart F., Yoon H., Basak J.M., Finn M.B., Holtzman D.M. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J. Neurosci. 2011;31:18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Eltorai A.E., Jiang H., Liao F., Verghese P.B., Kim J., Stewart F.R., Basak J.M., Holtzman D.M. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J. Exp. Med. 2012a;209:2149–2156. doi: 10.1084/jem.20121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Yoon H., Ramírez C., Lee S., Hoe H., Fernández-Hernando C., Kim J. miR-106b impairs cholesterol efflux and increases Aβ levels by repressing ABCA1 expression. Exp. Neurol. 2012b;235:476–483. doi: 10.1016/j.expneurol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnunen T., Raulo E., Nolo R., Maccarana M., Lindahl U., Rauvala H. Neurite outgrowth in brain neurons induced by heparin-binding growth-associated molecule (HB-GAM) depends on the specific interaction of HB-GAM with heparan sulfate at the cell surface. J. Biol. Chem. 1996;271:2243–2248. doi: 10.1074/jbc.271.4.2243. [DOI] [PubMed] [Google Scholar]
- Kinnunen T., Kaksonen M., Saarinen J., Kalkkinen N., Peng H.B., Rauvala H. Cortactin-Src kinase signaling pathway is involved in N-syndecan-dependent neurite outgrowth. J. Biol. Chem. 1998;273:10702–10708. doi: 10.1074/jbc.273.17.10702. [DOI] [PubMed] [Google Scholar]
- Kitamura H.W., Hamanaka H., Watanabe M., Wada K., Yamazaki C., Fujita S.C., Manabe T., Nukina N. Age-dependent enhancement of hippocampal long-term potentiation in knock-in mice expressing human apolipoprotein E4 instead of mouse apolipoprotein E. Neurosci. Lett. 2004;369:173–178. doi: 10.1016/j.neulet.2004.07.084. [DOI] [PubMed] [Google Scholar]
- Kornecook T.J., McKinney A.P., Ferguson M.T., Dodart J.C. Isoform-specific effects of apolipoprotein E on cognitive performance in targeted-replacement mice overexpressing human APP. Genes Brain Behav. 2010;9:182–192. doi: 10.1111/j.1601-183X.2009.00545.x. [DOI] [PubMed] [Google Scholar]
- Kowal R.C., Herz J., Weisgraber K.H., Mahley R.W., Brown M.S., Goldstein J.L. Opposing effects of apolipoproteins E and C on lipoprotein binding to low density lipoprotein receptor-related protein. J. Biol. Chem. 1990;265:10771–10779. [PubMed] [Google Scholar]
- Kramar E.A., Chen L.Y., Brandon N.J., Rex C.S., Liu F., Gall C.M., Lynch G. Cytoskeletal changes underlie estrogen’s acute effects on synaptic transmission and plasticity. J. Neurosci. 2009;29:12982–12993. doi: 10.1523/JNEUROSCI.3059-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszczyk M.A., Sanchez S., Pankiewicz J., Kim J., Duszczyk M., Guridi M., Asuni A.A., Sullivan P.M., Holtzman D.M., Sadowski M.J. Blocking the interaction between apolipoprotein E and Abeta reduces intraneuronal accumulation of Abeta and inhibits synaptic degeneration. Am. J. Pathol. 2013;182:1750–1768. doi: 10.1016/j.ajpath.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane-Donovan C., Philips Gary T., Herz J. More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron. 2014;83:771–787. doi: 10.1016/j.neuron.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz T.A., Carter D.B., Merchant K.M. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol. Dis. 2003;13:246–253. doi: 10.1016/s0969-9961(03)00079-2. [DOI] [PubMed] [Google Scholar]
- Liao F., Hori Y., Hudry E., Bauer A.Q., Jiang H., Mahan T.E., Lefton K.B., Zhang T.J., Dearborn J.T., Kim J., et al. Anti-ApoE antibody given after plaque onset decreases Abeta accumulation and improves brain function in a mouse model of Abeta amyloidosis. J. Neurosci. 2014;34:7281–7292. doi: 10.1523/JNEUROSCI.0646-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libeu C.P., Lund-Katz S., Phillips M.C., Wehrli S., Hernaiz M.J., Capila I., Linhardt R.J., Raffai R.L., Newhouse Y.M., Zhou F., et al. New insights into the heparan sulfate proteoglycan-binding activity of apolipoprotein E. J. Biol. Chem. 2001;276:39138–39144. doi: 10.1074/jbc.M104746200. [DOI] [PubMed] [Google Scholar]
- Mahley R.W., Huang Y. Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron. 2012;76:871–885. doi: 10.1016/j.neuron.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak A.C., Pullinger C.R., Tang L.F., Wong J.S., Deo R.C., Schwarz J.M., Gugliucci A., Movsesyan I., Ishida B.Y., Chu C., et al. Effects of the absence of apolipoprotein E on lipoproteins, neurocognitive function, and retinal runction. JAMA Neurol. 2014 doi: 10.1001/jamaneurol.2014.2011. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann K.M., Thorngate F.E., Katoh-Fukui Y., Hamanaka H., Williams D.L., Fujita S., Lamb B.T. Independent effects of APOE on cholesterol metabolism and brain Abeta levels in an Alzheimer disease mouse model. Hum. Mol. Genet. 2004;13:1959–1968. doi: 10.1093/hmg/ddh199. [DOI] [PubMed] [Google Scholar]
- Masliah E., Samuel W., Veinbergs I., Mallory M., Mante M., Saitoh T. Neurodegeneration and cognitive impairment in apoE-deficient mice is ameliorated by infusion of recombinant apoE. Brain Res. 1997;751:307–314. doi: 10.1016/s0006-8993(96)01420-5. [DOI] [PubMed] [Google Scholar]
- McGowan E., Pickford F., Kim J., Onstead L., Eriksen J., Yu C., Skipper L., Murphy M.P., Beard J., Das P., et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami S.S., Cordova A., Cirrito J.R., Tesoriero J.A., Babus L.W., Davis G.C., Dakshanamurthy S., Turner R.S., Pak D.T., Rebeck G.W., et al. ApoE mimetic peptide decreases Abeta production in vitro and in vivo. Mol. Neurodegener. 2010;5:16. doi: 10.1186/1750-1326-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba Y., Tomonaga M., Kawasaki H., Otomo E., Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541:163–166. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- Narita M., Bu G., Holtzman D.M., Schwartz A.L. The low-density lipoprotein receptor-related protein, a multifunctional apolipoprotein E receptor, modulates hippocampal neurite development. J. Neurochem. 1997;68:587–595. doi: 10.1046/j.1471-4159.1997.68020587.x. [DOI] [PubMed] [Google Scholar]
- Nathan B.P., Bellosta S., Sanan D.A., Weisgraber K.H., Mahley R.W., Pitas R.E. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264:850–852. doi: 10.1126/science.8171342. [DOI] [PubMed] [Google Scholar]
- Nathan B.P., Jiang Y., Wong G.K., Shen F., Brewer G.J., Struble R.G. Apolipoprotein E4 inhibits, and apolipoprotein E3 promotes neurite outgrowth in cultured adult mouse cortical neurons through the low-density lipoprotein receptor-related protein. Brain Res. 2002;928:96–105. doi: 10.1016/s0006-8993(01)03367-4. [DOI] [PubMed] [Google Scholar]
- Nichol K., Deeny S.P., Seif J., Camaclang K., Cotman C.W. Exercise improves cognition and hippocampal plasticity in APOE epsilon4 mice. Alzheimers Dement. 2009;5:287–294. doi: 10.1016/j.jalz.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu S., Yabut O., D’Arcangelo G. The reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J. Neurosci. 2008;28:10339–10348. doi: 10.1523/JNEUROSCI.1917-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osherovich L. The APOE4 conundrum. SciBX. 2009;2:1–3. [Google Scholar]
- Pankiewicz J.E., Guridi M., Kim J., Asuni A.A., Sanchez S., Sullivan P.M., Holtzman D.M., Sadowski M.J. Blocking the apoE/Abeta interaction ameliorates Abeta-related pathology in APOE2 and 4 targeted replacement Alzheimer model mice. Acta Neuropathol. Commun. 2014;2:75. doi: 10.1186/s40478-014-0075-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P., Jones K.A. Dendritic spine dynamics--a key role for kalirin-7. Trends Neurosci. 2008;31:419–427. doi: 10.1016/j.tins.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttfarcken P.S., Manelli A.M., Falduto M.T., Getz G.S., LaDu M.J. Effect of apolipoprotein E on neurite outgrowth and beta-amyloid-induced toxicity in developing rat primary hippocampal cultures. J. Neurochem. 1997;68:760–769. doi: 10.1046/j.1471-4159.1997.68020760.x. [DOI] [PubMed] [Google Scholar]
- Raber J., Wong D., Buttini M., Orth M., Bellosta S., Pitas R.E., Mahley R.W., Mucke L. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc. Natl. Acad. Sci. USA. 1998;95:10914–10919. doi: 10.1073/pnas.95.18.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber J., Wong D., Yu G.Q., Buttini M., Mahley R.W., Pitas R.E., Mucke L. Apolipoprotein E and cognitive performance. Nature. 2000;404:352–354. doi: 10.1038/35006165. [DOI] [PubMed] [Google Scholar]
- Ramaswamy G., Xu Q., Huang Y., Weisgraber K.H. Effect of domain interaction on apolipoprotein E levels in mouse brain. J. Neurosci. 2005;25:10658–10663. doi: 10.1523/JNEUROSCI.1922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex C.S., Gavin C.F., Rubio M.D., Kramar E.A., Chen L.Y., Jia Y., Huganir R.L., Muzyczka N., Gall C.M., Miller C.A. Myosin IIb regulates actin dynamics during synaptic plasticity and memory formation. Neuron. 2010;67:603–617. doi: 10.1016/j.neuron.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell D.R., Zhou H., Atchison K., Warwick H.K., Atkinson P.J., Jefferson J., Xu L., Aschmies S., Kirksey Y., Hu Y., et al. Impact of Apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J. Neurosci. 2008;28:11445–11453. doi: 10.1523/JNEUROSCI.1972-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz J., Kouiavskaia D., Migliorini M., Robinson S., Saenko E.L., Gorlatova N., Li D., Lawrence D., Hyman B.T., Weisgraber K.H., et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J. Lipid Res. 2005;46:1721–1731. doi: 10.1194/jlr.M500114-JLR200. [DOI] [PubMed] [Google Scholar]
- Sadowski M., Pankiewicz J., Scholtzova H., Ripellino J.A., Li Y., Schmidt S.D., Mathews P.M., Fryer J.D., Holtzman D.M., Sigurdsson E.M., et al. A synthetic peptide blocking the Apolipoprotein E/{beta}-amyloid binding mitigates {beta}-amyloid toxicity and fibril formation in vitro and reduces {beta}-amyloid plaques in transgenic mice. Am. J. Pathol. 2004;165:937–948. doi: 10.1016/s0002-9440(10)63355-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski M.J., Pankiewicz J., Scholtzova H., Mehta P.D., Prelli F., Quartermain D., Wisniewski T. Blocking the apolipoprotein E/amyloid-{beta} interaction as a potential therapeutic approach for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 2006;103:18787–18792. doi: 10.1073/pnas.0604011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D.J. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar G.M., Bloodgood B.L., Townsend M., Walsh D.M., Selkoe D.J., Sabatini B.L. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Mantuano E., Inoue G., Campana W.M., Gonias S.L. Ligand binding to LRP1 transactivates Trk receptors by a Src family kinase-dependent pathway. Sci. Signal. 2009;2:ra18. doi: 10.1126/scisignal.2000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel J.A., Haley G.E., Raber J. Apolipoprotein E isoform-dependent effects on anxiety and cognition in female TR mice. Neurobiol. Aging. 2012;33:345–358. doi: 10.1016/j.neurobiolaging.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford K.I., Bishop J.R., Foley E.M., Gonzales J.C., Niesman I.R., Witztum J.L., Esko J.D. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J. Clin. Invest. 2009;119:3236–3245. doi: 10.1172/JCI38251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter W.J., Saunders A.M., Goedert M., Weisgraber K.H., Dong L.M., Jakes R., Huang D.Y., Pericak-Vance M., Schmechel D., Roses A.D. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1994;91:11183–11186. doi: 10.1073/pnas.91.23.11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Wu S., Bu G., Onifade M.K., Patel S.N., LaDu M.J., Fagan A.M., Holtzman D.M. Glial fibrillary acidic protein-apolipoprotein E (apoE) transgenic mice: astrocyte-specific expression and differing biological effects of astrocyte-secreted apoE3 and apoE4 lipoproteins. J. Neurosci. 1998;18:3261–3272. doi: 10.1523/JNEUROSCI.18-09-03261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry R.D., Masliah E., Salmon D.P., Butters N., DeTeresa R., Hill R., Hansen L.A., Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Teter B., Xu P.T., Gilbert J.R., Roses A.D., Galasko D., Cole G.M. Human apolipoprotein E isoform-specific differences in neuronal sprouting in organotypic hippocampal culture. J. Neurochem. 1999;73:2613–2616. doi: 10.1046/j.1471-4159.1999.0732613.x. [DOI] [PubMed] [Google Scholar]
- Teter B., Xu P.T., Gilbert J.R., Roses A.D., Galasko D., Cole G.M. Defective neuronal sprouting by human apolipoprotein E4 is a gain-of-negative function. J. Neurosci. Res. 2002;68:331–336. doi: 10.1002/jnr.10221. [DOI] [PubMed] [Google Scholar]
- Trommer B.L., Shah C., Yun S.H., Gamkrelidze G., Pasternak E.S., Ye G.L., Sotak M., Sullivan P.M., Pasternak J.F., LaDu M.J. ApoE isoform affects LTP in human targeted replacement mice. Neuroreport. 2004;15:2655–2658. doi: 10.1097/00001756-200412030-00020. [DOI] [PubMed] [Google Scholar]
- Trommer B.L., Shah C., Yun S.H., Gamkrelidze G., Pasternak E.S., Stine W.B., Manelli A., Sullivan P., Pasternak J.F., LaDu M.J. ApoE isoform-specific effects on LTP: blockade by oligomeric amyloid-beta1-42. Neurobiol. Dis. 2005;18:75–82. doi: 10.1016/j.nbd.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Veinbergs I., Mallory M., Mante M., Rockenstein E., Gilbert J.R., Masliah E. Differential neurotrophic effects of apolipoprotein E in aged transgenic mice. Neurosci. Lett. 1999;265:218–222. doi: 10.1016/s0304-3940(99)00243-8. [DOI] [PubMed] [Google Scholar]
- Vitek M.P., Brown C.M., Colton C.A. APOE genotype-specific differences in the innate immune response. Neurobiol. Aging. 2007;30:1350–1360. doi: 10.1016/j.neurobiolaging.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle S.E., Jiang H., Parsadanian M., Kim J., Li A., Knoten A., Jain S., Hirsch-Reinshagen V., Wellington C.L., Bales K.R., et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Invest. 2008;118:671–682. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D.M., Selkoe D.J. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Wang C., Wilson W.A., Moore S.D., Mace B.E., Maeda N., Schmechel D.E., Sullivan P.M. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol. Dis. 2005;18:390–398. doi: 10.1016/j.nbd.2004.10.013. [DOI] [PubMed] [Google Scholar]
- White F., Nicoll J.A., Roses A.D., Horsburgh K. Impaired neuronal plasticity in transgenic mice expressing human apolipoprotein E4 compared to E3 in a model of entorhinal cortex lesion. Neurobiol. Dis. 2001;8:611–625. doi: 10.1006/nbdi.2001.0401. [DOI] [PubMed] [Google Scholar]
- Wilsie L.C., Gonzales A.M., Orlando R.A. Syndecan-1 mediates internalization of apoE-VLDL through a low density lipoprotein receptor-related protein (LRP)-independent, nonclathrin-mediated pathway. Lipids Health Dis. 2006;5:23. doi: 10.1186/1476-511X-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]