

Abstract
Epidemiologic studies have shown that low-density lipoprotein cholesterol (LDL-C) is a strong risk factor, whilst high-density lipoprotein cholesterol (HDL-C) reduces the risk of coronary heart disease (CHD). Therefore, strategies to manage dyslipidemia in an effort to prevent or treat CHD have primarily attempted at decreasing LDL-C and raising HDL-C levels. Cholesteryl ester transfer protein (CETP) mediates the exchange of cholesteryl ester for triglycerides between HDL and VLDL and LDL. We have published the first report indicating that a group of Japanese patients who were lacking CETP had extremely high HDL-C levels, low LDL-C levels and a low incidence of CHD. Animal studies, as well as clinical and epidemiologic evidences, have suggested that inhibition of CETP provides an effective strategy to raise HDL-C and reduce LDL-C levels. Four CETP inhibitors have substantially increased HDL-C levels in dyslipidemic patients. This review will discuss the current status and future prospects of CETP inhibitors in the treatment of CHD. At present anacetrapib by Merck and evacetrapib by Eli Lilly are under development. By 100mg of anacetrapib HDL-C increased by 138%, and LDL-C decreased by 40%. Evacetrapib 500 mg also showed dramatic 132% increase of HDL-C, while LDL-C decreased by 40%. If larger, long-term, randomized, clinical end point trials could corroborate other findings in reducing atherosclerosis, CETP inhibitors could have a significant impact in the management of dyslipidemic CHD patients. Inhibition of CETP synthesis by antisense oligonucleotide or small molecules will produce more similar conditions to human CETP deficiency and may be effective in reducing atherosclerosis and cardiovascular events. We are expecting the final data of prospective clinical trials by CETP inhibitors in 2015.
Keywords: CETP deficiency, cholesteryl ester transfer protein (CETP), HDL & LDL, hyper-HDL-cholesterolemia, inhibitors of CETP
INTRODUCTION
Epidemiologic studies have shown that low-density lipoprotein cholesterol (LDL-C) is a strong coronary risk factor, whilst high-density lipoprotein cholesterol (HDL-C) reduces the risk of coronary heart disease (CHD). Therefore, strategies to manage dyslipidemia in an effort to prevent or treat CHD have primarily attempted at decreasing LDL-C and raising HDL-C levels. Despite evidence showing that treatments with 3-hydroxy-3-methylglutaryl Co-enzyme A (HMG Co-A) reductase inhibitors (statins) reduce LDL-C levels and decrease CHD events, they have not been able to eradicate the residual CHD risk (Fig. 1). Although LDL reduction remains the first priority in lipid management, it is essential to target HDL-C levels. Strategies for intervention against CHD have usually entailed LDL-C lowering therapies using statins (Downs et al., 1998; Pedersen et al., 1994; Sacks et al., 1996; Shepherd et al., 1995). However, for effective prophylactic efforts, the search for better therapeutic targets has recently shifted toward boosting HDL-C levels, based on epidemiologic findings that a low HDL-C is a strong and independent risk factor for CHD (Gordon et al., 1981).
Fig. 1.
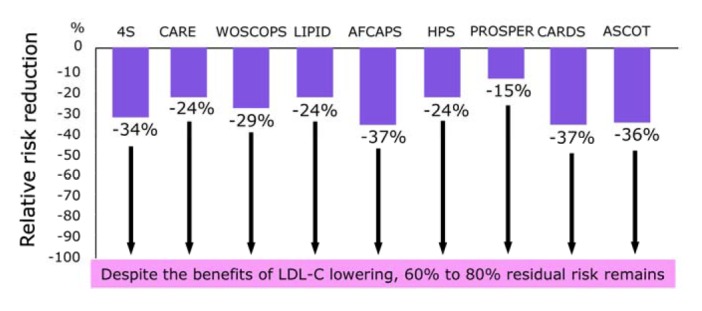
Therapies based on LDL-C lowering by statins reduce the risk of coronary heart disease.
Cholesteryl ester transfer protein (CETP) inhibitors have proven to be effective in achieving both a reduction in LDL-C and an increase in HDL-C. Here we will discuss the current status and future prospects of CETP deficiency and CETP inhibitors in the treatment of CHD. CETP mediates the exchange of cholesteryl-ester (CE) for triglycerides between HDL and very-low-density lipoprotein (VLDL) and LDL (Tall, 1993). It may be proatherogenic if the CETP-mediated VLDL-LDL CE is taken up by arterial macrophages, but antiatherogenic if the CE is returned to the liver through the LDL receptor.
We have published the first report indicating that a group of Japanese patients who were lacking CETP had extremely high HDL-C levels, low LDL-C levels and a low incidence of CHD (Inazu and Mabuchi, 2003). Animal studies, as well as clinical and epidemiologic evidence, have suggested that inhibition of CETP provides an effective strategy to raise HDL-C. Indeed, a number of CETP inhibitors are now in the developing stages of clinical trial. Four CETP inhibitors have substantially increased HDL-C and modestly reduced LDL-C levels in dyslipidemic patients. If larger, long-term, randomized, clinical end point trials, particularly in combination with statins, could corroborate other findings in reducing atherosclerosis, they could have a significant impact in the management of dyslipidemic CHD patients.
LIPOPROTEIN METABOLISM ASSOCIATED WITH CETP, AND CHD
LDL-C lowering therapies using statins are well established in both primary (Downs et al., 1998; Shepherd et al., 1995) and secondary prevention (Pedersen et al., 1994; Sacks et al., 1996) of CHD. However, a large fraction of the population treated with statins still develop coronary events, suggesting a therapeutic limitation of LDL-lowering therapies with statins in CHD (Fig. 1). In the search for additional therapeutic targets, attention has recently shifted toward strategies for increasing HDL-C (Barter et al., 2003; Gordon et al., 1981; Linsel-Nitschke et al., 2005), because most prospective epidemiological studies have clearly shown that a low HDL-C level is a strong and independent risk factor for CHD (Gordon et al., 1981). CETP mediates the exchange of CE for triglycerides between HDL and VLDL-LDL and may be proatherogenic if the CETP mediated VLDL-LDL CE is taken up by arterial macrophages, or may be antiatherogenic if this CE is returned to the liver through the LDL receptor (Tall, 1993)(Fig. 2). CETP inhibitors are a new class of compounds that can inhibit CETP activity and increase HDL-C levels and decrease LDL-C levels. In our previous review, the development and clinical use of CETP inhibitors were described based on findings in clinical and epidemiological studies of genetic CETP deficiency and rabbit models (Inazu and Mabuchi, 2003). Here we describe the current status and future prospects of orally active small compounds of CETP inhibitors as potential new treatment for CHD.
Fig. 2.
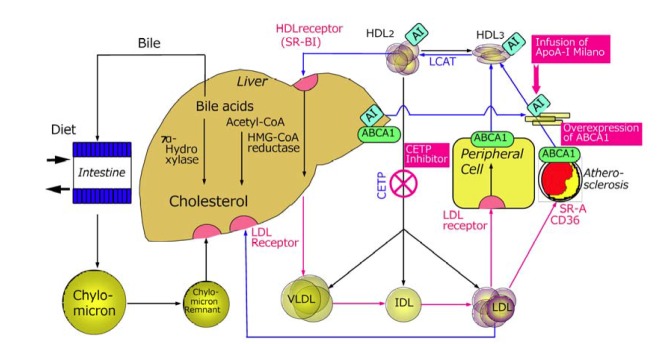
Lipoprotein metabolism in CETP deficiency and principles of increasing HDL-cholesterol
PRINCIPLES OF HDL-CHOLESTEROL RAISING THERAPIES
Numerous epidemiologic studies have shown that LDL-C levels are positively associated with the incidence of CHD, while HDL-C levels are negatively correlated with CHD. Those studies suggested that lowering LDL-C and increasing HDL-C might decrease morbidity and mortality of CHD additively (Brewer, 2004; Wolfe and Rader, 2004). Statins have gotten the triumphs of modern medicine in preventing and treating CHD, and definitely reduced CHD by 24–37% in the primary (Downs et al., 1998; Shepherd et al., 1995), and secondary (Pedersen et al., 1994; Sacks et al., 1996), prevention studies, as well as reducing all-cause of mortality by up to 30% (Fig. 1). Prospective clinical and epidemiological studies have shown that 1 mg/dl of LDL-C decrease reduces 1% of relative incidence of CHD, while 1 mg/dl of HDL-C increase reduces 3% relative incidence of CHD (Gordon et al., 1981). National Cholesterol Education Program (NCEP) guidelines (ATP III) (NCEP report, 2002), although focusing on LDL-C lowering with statin therapy as the primary therapeutic intervention, have also highlighted the importance of HDL-C in the prevention and treatment of CHD (Olsson et al., 2005). Further reductions in CHD incidence can be predicted by increasing HDL-C levels. However, there have been no definite clinical evidences that increasing HDL-C levels reduces CHD incidence. The MIRACLE study has shown in patients with acute coronary syndrome that baseline LDL-C and LDL-C changes after atorvastatin treatment were not predictive of subsequent events (Olsson et al., 2005). In contrast, HDL-C levels at baseline predicted a short-term risk reduction of 1.4% for each 1 mg/dl increment of HDL-C. Thus, low HDL-C levels become crucial issue in clinical practice.
HUMAN MODELS OF ELEVATED HDL-C CONCENTRATIONS AND ATHEROSCLEROSIS
Familial hyperalphalipoproteinemia first reported by Glueck and colleagues, has been known to be associated with longevity (Glueck et al., 1976). However, the basic mechanisms underlying the condition remain unknown. We found unique probands with prominent hyper-HDL-cholesterolemia, showing a CETP deficiency (Koizumi et al., 1985). Discovery of the deficiency in Japanese patients opened the gates of study for the role of CETP in HDL metabolism. In a male subject aged 58 years, lacking CETP due to genetic aberration (Brown et al., 1989, Inazu et al., 1990), the serum lipoprotein profile was similar to that of rodents: deficient CETP, high HDL-C and low LDL-C concentrations (Table 1). The serum HDL-C level was 301 mg/dl. Subsequently, several patients deficient in CETP in Japan and other Southeast Asian countries, such as Korea (Han et al., 1996) and Vietnam, showed a low incidence of CHD. Studies of lipids and lipoprotein metabolism in these individuals have elucidated the role of CETP as well as the routes of human reverse cholesterol transport. CETP deficiency arising from two common specific mutations in the CETP gene occurs frequently (9% in the Japanese population) (Inazu et al., 1990; 1994). There was no cardiovascular disease in 44 homozygous patients with CETP deficiency (Unpublished data). In fact, some Japanese family members reached the age of 90 years (Koizumi et al., 1985). The Japanese population genetically deficient in CETP showed markedly elevated HDL-C, as much as three- to six-fold higher than the normal range (Koizumi et al., 1985). In addition, a modest reduction in LDL-C level was observed in these patients (Fig. 3)(Inazu et al., 1990).
Table 1.
Lipoprotein metabolism and atherogenesis in different animals
| Rat & Mouse | Human | Rabbit | |
|---|---|---|---|
| Atherogenesis | Barely | Intermediate | Easy |
| CETP Activity | Low or no (Hyper-HDL-emia) | Intermediate | High (Low-HDL-emdia) |
| PLTP Activity | High | Intermediate | Low |
| Intestinal ApoB Editing | 90% | > 90% | > 90% |
| 60–70% | None | < 1% | |
| Hepatic ApoB Editing | B-48 mainly | B-100 mainly | B-100 mainly |
| Low-LDL-emia | No Apo A-II | ||
| Low Hepatic Lipase Activity | |||
| Other Findings | High Remnant Lipoprotein |
Fig. 3.
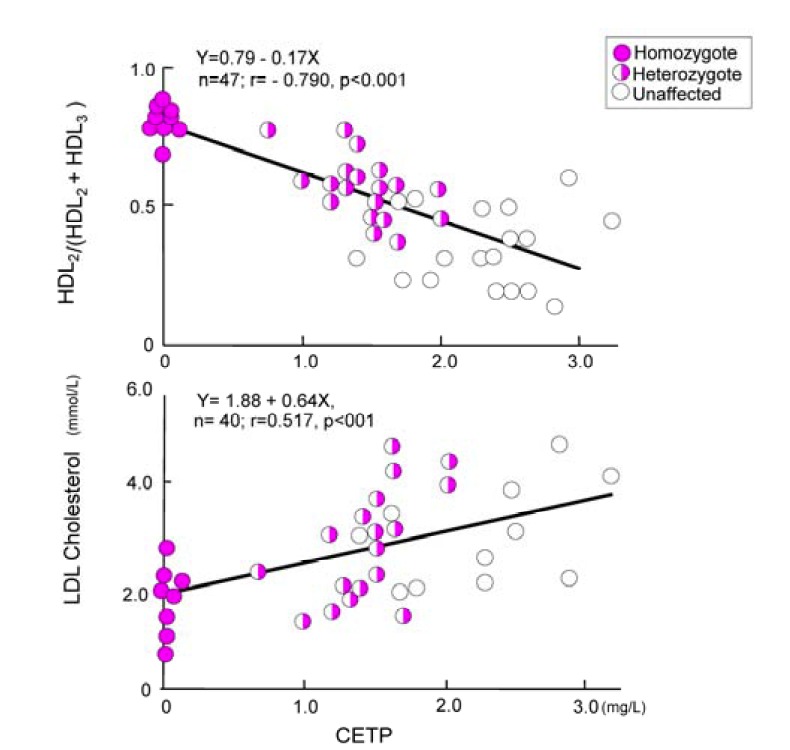
Correlation between CETP and LDL-C, and HDL2/(HDL2 + HDL3) (Inazu et al., 1990).
CETP DEFICIENCY & ATHEROSCLEROTIC CARDIOVASCULAR DISEASE
CETP deficiency is associated with elevated plasma levels of HDL-C and decreased LDL-C levels. Among the major subfractions, HDL2 levels are significantly increased. Recent results from Tall’s laboratory (Matsuura et al., 2006), and also data from our laboratory (Miwa et al., 2009), indicated that SR-B1 or ABCG1-mediated cholesterol efflux to HDL isolated from patients with genetic CETP deficiency is increased when compared with that of control subjects. These observations suggest that CETP inhibition and ABCG1-mediated cholesterol efflux to the larger HDL particles provides an effective clinical mechanism for preventing CHD.
Pure CETP deficiency, with increased HDL-C levels and low levels of LDL-C, without any concurrent dyslipidemic disorders, should substantially decrease the risk of CHD. However, the increased HDL-C concentrations are not strong enough to reverse the CHD risk if the CETP-deficient heterozygotes are complicated with other dyslipidemias, such as familial hypercholesterolemia (FH) with a high risk for premature CHD due to defective LDL receptor function, HDL deficiency and other dyslipidemias. We reported double heterozygotes with FH and CETP deficiency (Haraki et al., 1997). Partial CETP deficiency produced mildly elevated HDL-C levels in heterozygous FH patients. But, the antiatherogenicity of high HDL levels caused by heterozygous CETP deficiency is insufficient to prevent CHD in FH whose LDL-C levels are extraordinarily high. Similarly, patients with combined reduction of CETP and HL only increases the risk for atherosclerotic diseases (Hirano et al., 1995). de Grooth et al. (2004) studied the relationship between CETP levels and atherosclerosis in patients with FH. In FH with compromised LDL receptor function, CETP-induced transfer of CE from HDL to VLDL–LDL for final transport to the liver via the LDL receptor is significantly inhibited. In this context, high CETP can be hypothesized to be especially unfavorable in FH patients since CE-enriched LDL cannot be efficiently cleared. In 281 patients with FH, two statins were compared in a 2-year, randomized, double-blind study (de Grooth et al., 2004). Statins improved the lipoprotein profile in FH patients, but did not sufficiently correct the atherogenic lipid profile in FH patients with the highest CETP levels.
Rodents deficient in CETP are highly resistant to dietary cholesterol-induced atherosclerosis (Ha and Barter, 1982) (Table 1). They have high HDL and low LDL and VLDL concentrations (Table 1). CETP-transgenic mice revealed profoundly reduced HDL-C levels and a concomitant rise in atherosclerotic lesions (Agellon et al., 1991; Marotti et al., 1993). By contrast, animals such as rabbits naturally express high CETP activity and are particularly susceptible to diet-induced atherosclerosis. Numerous animal studies support the hypothesis that inhibition of CETP activity has a beneficial effect on HDL-C levels and attenuate atherosclerosis (Sikorski, 2006).
Molecular approaches including the administration of TP-2, a CETP-neutralizing monoclonal antibody, to hamsters (Gaynor et al., 1994); antisense oligodeoxynucleotides against CETP to rabbits (Sugno et al., 1998), and CETP peptide-based vaccine to rabbits (Rittershaus et al., 2000), reduced plasma CETP activity. The results, raised HDL-C levels and lowered LDL-C, reduced aortic atherosclerosis, underscoring the need for therapeutically efficacious CETP inhibitors.
CETP DEFICIENCY: GENETIC & EPIDEMIOLOGICAL SURVEYS
There is a lack of consensus over the prevalence of cardiovascular disease in the relatively small number of homozygous CETP-deficient subjects, limited to Japan and other Asian countries. Therefore, most efforts to assess the relationship between CETP deficiency and CHD have focused on heterozygotes. The cross-sectional report that subjects in the Honolulu Heart Program (HHP) with partial CETP deficiency had an apparent increase in CHD suggested that lowered CETP levels might not be atheroprotective (Zhong et al., 1996). However, more recent analysis of prospective data from the same group revealed that in a 7-year follow up of 2340 men between 71 and 93 years in the HHP, the age-adjusted CHD incidence rates were significantly lower in men with high versus low HDL-C levels (Curb et al., 2004).
After adjustment for age, hypertension, smoking and total cholesterol, the relative risk of CHD for those with HDL-C levels of 60 mg/dl or more, compared with those with HDL-C levels less than 40 mg/dl, was 0.6. Men with a CETP mutation had the lowest rates of CHD, although this was not statistically significant. These data indicate that HDL-C remains an important risk factor for CHD in the elderly. Whether a CETP mutation offers additional protection against CHD warrants further investigation (Moriyama et al., 1998). The results of the HHP are consistent with those of a large population study of Japanese men and women, suggesting that an HDL-C of 60 mg/dl or greater is protective regardless of whether it is associated with CETP deficiency (Moriyama et al., 1998). In a community-based survey in Japan, Moriyama and colleagues reported a decreased incidence of CHD among individuals with very high HDL-C levels (> 80 mg/dl) as well as mild-to-moderate HDL elevations (60–79 mg/dl). This occurred irrespective of the presence of CETP deficiency, and was particularly the case in men (Moriyama et al., 1998). However, this does not directly indicate that the CETP heterozygous state is protective.
Human studies of patients with CETP deficiency have provided equally conflicting and confusing data (Hirano et al., 1997; Moriyama et al., 1998). Our studies indicated that elevated levels of plasma HDL-C in CETP-deficient subjects strongly suggest that the absence of CETP activity may exert a protective effect against atherosclerosis (Inazu et al., 1990; Moriyama et al., 1998). Hirano and coworkers have shown that reduced CETP function in conjunction with reduced HL activity is associated with an increased risk for CHD (Hirano et al., 1997). This indicates that the metabolic milieu of the individual is a major factor underlying the CETP effect on atherosclerosis.
RATIONALE OF DEVELOPING CETP INHIBITORS AND ITS PHENOTYPIC SIMILARITY TO HUMAN CETP DEFICIENCY
Cholesterol is an essential molecule for the structure and function of the cells and tissues in humans as well as animals. All cells can synthesize cholesterol by themselves, and in addition cholesterol is absorbed from diet and transported in the blood as lipoprotein cholesterols, which are taken up through the specific lipoprotein receptors and other mechanisms into liver and peripheral tissues. The cholesterol transported into the peripheral tissue is never degraded there, and usually is transported back to the liver, where a part of the cholesterol is degraded into the bile acids or most of them can be reutilized as constituent of VLDL and secreted into the blood (Fig. 2). HDL-C is involved in transport of cholesterol away from arterial walls to the liver to be subsequently excreted either directly or after conversion to bile acids. The RCT from peripheral tissues to the liver is limited by receptor-mediated VLDL-remnant uptake via LDLR and LDLR-related protein (LRP). Therefore, if uptake of remnant particles is saturated, CE-enriched VLDL will act as an LDL precursor or CE donor to LDL, resulting in a proatherogenic phenotype (Inazu et al., 1994).
Cholesterol and bile acids in the bile are reabsorbed into the portal vein (entero-hepatic cycle). In humans as well as other animals, recycling pathways of cholesterol have evolved and utilize cholesterol highly efficiently. In humans the following cholesterol pathways have evolved, which make them resistant to cholesterol deficiency by starvation.
Efficient intestinal absorption and reabsorption of dietary cholesterol,
Smooth secretion of VLDL from liver and specific uptake into the peripheral organs through receptors (cholesterol transport system),
No degradation of sterol structure in the peripheral organs,
Development of LDL-receptor reducing system (i.e. PCSK9, et al.) in the liver.
After industrial and agricultural revolutions food situation remarkably changed from malnutrition (cholesterol deficiency) to its excess in highly industrialized countries. Therefore, therapies blocking the physiological pathways are the principles of management of dyslipidemic patients by restricting dietary cholesterol content, blocking the cholesterol and bile acids absorption, blocking biosynthesis of cholesterol by HMG-CoA reductase inhibitors, and blocking the reutilization of CE by blocking CETP.
HDL metabolism is therefore a major emerging target for drug discovery. Consequently, CETP inhibitors may be particularly beneficial for CHD patients whose plasma CETP protein exceeds levels that are required to maintain the normal RCT (TPL1) process. Rationale of developing CETP inhibitors is summarized in Table 2.
Table 2.
Rationale of developing CETP inhibitors
| 1 | Naturally CETP-deficient mice do not produce atherosclerosis, but CETP-transgenic mice produce severe atherosclerosis. |
| 2 | Homozygous CETP-deficient patients do not show atherosclerotic cardiovascular disease. |
| 3 | High HDL-cholesterol subjects due to CETP-deficiency do not show atherosclerotic cardiovascular disease in Japan and in Hawaii. |
| 4 | In Japan the frequency of CETP-deficiency is very high, but the incidence of atherosclerotic cardiovascular disease is low. |
| 5 | CETP-inhibitor increase HDL levels and decrease LDL levels as observed in homozygous CETP-deficiency. |
| 6 | In human CETP-deficiency, plasma CETP mass as well as CETP activity are decreased, but in subjects treated by CETP inhibitors increase CETP complex but decrease CETP activity. |
| 7 | Anti-synthetic drug of CETP like antisense oligonucleotide inhibitor of human CETP will reduce plasma CETP mass as well as CETP activity, and decrease atherosclerosis by enhancing fecal cholesterol excretion. |
Until now, a number of potent, selective, and orally active CETP inhibitors in preclinical models of atherosclerosis were identified (Fig. 4). Similar to simvastatin, JTT-705 (dalcetrapib) reduced the area of atherosclerotic lesions by 80% in rabbits (Okamoto et al., 2000). However, rabbits with severe hypercholesterolemia, treated with various dosages of the JTT-705, showed no effect on aortic cholesterol content, despite a 70% inhibition of CETP activity and a concomitant 200% increase of HDL-C levels. Instead, TG and cholesterol levels increased, which may explain the lack of effect on atherosclerosis (Huang et al., 2002). Indeed, the effect of CETP inhibition on atherosclerosis is still a matter of debate (Tall, 1993) and pharmacological intervention depends on individual characteristics.
Fig. 4.
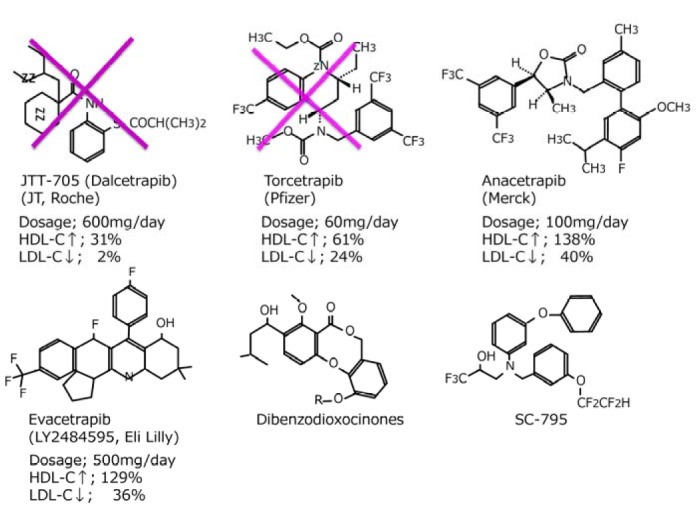
Structure and effects of CETP inhibitors.
Such patients are at an increased risk for cardiovascular diseases (Brousseau et al., 2004). Therefore, partial inhibition of CETP might represent a therapeutic approach for raising the plasma concentration of HDL-C. However, CETP-deficient, homozygous patients and animals are healthy in spite of the complete absence of CETP activity.
There are 3 compounds which have been developed in clinical trials; these are torcetrapib (CP-529,414), anacetrapib (MK-859) and dalcetrapib (JTT-705/ Roche R1658). The phase III of torcetrapib was terminated on December, 2006 due to unexpected excess of mortality in the torcetrapib arm (Nissen et al., 2007). The early termination was partially explained by hypertension due to aldosterone excess. However, role of CETP inhibition on the increased mortality was not clearly shown, but it may be rather associated with infection or malignancy than coronary heart disease. However, vascular end-points of carotid atherosclerosis and coronary atheroma volume assessed by intravascular ultrasound showed no further benefit from torcetrapib in a background of atorvastatin despite increased levels of HDL and further decreased levels of LDL and TG (Kastelein et al., 2007; Nissen et al., 2007).
CLINICAL EFFICACY OF CETP INHIBITORS
At least two promising CETP inhibitors are in the preliminary stages of clinical development: anacetrapib and evacetrapib (Table 3, Fig. 4). There is an urgent need for larger, long-term, randomized, clinical end point trials to investigate their safety and efficacy in atherosclerosis.
Table 3.
Percent changes from baseline levels of lipoprotein and apoprotein by different CETP inhibitors
| % Changes from baseline by different CETP inhibitors | ||||
|---|---|---|---|---|
|
| ||||
| Dalcetrapib 600 mg | Torcetrapib 60 mg | Anacetrapib 100 mg | Evacetrapib 500 mg | |
| HDL-C | +31 | +61 | +138 | +132 |
| Apo-A1 | +11 | +25 | +45 | +50 |
| LDL-C | −2 | −24 | −40 | −40 |
| Apo-B | +4 | −12 | −21 | −26 |
| TG | −3 | −9 | −7 | −20 |
| TC | +8 | +4 | +16 | +16 |
The clinical data due to JTT-705 have been obtained in only one clinical study conducted by Kastelein et al. (2007). In October 2004, Japan Tobacco announced a joint Phase III clinical development program for JTT-705 in partnership with Roche to evaluate its therapeutic utility in reducing cardiovascular events. If approved, JTT-705 will probably be prescribed in combination with one of the statins for a competitive advantage. However, safety and efficacy issues of such combination therapy remain to be addressed.
In May 2012, Hoffman-La Roche announced that the clinical endpoint study dal-OUTCOMES was terminated early, after the Independent Data and Safety Monitoring Board had recommended stopping the trial. The second interim analysis of the study showed that dalcetrapib lacked clinically meaningful efficacy. The dal-OUTCOMES study was a randomized placebo controlled trial that was designed to include approximately 156,600 patients with stable CHD following an acute coronary syndrome.
The CETP inhibitor (torcetrapib) increased with escalating dose, leading to elevations of HDL-C of 16–91%. Increases in CETP mass were consistent with the proposed mechanism of inhibition. Total cholesterol did not change significantly because of a reduction in non-HDL, including a 21–42% lowering of LDL-C at the higher doses. Apo A-I and E were elevated 27 and 66%, respectively, and Apo B was reduced 26% with 120 mg twice daily. These effects of CETP inhibition resemble those observed in partial CETP deficiency.
Brousseau et al. (2004) conducted a single-blind, placebo-controlled study to examine the effects of torcetrapib on plasma lipoprotein levels in 19 subjects with low levels of HDL-C (< 40 mg/dl), nine of whom were also treated with 20 mg of atorvastatin daily. Treatment with 120 mg of torcetrapib daily increased plasma concentrations of HDL-C by 61% (p < 0.001) and 46% (p = 0.001) in the atorvastatin and non-atorvastatin cohorts, respectively, and treatment with 120 mg twice daily increased HDL-C by 106% (p < 0.001). Torcetrapib also reduced LDL-C levels by 17% in the atorvastatin cohort (p = 0.02). However, torcetrapib therapy resulted in an increased risk of mortality and morbidity of unknown mechanism. In December 2006, the study was terminated early due to increased risk of death from any cause, namely 93 deaths in the torcetrapib treated group versus 59 in the control group. The major cardiovascular event rate was also higher, with 464 events in the torcetrapib group versus 373 events in the control group (Kastelein et al., 2007; Nissen et al., 2007).
Despite CETP inhibition’s efficacy in raising HDL in humans, there is no unequivocal evidence directly supporting the benefits of CETP inhibition for atherosclerosis. Large, double-blind, placebo-controlled, randomized controlled trials of CETP inhibitors should offer interesting new data for pharmacologic intervention in atherosclerosis and cardiovascular events.
FUTURE PERSPECTIVE
At present anacetrapib by Merck and evacetrapib by Eli Lilly are under development (Table 3). By 100 mg of anacetrapib HDL-C increased by 138%, and LDL-C decreased by 40% by anacetrapib alone. When anacetrapib was given to add-on the atorvastatin 20mg, the effects were same in monotherapy and add-on therapy (Table 3, Fig. 4). Effects of evacetrapib being developed by Eli-Lilly also showed dramatic 132% increase of HDL-C while LDL-C decreased by 40%. Percent changes of HDL-C and LDL-C by different CETP inhibitors are shown in Table 3.
Schaefer published a review article entitled “Effects of cholesteryl ester transfer protein inhibitors on human lipoprotein metabolism: why have they failed in lowering CHD risk?” (Schaefer, 2013) He suggested that as CETP inhibitors form a complex between themselves, CETP, and HDL particles, which may interfere with the many physiologic functions of HDL, including reverse cholesterol transport. Available data would suggest that CETP inhibitors would fail as lipid-altering medications to reduce CHD risk because of interference with normal human HDL metabolism.
Recently an excellent original paper was published in J Lipid Res by Bell III from ISIS company, entitled “Antisense oligonucleotide inhibition of cholesteryl ester transfer protein enhances RCT in hyperlipidemic, CETP transgenic, LDLr−/− mice” (Bell et al., 2013). The purpose of these studies was to evaluate an antisense oligonucleotide (ASO) inhibitor of CETP and to compare and contrast its pharmacological effects to those of the anacetrapib. The small molecule inhibitor, SMI, such as anacetrapib, are reported to bind and inactivate CETP associated with the HDL particle, whereas the CETP ASO specifically targets and degrades CETP mRNA, thus significantly lowering the amount of protein that is synthesized in the liver (Sugano et al., 1998). This difference in mechanism of action could have significant implications on pharmacology because the HDL-SMI complex could affect HDL function and interfere normal HDL metabolism and functions. ASO-mediated reductions in CETP mRNA were associated with lower CETP mRNA and CETP protein. However, by anacetrapib CETP mRNA and protein were increased. Thus, treatment by ASO is supposed to produce similar phenotypic features to human CETP deficiency (Inazu et al., 1990). In CETP tg LDLr-/- mice, antisense oligonucleotide decreased CETP protein, but by anacetrapib CETP protein was greatly increased and made a complex with HDL fraction. Fecal excretion of 3H-cholesterol was increased in CETP-ASO, but not by anacetrapib. In human CETP-deficiency, plasma CETP mass as well as CETP activity are decreased, but in subjects treated by CETP inhibitors increase CETP complex but decrease CETP activity. Aortic FC and CE accumulations were proportionally increased with CETP mRNA concentrations.
Both drugs (CETP-ASO) and small molecule inhibitor provided comparable reductions in total plasma cholesterol, decreases in CETP activity, and increases in HDL cholesterol. Additionally, the ASO-mediated reductions in CETP mRNA were associated with less accumulation of aortic cholesterol (Sugano et al., 1998). These preliminary findings suggest that CETP ASOs may represent an alternative means to inhibit that target and to support their continued development as a treatment for cardiovascular disease in man.
DISCUSSION
Our discovery that genetic deficiency in CETP is associated with elevated levels of HDL-C has led to the concept that CETP inhibition might be a therapeutic strategy for raising HDL-C levels.
To date, there is considerable evidence to support the concept of CETP inhibition as a therapeutically efficacious approach to raise HDL-C.
Low LDL-C levels in addition to high HDL-C levels are important for antiatherogenesis of CETP deficiency.
However, large-scale, randomized, clinical trials are ultimately required to assess the impact of CETP inhibitors on atherosclerosis and cardiovascular events.
Just as the statins revolutionized the treatment of hyperlipidemic CHD patients, these CETP inhibitors have the potential to dramatically impact the management of CHD patients, particularly by the combination with statins.
CONCLUSION
Inhibition of CETP synthesis by ASO or small molecules will produce more similar conditions to human CETP deficiency and may be effective in reducing atherosclerosis and cardiovascular events.
REFERENCES
- Agellon L.B., Walsh A., Hayek T., Moulin P., Jiang X.C., Shelanski S.A., Breslow J.L., Tall A.R. Reduced high-density lipoprotein cholesterol in human cholesteryl ester transfer protein transgenic mice. J. Biol. Chem. 1991;266:10796–10801. [PubMed] [Google Scholar]
- Barter P.J., Brewer H.B., Jr., Chapman M.J., Hennekens C.H., Rader D.J., Tall A.R. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003;23:160–167. doi: 10.1161/01.atv.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- Bell T.A., Graham M.J., Lee R.G., Mullick A.E., Fu W., Norris D., Crooke R.M. Antisense oligonucleotide inhibition of cholesteryl ester transfer protein enhances RCT in hyperlipidemic, CETP transgenic, LDLr-/- mice. J. Lipid Res. 2013;54:2647–2657. doi: 10.1194/jlr.M036509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer H.B., Jr. Increasing HDL cholesterol levels. N. Engl. J. Med. 2004;350:1491–1494. doi: 10.1056/NEJMp048023. [DOI] [PubMed] [Google Scholar]
- Brousseau M.E., Schaefer E.J., Wolfe M.L., Bloedon L.T., Digenio A.G., Clark R.W., Mancuso J.P., Rader D.J. Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. N. Engl. J. Med. 2004;350:1505–1515. doi: 10.1056/NEJMoa031766. [DOI] [PubMed] [Google Scholar]
- Brown M.L., Inazu A., Hesler C.B., Agellon L.B., Mann C., Whitlock M.E., Marcel Y.L., Milne R.W., Koizumi J., Mabuchi H., et al. Molecular basis of lipid transfer protein deficiency in a family with increased high-density lipoproteins. Nature. 1989;342:448–451. doi: 10.1038/342448a0. [DOI] [PubMed] [Google Scholar]
- Curb J.D., Abbott R.D., Rodriguez B.L., Masaki K., Chen R., Sharp D.S., Tall A.R. A prospective study of HDL-C and cholesteryl ester transfer protein gene mutations and the risk of coronary heart disease in the elderly. J. Lipid Res. 2004;45:948–953. doi: 10.1194/jlr.M300520-JLR200. [DOI] [PubMed] [Google Scholar]
- de Grooth G.J., Smilde T.J., Van Wissen S., Klerkx A.H., Zwinderman A.H., Fruchart J.C., Kastelein J.J., Stalenhoef A.F., Kuivenhoven J.A. The relationship between cholesteryl ester transfer protein levels and risk factor profile in patients with familial hypercholesterolemia. Atherosclerosis. 2004;173:261–267. doi: 10.1016/j.atherosclerosis.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Downs J.R., Clearfield M., Weis S., Whitney E., Shapiro D.R., Beere P.A., Langendorfer A., Stein E.A., Kruyer W., Gotto A.M., Jr, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279:1615–1622. doi: 10.1001/jama.279.20.1615. [DOI] [PubMed] [Google Scholar]
- Gaynor B.J., Sand T., Clark R.W., Aiello R.J., Bamberger M.J., Moberly J.B. Inhibition of cholesteryl ester transfer protein activity in hamsters alters HDL lipid composition. Atherosclerosis. 1994;110:101–109. doi: 10.1016/0021-9150(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Glueck C.J., Gartside P., Fallat R.W., Sielski J., Steiner P.M. Longevity syndromes: familial hypo-β and familial hyper-α-lipoproteinemia. J. Lab. Clin. Med. 1976;88:941–957. [PubMed] [Google Scholar]
- Gordon T., Kannel W.B., Castelli W.P., Dawber T.R. Lipoproteins, cardiovascular disease, and death. The Framingham study. Arch. Intern. Med. 1981;141:1128–1131. [PubMed] [Google Scholar]
- Ha Y.C., Barter P.J. Differences in plasma cholesteryl ester transfer activity in sixteen vertebrate species. Comp. Biochem. Physiol. B. 1982;71:265–269. doi: 10.1016/0305-0491(82)90252-8. [DOI] [PubMed] [Google Scholar]
- Han K.H., Park Y.B., Chae I.H., Kim H.S., Sohn D.W., Oh B.H., Lee MM, Choi Y.S., Seo J.D., Lee Y.W., et al. CETP (cholesteryl esters transfer protein) deficiency caused by genetic mutation in the CETP gene in normal Korean population. Korean Circ. J. 1996;26:500–506. [Google Scholar]
- Haraki T., Inazu A., Yagi K., Kajinami K., Koizumi J., Mabuchi H. Clinical characteristics of double heterozygotes with familial hypercholesterolemia and cholesteryl ester transfer protein deficiency. Atherosclerosis. 1997;132:229–236. doi: 10.1016/s0021-9150(97)00093-2. [DOI] [PubMed] [Google Scholar]
- Hirano K., Yamashita S., Kuga Y., Sakai N., Nozaki S., Kihara S., Arai T., Yanagi K., Takami S., Menju M., et al. Atherosclerotic disease in marked hyperalphalipoproteinemia. Combined reduction of cholesteryl ester transfer protein and hepatic triglyceride lipase. Arterioscler. Thromb. Vasc. Biol. 1995;15:1849–1856. doi: 10.1161/01.atv.15.11.1849. [DOI] [PubMed] [Google Scholar]
- Hirano K., Yamashita S., Nakajima N., Arai T., Maruyama T., Yoshida Y., Ishigami M., Sakai N., Kameda-Takemura K., Matsuzawa Y. Genetic cholesteryl ester transfer protein deficiency is extremely frequent in the Omagari area of Japan. Marked hyperalphalipoproteinemia caused by CETP gene mutation is not associated with longevity. Arterioscler. Thromb. Vasc. Biol. 1997;17:1053–1059. doi: 10.1161/01.atv.17.6.1053. [DOI] [PubMed] [Google Scholar]
- Huang Z., Inazu A., Nohara A., Higashikata T., Mabuchi H. Cholesteryl ester transfer protein inhibitor (JTT-705) and the development of atherosclerosis in rabbits with severe hypercholesterolaemia. Clin. Sci. (Lond) 2002;103:587–594. doi: 10.1042/cs1030587. [DOI] [PubMed] [Google Scholar]
- Inazu A.1., Mabuchi H. Therapeutic implications of cholesteryl ester transfer protein inhibitors in hyperlipidemia and low high-density lipoprotein-cholesterolemia. Curr. Opin. Investig. Drugs. 2003;4:291–297. [PubMed] [Google Scholar]
- Inazu A.1., Brown M.L., Hesler C.B., Agellon L.B., Koizumi J., Takata K., Maruhama Y., Mabuchi H., Tall A.R. Increased high-density lipoprotein levels caused by a common cholesterylester transfer protein gene mutation. N. Engl. J. Med. 1990;323:1234–1238. doi: 10.1056/NEJM199011013231803. [DOI] [PubMed] [Google Scholar]
- Inazu A.1., Jiang X.C., Haraki T., Yagi K., Kamon N., Koizumi J., Mabuchi H., Takeda R., Takata K., Moriyama Y., et al. Genetic cholesteryl ester transfer protein deficiency caused by two prevalent mutations as a major determinant of increased levels of high-density lipoprotein cholesterol. J. Clin. Invest. 1994;94:1872–1882. doi: 10.1172/JCI117537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastelein J.J., van Leuven S.I., Burgess L., Evans G.W., Kuivenhoven J.A., Barter P.J., Revkin J.H., Grobbee D.E., Riley W.A., Shear C.L., et al. Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N. Engl. J. Med. 2007;356:1620–1630. doi: 10.1056/NEJMoa071359. [DOI] [PubMed] [Google Scholar]
- Koizumi J., Mabuchi H., Yoshimura A., Michishita I., Takeda M., Itoh H., Sakai Y., Sakai T., Ueda K., Takeda R. Deficiency of serum cholesteryl-ester transfer activity in patients with familial hyperalphalipoproteinaemia. Atherosclerosis. 1985;58:175–186. doi: 10.1016/0021-9150(85)90064-4. [DOI] [PubMed] [Google Scholar]
- Linsel-Nitschke P.I., Tall A.R. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat. Rev. Drug Discov. 2005;4:193–205. doi: 10.1038/nrd1658. [DOI] [PubMed] [Google Scholar]
- Marotti K.R., Castle C.K., Boyle T.P., Lin A.H., Murray R.W., Melchior G.W. Severe atherosclerosis in transgenic mice expressing simian cholesteryl ester transfer protein. Nature. 1993;364:73–75. doi: 10.1038/364073a0. [DOI] [PubMed] [Google Scholar]
- Matsuura F., Wang N., Chen W., Jiang X.C., Tall A.R. HDL from CETP-deficient subjects shows enhanced ability to promote cholesterol efflux from macrophages in an apoE- and ABCG1-dependent pathway. J. Clin. Invest. 2006;116:1435–1442. doi: 10.1172/JCI27602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa K., Inazu A., Kawashiri M., Nohara A., Higashikata T., Kobayashi J., Koizumi J., Nakajima K., Nakano T., Niimi M., et al. Cholesterol efflux from J774 macrophages and Fu5AH hepatoma cells to serum is preserved in CETP-deficient patients. Clin. Chim. Acta. 2009;402:19–24. doi: 10.1016/j.cca.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Moriyama Y., Okamura T., Inazu A., Doi M., Iso H., Mouri Y., Ishikawa Y., Suzuki H., Iida M., Koizumi J., et al. A low prevalence of coronary heart disease among subjects with increased high-density lipoprotein cholesterol levels, including those with plasma cholesteryl ester transfer protein deficiency. Prev. Med. 1998;27:659–667. doi: 10.1006/pmed.1998.0340. [DOI] [PubMed] [Google Scholar]
- National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) Third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III) final report. Circulation. 2002;106:3143–3421. [PubMed] [Google Scholar]
- Nissen SE., Tardif J.C., Nicholls S.J., Revkin J.H., Shear C.L., Duggan W.T., Ruzyllo W., Bachinsky W.B., Lasala G.P., Tuzcu E.M., et al. Effect of torcetrapib on the progression of coronary atherosclerosis. N. Engl. J. Med. 2007;356:1304–1316. doi: 10.1056/NEJMoa070635. [DOI] [PubMed] [Google Scholar]
- Okamoto H., Yonemori F., Wakitani K., Minowa T., Maeda K., Shinkai H. A cholesteryl ester transfer protein in hibitor attenuates atherosclerosis in rabbits. Nature. 2000;406:203–207. doi: 10.1038/35018119. [DOI] [PubMed] [Google Scholar]
- Olsson A.G., Schwartz G.G., Szarek M., Sasiela W.J., Ezekowitz M.D., Ganz P., Oliver M.F., Waters D., Zeiher A. High-density lipoprotein, but not low-density lipoprotein cholesterol levels influence short-term prognosis after acute coronary syndrome: results from the MIRACLE trial. Eur. Heart J. 2005;26:890–896. doi: 10.1093/eurheartj/ehi186. [DOI] [PubMed] [Google Scholar]
- Pedersen T.R., Kjekshus J., Berg K., Haghfelt T., Faergeman O., Faergeman G., Pyörälä K., Miettinen T., Wilhelmsen L., Olsson A.G., et al. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–1389. doi: 10.1016/j.atherosclerosissup.2004.08.027. [DOI] [PubMed] [Google Scholar]
- Rittershaus C.W., Miller D.P., Thomas L.J., Picard M.D., Honan C.M., Emmett C.D., Pettey C.L., Adari H., Hammond R.A., Beattie D.T., et al. Vaccine-induced antibodies inhibit CETP activity in vivo and reduce aortic lesions in a rabbit model of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2000;20:2106–2112. doi: 10.1161/01.atv.20.9.2106. [DOI] [PubMed] [Google Scholar]
- Sacks F.M., Pfeffer M.A., Moye L.A., Rouleau J.L., Rutherford J.D., Cole T.G., Brown L., Warnica J.W., Arnold J.M., Wun C.C., et al. The effect of pravastatin on coronary events after myocardial infarction in participants with average cholesterol levels. N. Engl. J. Med. 1996;335:1001–1009. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- Schaefer E.J. Effects of cholesteryl ester transfer protein inhibitors on human lipoprotein metabolism: why have they failed in lowering coronary heart disease risk? Curr. Opin. Lipidol. 2013;24:259–264. doi: 10.1097/MOL.0b013e3283612454. [DOI] [PubMed] [Google Scholar]
- Shepherd J., Cobbe S.M., Ford I., Isles C.G., Lorimer A.R., MacFarlane P.W., McKillop J.H., Packard C.J. For the West of Scotland Coronary Prevention Study Group: Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N. Engl. J. Med. 1995;333:1301–1307. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- Sikorski J.A. Oral cholesteryl ester transfer protein (CETP) inhibitors: a potential new approach for treating coronary artery disease. J. Med. Chem. 2006;49:1–22. doi: 10.1021/jm058224l. [DOI] [PubMed] [Google Scholar]
- Sugano M., Makino N., Sawada S., Otsuka S., Watanabe M., Okamoto H., Kamada M., Mizushima A. Effect of antisense oligonucleotides against cholesteryl ester transfer protein on the development of atherosclerosis in cholesterol-fed rabbits. J. Biol. Chem. 1998;273:5033–5036. doi: 10.1074/jbc.273.9.5033. [DOI] [PubMed] [Google Scholar]
- Tall A.R. Plasma cholesteryl ester transfer protein. J. Lipid Res. 1993;34:1255–1274. [PubMed] [Google Scholar]
- Wolfe M.L., Rader D.J. Cholesteryl ester transfer protein and coronary artery disease: an observation with therapeutic implications. Circulation. 2004;110:1338–1340. doi: 10.1161/01.CIR.0000143047.52724.BB. [DOI] [PubMed] [Google Scholar]
- Zhong S., Sharp D.S., Grove J.S., Bruce C., Yano K., Curb J.D., Tall A.R. Increased coronary heart disease in Japanese-American men with mutation in the cholesteryl ester transfer protein gene despite increased HDL levels. J. Clin. Invest. 1996;97:2917–2923. doi: 10.1172/JCI118751. [DOI] [PMC free article] [PubMed] [Google Scholar]