

Abstract
The protease-activated receptor (PAR)-2 is highly expressed in endothelial cells and vascular smooth muscle cells. It plays a crucial role in regulating blood pressure via the modulation of peripheral vascular tone. Although several mechanisms have been suggested to explain PAR-2-induced hypotension, the precise mechanism remains to be elucidated. To investigate this possibility, we investigated the effects of PAR-2 activation on N-type Ca2+ currents (ICa-N) in isolated neurons of the celiac ganglion (CG), which is involved in the sympathetic regulation of mesenteric artery vascular tone. PAR-2 agonists irreversibly diminished voltage-gated Ca2+ currents (ICa), measured using the patch-clamp method, in rat CG neurons, whereas thrombin had little effect on ICa. This PAR-2-induced inhibition was almost completely prevented by ω-CgTx, a potent N-type Ca2+ channel blocker, suggesting the involvement of N-type Ca2+ channels in PAR-2-induced inhibition. In addition, PAR-2 agonists inhibited ICa–N in a voltage-independent manner in rat CG neurons. Moreover, PAR-2 agonists reduced action potential (AP) firing frequency as measured using the current-clamp method in rat CG neurons. This inhibition of AP firing induced by PAR-2 agonists was almost completely prevented by ω-CgTx, indicating that PAR-2 activation may regulate the membrane excitability of peripheral sympathetic neurons through modulation of N-type Ca2+ channels. In conclusion, the present findings demonstrate that the activation of PAR-2 suppresses peripheral sympathetic outflow by modulating N-type Ca2+ channel activity, which appears to be involved in PAR-2-induced hypotension, in peripheral sympathetic nerve terminals.
Keywords: celiac ganglion, hypotension, N-type Ca2+ channel, peripheral sympathetic output, protease-activated receptor 2
INTRODUCTION
Protease-activated receptors (PARs) are a group of G-protein coupled receptors (GPCRs) with seven transmembrane domains. To date, four members of the PAR family (PAR 1–4) have been identified (Kawabata et al., 1999). The activation of PARs by proteases involves cleavage of the amino terminal sequence of the PAR extracellular N-terminal domain at specific sites. The new N-terminus exposed by cleavage acts as a ‘tethered ligand’ and binds to a site located in the second extracellular loop, triggering G-protein coupling and intracellular signaling.
Recently, it has been reported that PAR-2 is highly expressed in endothelial cells and vascular smooth muscle, suggesting a crucial role in regulating vascular tone (al-Ani et al., 1995; Hollenberg et al., 1996; Magazine et al., 1996; Saifeddine et al., 1996). In fact, PAR-2 activation by receptor-activating peptides decreases blood pressure in anesthetized rats or mice in vivo (Cheung et al., 1998; Cicala et al., 1999; 2001; Damiano et al., 1999; Emilsson et al., 1997; Kawabata et al., 2003). This hypotension is only partially inhibited by the nitric oxide synthase inhibitor, L-NAME, suggesting that a nitric oxide (NO)-independent mechanism as well as a NO-dependent mechanism are involved in hypotension induced by PAR-2 activation (Cicala et al., 2001; Emilsson et al., 1997). However, the effect of PAR-2 on peripheral sympathetic activity, which regulates peripheral vascular resistance, is not fully understood. According to previous reports, hypotension induced by PAR-2 activation is maintained for around 2–3 min, and the ganglion-blocking agent chlorisondamine significantly increases the duration of hypotension induced by PAR-2 activation, suggesting that PAR-2-induced hypotension may be maintained by modulating peripheral sympathetic tone (Cheung et al., 1998; Cicala et al., 2001; Emilsson et al., 1999). However, a detailed mechanism for PAR-2 induced suppression of peripheral sympathetic outflow has not yet been elucidated.
It is well documented that voltage-gated N-type Ca2+ channels play an important role in regulating peripheral sympathetic tone in postganglionic sympathetic neurons (Molderings et al., 2000; Shimosawa et al., 2004). In fact, decreases in blood pressure activate a peripheral sympathetic reflex. This enhanced peripheral sympathetic output increases Ca2+ influx through N-type Ca2+ channels located in peripheral sympathetic nerve terminals, which in turn triggers the release of noradrenaline (NA) from these terminals. Thus, any substance that modulates N-type Ca2+ channel activity can affect NA release from peripheral sympathetic nerve terminals and significantly alter sympathetic tone (Hille, 1994). It is therefore possible that PAR-2-mediated hypotension is associated, in part, with the inhibition of peripheral sympathetic output by modulating N-type Ca2+ channels that are located on sympathetic nerve terminals.
To investigate this hypothesis, we directly examined the effect of PAR-2 activation on N-type Ca2+ currents in isolated neurons of the celiac ganglion (CG), which regulates vascular sympathetic tone of the mesenteric artery. Our results demonstrate that PAR-2 may induce hypotension by suppressing peripheral vascular sympathetic activity via the inhibition of N-type Ca2+ channels located in peripheral sympathetic nerve terminals in rat CG neurons. Our findings provide new and significant evidence that proves a direct relationship between PAR-2 activation and peripheral sympathetic activity.
MATERIALS AND METHODS
Animals used
Male Sprague-Dawley rats were used in all of the experiments conducted. They were purchased from Orient Co., Seoul, Republic of Korea. All procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the Yonsei University Health System (approval reference number: 2014-0052).
Preparation of celiac ganglion neurons
Celiac ganglion (CG) neurons were enzymatically dissociated using modifications of methods described previously (Chung et al., 2010; Schofield and Ikeda, 1988). Briefly, adult (200–300 g) male rats were deeply anesthetized with isoflurane (3%, 2–3 min) and blood was removed from blood vessels by perfusing the rats transcardially with cold Dulbecco’s phosphate buffer saline (DPBS), in the same manner as described in the preparation of mesenteric artery strips. Ganglia were dissected out from the lateral side of mesenteric artery bifurcation and placed in cold DPBS. The CG is surrounded by several small ganglia, which were removed, and it was dissected into small pieces (Sosa et al., 2000), and incubated in Earle’s balanced salt solution (EBSS), containing 0.6 mg/ml papain (Sigma-Aldrich Co., Korea) and 0.3 mg/ml trypsin type 1A (Sigma-Aldrich Co., Korea), at 35°C for 45 min in a shaking water bath. After incubation, ganglia were dispersed into single neurons by vigorous shaking of the culture flask containing the ganglia. After centrifugation at 1000 rpm, the neurons were resuspended in Dulbecco’s modified eagle medium (DMEM), containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (all from Invitrogen, USA). The neurons were plated onto poly-L-lysine-coated 12-mm glass cover slips and incubated in a humidified incubator with 95% air and 5% CO2. Neurons were used within 12 h after plating.
Electrophysiology
Voltage-gated Ca2+ currents (ICa) were recorded using conventional whole-cell techniques. Electrode resistances varied from 3-5 MΩ when filled with internal solution. Measurements were performed using an Axopatch 200A patch-clamp amplifier (Molecular Devices, USA). Voltage and current commands and the digitization of membrane voltages and currents were controlled using a Digidata 1322A interfaced with Clampex 10.2 (pClamp Software, Molecular Devices, USA) on an IBM-compatible computer. Data were analyzed using Clampfit (Molecular Devices, USA) and Prism 5.0 (GraphPad, USA). The cells were moved from the incubator to a recording chamber, visualized using an inverted microscope, and subjected to whole-cell voltage clamp recordings. Currents were low-pass filtered at 2 kHz using the four-pole Bessel filter of the amplifier. Capacitance (Cm) values were automatically calculated during recordings by the pClamp software. Action potentials were recorded in current clamp mode. Membrane potential measurements were low-pass filtered at 10 kHz. Only cells with a resting membrane potential < −50 mV were included in the analysis. Multiple independently controlled syringes served as reservoirs for a gravity-driven fast drug perfusion system. Switching between solutions was accomplished by manually controlled valves. All experiments were conducted at room temperature.
Western blot analysis
CG, liver, and lungs from rats were rapidly removed, immediately frozen in liquid nitrogen, and stored at −80°C until used. The homogenates were prepared from these samples. Samples were lysed in buffer, which contained Complete Miniprotease inhibitors. The protein concentration was determined using a Bradford protein assay kit (Bio-Rad, USA). An aliquot containing 100 μg of protein from the total lysate was electrophoresed on a 10% SDS-PAGE gel and then transferred to Immobilon-P (Millipore, USA). After blocking with 5% nonfat dry milk in Trisbuffered saline (TBS), the membrane was incubated with a PAR-2 antibody (Santa Cruz Biotechnology Inc., USA) at an appropriate dilution overnight, at 4°C, and then washed four times in TBS-Tween (TBS-T). The membrane was then incubated with a goat anti-mouse Ig (Thermo Scientific, USA) at a dilution of 1:5000 at room temperature for 1 h, and then again washed four times in TBS-T. The blots were visualized using ECL reagents (Thermo Scientific, USA).
Solutions and drugs
The internal (pipette) solution contained the following (in mM): 140 CsCl2, 1.2 MgCl2, 4 MgATP, 0.4 Na2GTP, 10 phosphocreatine, 10 HEPES, and 10 EGTA; the solution was adjusted to pH 7.2 with CsOH. The external (bath) solution contained (in mM) 155 tetraethylammonium (TEA)-Cl, 2.5 CaCl2, 1.2 MgCl2, 14 glucose, and 10.5 HEPES; the solution was adjusted to pH 7.4 with TEA-OH. The external solution for current clamp recordings contained the following (in mM): 143 NaCl, 5.4 KCl, 2.5 CaCl2, 1.2 MgCl, 10 glucose, and 10 HEPES; the solution was adjusted to pH 7.4 with NaOH. The pipette solution used for current clamp recordings contained the following (in mM): 113 K-gluconate, 30 KCl, 1.2 MgCl2, 4 MgATP, 0.4 Na2GTP, 10 phosphocreatine, 10 HEPES, and 0.05 EGTA; the solution was adjusted to pH 7.2 with KOH. Sample lysis buffer consisted of 20 mM Tris–HCl, pH 7.4, 1 mM EDTA, 150 mM NaCl, 0.1% (w/v) SDS. ω-conotoxin GVIA (CgTx) was purchased from Alomone (Alomone Laboratories, Israel). SLIGRL-NH2 (SL-NH2, a synthetic PAR-2 agonist peptide) and LRGILS-NH2 (control peptide with the reverse sequence) (Nystedt et al., 1994) were purchased from COSMO Genetech (COSMO Genetech Inc., Korea). All other drugs were purchased from Sigma-Aldrich. All drugs were dissolved in distilled water as stock solutions (1–100 mM).
Data analysis
Data are presented as the means ± SEM, with the number of experiments denoted within parentheses. The concentration-response curves of trypsin and SR-NH2 for ICa inhibition were calculated by fitting the data to a single-site binding isotherm with least-squares nonlinear regression using Prism 5.0 (GraphPad, USA). We used unpaired Student’s t-tests to compare data from two groups. Differences were considered statistically significant at P < 0.05.
RESULTS
PAR-2 agonists inhibit ICa in rat CG neurons
To determine the relationship between PAR-2 and N-type voltage-gated Ca2+ channels directly, we examined the effects of PAR-2 agonists on ICa using the conventional voltage clamp method in dissociated CG neurons. These neurons contribute to regulating the peripheral vascular sympathetic tone of the mesenteric artery (Whorlow et al., 1996). We first performed immunoblotting to confirm the presence of PAR-2 protein in CG neurons. Consistent with previous data (Chien et al., 2003), the expression of PAR-2 protein, with a predicted weight of 52 kDa, was identified in rat lung tissue, but was not detected in rat liver tissue. In addition, immunoreactivity of PAR-2 protein was also detected, indicating the presence of PAR-2 protein in rat CG neurons (Supplementary Fig. S1).
To measure ICa in rat CG neurons, the current was evoked by 200 ms depolarizing step pulsesto a test potential of 0 mV from a holding potential of −80 mV. The average membrane capacitance of the CG neurons was 30.6 ± 0.3 pF (n = 56). Figure 1A, left shows a typical example of the effect of trypsin on the ICa in CG neurons. Application of 30 nM trypsin irreversibly diminished ICa by 47.5 ± 0.6% (1017.7 ± 134.9 pA for control; 529.3 ± 64.1 pA for trypsin, n = 7) (Figs. 1A and 1C). Likewise, 100 μM SL-NH2 (PAR-2 activating peptide) also decreased ICa by 45 ± 1.5% in an irreversible manner (1216.1 ± 118.4 pA for control, 688.1 ± 56.1 pA for SL-NH2, n = 7) (Figs. 1D and 1F). The PAR-2 agonists inhibited ICa over the voltage range of −40 mV to +40 mV according to the current-voltage (I–V) relationship (Figs. 1A, right and 1D, right). BT (boiled trypsin) (30 nM) (Figs. 1B and 1C) did not affect ICa (inhibition 3.3 ± 1.1%; 1099.8 ± 151.3 pA for control, 1057.1 ± 132.7 pA for BT, n = 7, P > 0.05). Similarly, 100 μM LR-NH2 (inactive peptide) (Figs. 1E and 1F) did not affect ICa (inhibition 2.3 ± 2.2%; 1069.7 ± 110.8 pA for control, 1040.4 ± 103.5 pA for LRNH2, n = 7, P > 0.01). We generated concentration–response curves for the PAR-2 agonist-induced ICa inhibition. The concentration at which the PAR-2 agonists inhibited ICa in CG neurons by 50% was approximately 30 nM for trypsin, and 100 μM for SL-NH2 (Figs. 1G and 1H). For comparison, thrombin (a PAR-1 activator; 30 nM) had little effect on ICa in rat CG neurons (6.3 ± 1% inhibition; control, 1147.7 ± 287.4 pA; thrombin, 1061.1 ± 235.1 pA, n = 7) (Supplementary Fig. S2). These results suggest that voltage-gated Ca2+ channels are modulated primarily by PAR-2 activation, rather than by PAR-1 activation.
Fig. 1.
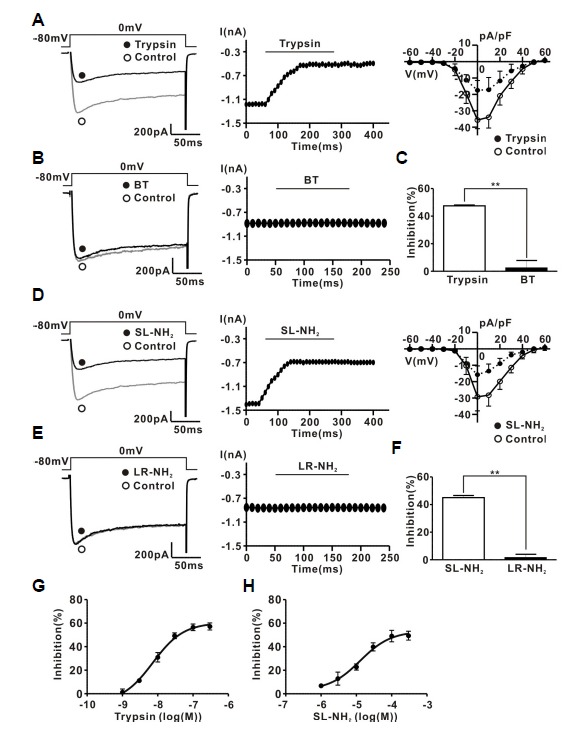
PAR-2 agonists inhibit ICa in rat CG neurons. (A) Left, a representative trace of ICa in the presence (●) and absence (○) of 30 nM trypsin. Midle, time course of the effect of 30 nM trypsin on ICa. Right, I–V relationship curve of ICa measured 10 ms after the onset of the depolarizing pulses in the absence (○) and presence (●) of 30 nM trypsin. (B) Left, a representative trace of ICa in the presence (●) and absence (○) of 30 nM BT. Right, time course of BT (30 nM)-induced ICa inhibition. (C) Summary of ICa inhibition by trypsin or BT. (D) Left, representative traces of ICa in the presence (●) and absence (○) of 100 μM SL-NH2. Middle, time course of 100 μM SL-NH2 -induced ICa inhibition. Right, I–V relationship curve of ICa measured 10 ms after the onset of the depolarizing pulses in the absence (○) and presence (●) of 100 μM SL-NH2. (E) Left, representative traces of ICa in the presence (●) and absence (○) of 100 μM LR-NH2. Right, time course of the 100 μM LR-NH2 effect on ICa. (F) Summary of ICa inhibition by SL-NH2 or LR-NH2. (G) Concentration response curve for trypsin- or SL-NH2-induced ICa inhibition. n = 7 in all groups.
Intracellular GDPβS prevents ICa inhibition by PAR-2 agonists
We determined the involvement of G proteins in PAR-2 agonist-induced ICa inhibition using GDPβS, a hydrolysis-resistant GDP analogue known to prevent G protein activation (Holz et al., 1986). As shown in Figs. 2A and 2B, intracellular dialysis of GDPβS (2 mM) significantly decreased ICa inhibition induced by 30 nM trypsin (47.3 ± 1.7% for control group, 7.3 ± 1% for GDPβS group, n = 7 respectively, P < 0.01). Similarly, SL-NH2 (100 μM) had little effect on ICa in the presence of GDPβS (47.6 ± 3.5% for control group, 4.9 ± 2.5% for GDPβS group, n = 7 respectively, P < 0.01) (Figs. 2C and 2D). These results suggest that PAR-2 agonists inhibited ICa mainly through activation of PAR-2 in rat CG neurons.
Fig. 2.
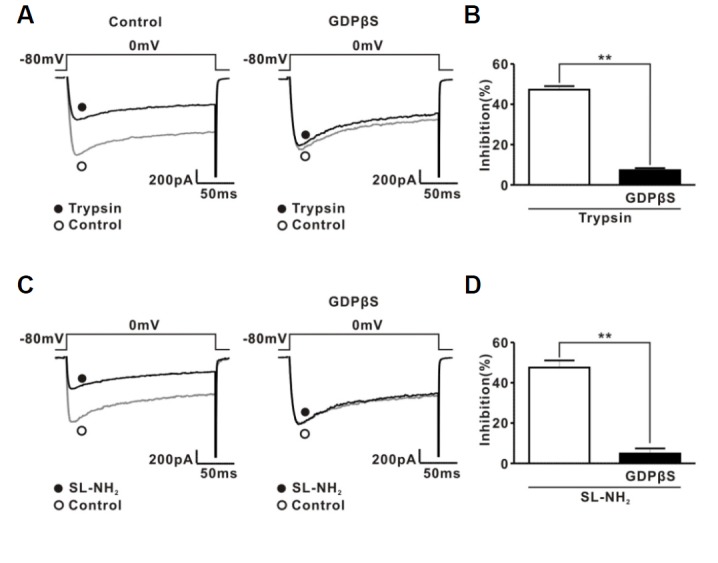
Effect of GDPβS on PAR-2 agonist-induced ICa inhibition. (A) Left, a representative trace showing the effect of trypsin (30 nM) on ICa. Open (○) and filled circles (●) indicate ICa before and after trypsin application, respectively. Right, a representative trace showing the effect of trypsin (30 nM) on ICa with GDPβS (2 mM) in the pipette solution. Trypsin was applied to rat CG neurons after 7 min dialysis with GDPβS in the pipette solution. (B) Summary showing the effect of GDPβS on trypsin-induced ICa inhibition.(C) Left, a representative trace showing the effect of SL-NH2 (100 μM) on ICa. Open (○) and filled circles (●) indicate ICa before and after SL-NH2 application, respectively. Right, a representative trace showing the effect of SL-NH2 (100 μM) on ICa with GDPβS (2 mM) in the pipette solution. SL-NH2 was applied to rat CG neurons after 7 min dialysis with GDPβS in the pipette solution. (D) Summary showing the effect of GDPβS on SL-NH2-induced ICa inhibition.
Characterization of PAR-2 agonists-induced ICa inhibition
Next, we investigated the characteristics of PAR-2 agonist-induced ICa inhibition in rat CG neurons. Consistent with previous results (Carrier and Ikeda, 1992), rat CG neurons displayed a large ICa, with about 60% attributable to the ω-CgTx-sensitive N-type Ca2+ current (ICa-N) (65.1 ± 3.4% for ω-CgTx; 35.5 ± 3.8% for nifedipine, n = 5) (Figs. 3A and 3B). To exclude a possible synergic effect of ω-CgTx and nifedipine, we also investigated the effect of nifedipine and ω-CgTx on ICa separately. As shown in Supplementary Fig. S5, nifedipine (1 μM) decreased ICa by about 37.1% (785.5 ± 57.1 pA for control; 494.5 ± 49.9 pA for nifedipine, n = 5). In addition, ω-CgTx (1 μM) inhibited ICa by 67.3% (703.2 ± 29.7 pA for control; 227.0 ± 22.5 pA for ω-CgTx, n = 5). Next, we determined whether N-type Ca2+ channels were modulated by PAR-2 agonists using a ω-CgTx prevention experiment. Figure 3C shows a representative trace of the effects of trypsin (30 nM) on ICa in the presence of ω-CgTx (1 μM). Trypsin-induced ICa inhibition was almost completely prevented by application of ω-CgTx (−0.6 ± 2.5%, n = 7, P < 0.01) (Figs. 3C and 3D). Likewise, SL-NH2-induced ICa inhibition was also almost completely prevented by application of ω-CgTx (-2.1 ± 4.3%, n = 7, P < 0.01) (Figs. 3E and 3F). To ensure that PAR-2 agonists inhibit mainly ICa-N in CG neurons, we reinvestigated the effect of PAR-2 agonists on ICa in the presence of nifedipine, which blocks L-type Ca2+ channels exclusively. These channels represent another major Ca2+ channel population in rat CG neurons. As shown in Supplementary Figs. S6A and S6C, trypsin inhibited ICa in the presence of nifedipine (1 μM), similar to trypsin’s effect on ICa-N without nifedipine (75.2 ± 3.4%, n = 5). Likewise, SL-NH2 also inhibited ICa in the presence of nifedipine (1 μM) (70.2 ± 1.3%, n = 5) (Supplementary Figs. S6D and S6F). In addition, Supplementary Figs. S6B and S6E, shows a representative trace of the effect of ω-CgTx on inhibition of ICa by PAR-2 agonists in the presence of nifedipine. In the presence of nifedipine (1 μM), trypsin-induced ICa inhibition was still abolished by application of ω-CgTx, similar to the data in Fig. 3 (5.0 ± 1.2%, n = 5, P < 0.001) (Supplementary Figs. S6B and S6C). Likewise, SL-NH2-induced ICa inhibition was also almost completely abolished by application of ω-CgTx in the presence of nifedipine (5.4 ± 0.8%, n = 5, P < 0.001) (Supplementary Figs. S6E and S6F). Many neurotransmitters, such as NA, inhibit ICa-N in a voltage-dependent manner (Elmslie et al., 1990). Hallmarks of this form of voltage-dependent inhibition include kinetic slowing, prepulse facilitation, and relief of current inhibition by conditioning depolarizing pulses. Consistent with previous data (Schofield, 1991; Shapiro et al., 1994), NA-induced ICa inhibition displayed Prepulse facilitation, which is defined as the ratio of the postpulse to prepulse current amplitude, also increased from 1.1 ± 1.7 to 1.7 ± 0.1 (P < 0.05, n = 6) after NA application (Fig. 4A). However, PAR-2 agonist-induced ICa inhibition did not show any characteristics of voltage-dependent inhibition (Fig. 4B). Moreover, prepulse facilitation was not significantly affected by 30 nM trypsin (control, 1.2 ± 0.09; trypsin, 1.2 ± 0.1, n = 7, P > 0.05) or 100 μM SL-NH2 (control, 1.2 ± 0.04, SL-NH2, 1.3 ± 0.08, P > 0.05) (Fig. 4C). Taken together, these results suggest that PAR-2 agonists inhibited ω-CgTx-sensitive ICa–N in a voltage-independent manner in rat CG neurons.
Fig. 3.
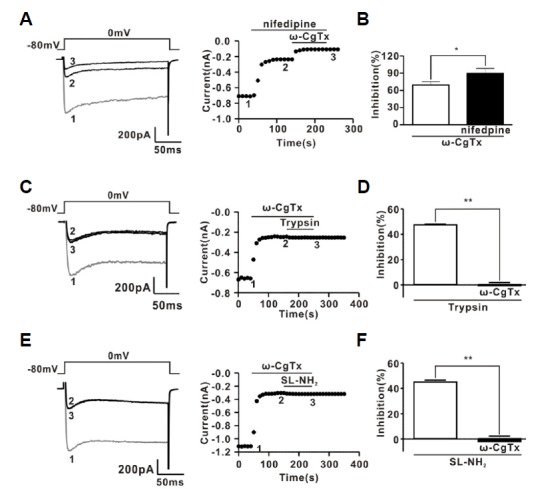
Effect of ω-CgTx on ICa and PAR-2 agonist-induced ICa inhibition. (A) Left, a representative trace showing the effect of consecutive application of ω-CgTx (1 μM) and nifedipine (1 μM) on ICa. Right, time course of the effects induced by consecutive application of ω-CgTx (1 μM) and nifedipine (1 μM) on ICa. Trace 1, 2, and 3 in the left panel represent the traces recorded at the corresponding time indicated in the right panel, respectively. (B) Contribution of N-type (ω-CgTx-sensitive) and L-type (nifedipine-sensitive) currents to the total ICa. (C) Left, a representative trace of the effects induced by consecutive application of ω-CgTx (1 μM) and trypsin (30 nM) on ICa. Right, time course of effects induced by consecutive application of ω-CgTx (1 μM) and trypsin (30 nM) on ICa.Trace 1, 2, and 3 in the left panel represent the traces recorded at the corresponding time indicated in the right panel, respectively. (D) Summary of ICa inhibition by trypsin in the absence and presence of ω-CgTx. (E) Left, a representative trace of the effects induced by consecutive application of ω-CgTx (1 μM) and SL-NH2 (100 μM) on ICa. Right, time course of effects induced by consecutive application of ω-CgTx (1 μM) and SL-NH2 (100 μM) on ICa. Trace 1, 2, and 3 in the left panel represent the traces recorded at the corresponding time indicated in the right panel, respectively. (F) Summary of ICa inhibition by SL-NH2 in the absence and presence of ω-CgTx.
Fig. 4.
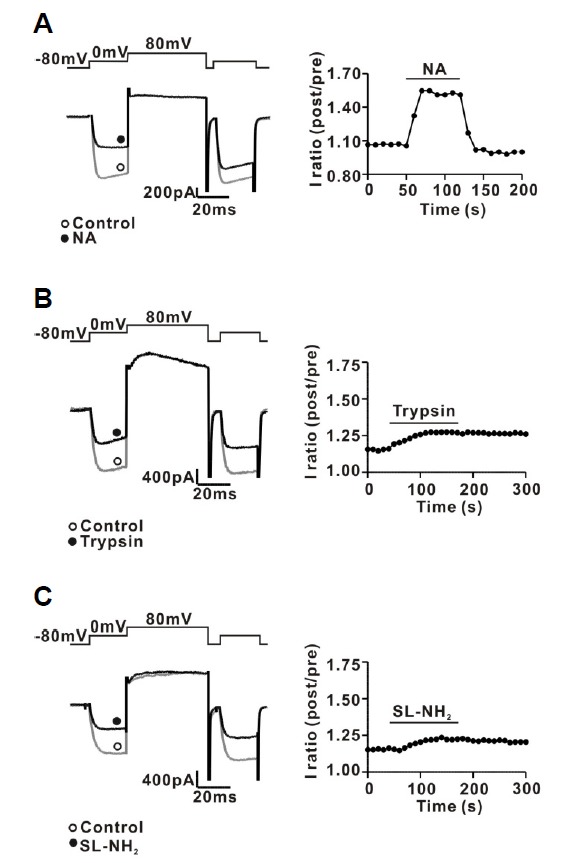
Characteristics of PAR-2 agonist-induced ICa inhibition in rat CG neurons. (A) Left, a representative trace of ICa inhibition by NA (1 μM) application. Right, time course of prepulse facilitation in the absence and presence of 1 μM NA. The ICa was evoked every 10 s by a double-pulse voltage protocol consisting of two identical test pulses (0 mV from a holding potential of −80 mV) separated by a large depolarizing conditioning pulse to +80 mV. Prepulse facilitation was calculated as the ratio of the postpulse to prepulse current amplitudes (post/pre) measured isochronally at 10 ms after the start of the test pulse. (B) Left, a representative trace of ICa inhibition by trypsin (30 nM) application. Right, time course of prepulse facilitation in the absence and presence of 30 nM trypsin. (A) Left, a representative trace of ICa inhibition by SL-NH2 (100 μM) application. Right, time course of prepulse facilitation in the absence and presence of 100 μM SL-NH2.
Effects of PAR-2 agonists on the repetitive firing of action potentials (APs) in CG neurons
The role of PAR-2 in regulating neuronal excitability was determined by measuring AP firing frequency in rat CG neurons. APs were evoked by constant-current injection through the patch pipette in current clamp mode. As shown in Fig. 5, Supplementary Figs. S3 and S4, injection of positive current (100–300 pA) for 300 ms evoked APs in rat CG neurons with a frequency of 16.8 ± 0.8 Hz (n = 48). Figure 5A shows a typical example of the effect of trypsin on the AP firing frequency in rat CG neurons. Application of 30 nM trypsin reduced AP firing frequency by 47.5 ± 1.6% (17.1 ± 2.8 Hz for control, 11.4 ± 2.2 Hz for trypsin, n = 7, P < 0.05). However, BT (30 nM) did not have any effect on the frequency (16.2 ± 2.4 Hz for control, 15.2 ± 2.0 Hz for BT, n = 7, P > 0.05) (Supplementary Fig. S3A). Likewise, 100 μM SL-NH2 hallmarks of voltage-dependent inhibition, namely kinetic slowing and relief of current inhibition by conditioning depolarizing pulses. also decreased AP firing frequency by 45.2 ± 2.6% (16.6 Hz ± 2.1 Hz for control, 10.2 ± 1.1 Hz for SL-NH2, n = 7, P < 0.05) (Fig. 5B). However, 100 μM LR-NH2 had no influence on frequency (15.2 ± 2.0 Hz for control, 14.3 ± 2.3 Hz for LR-NH2, n = 7, P > 0.05) (Supplementary Fig. S3B).
Fig. 5.
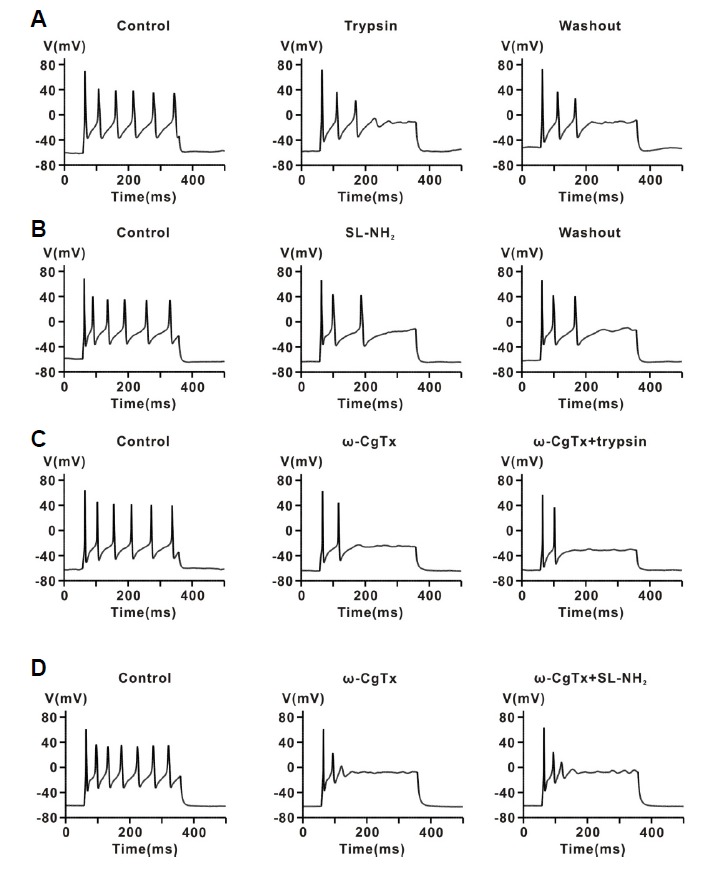
Effect of a PAR-2 agonist on repetitive firing of APs in celiac ganglia neurons. (A) Representative traces showing the effect of trypsin (30 nM) on repetitive APs evoked by current injection (100–200 pA, 300 ms) in current-clamp mode. (B) Representative traces showing the effect of SL-NH2 (100 μM) on repetitive APs evoked by current injection in current-clamp mode. (C) Representative traces showing the effect of consecutive application of ω-CgTx (1 μM) and trypsin (30 nM) on repetitive AP firing evoked by current injection. (D) Representative traces showing the effect of consecutive application of ω-CgTx (1 μM) and trypsin SL-NH2 (100 μM) on repetitive AP firing evoked by current injection.
To determine whether inhibition of AP firing by PAR-2 agonists is mediated by N-type Ca2+ channels, we examined the effect of ω-CgTx on AP firing evoked by current injection in rat CG neurons. As shown in Figs. 5C and 5D, ω-CgTx (1 μM) significantly suppressed the AP firing rate (18.1 ± 1.2 Hz for control, 8.6 ± 0.7 Hz for ω-CgTx, n = 14, P < 0.001), indicating that membrane excitability may be regulated by N-type Ca2+ channel activity. In addition, ω-CgTx (1 μM) almost completely abolished the inhibitory effect of trypsin (30 nM) on AP firing in rat CG neurons (3.7 ± 1.9%, n = 7, P > 0.05) (Fig. 5C). Likewise, SL-NH2 (100 μM) had no influence on AP firing rate in the presence of ω-CgTx (4.2 ± 3.0%, n = 7, P > 0.05) (Fig. 5D). For comparison, thrombin (30 nM) had little effect on AP firing in rat CG neurons (control, 15.6 ± 0.7 Hz; thrombin, 15.6 ± 0.7 Hz, P > 0.05, n = 6) (Supplementary Fig. S4). These results suggest that PAR-2 activation may regulate the membrane excitability of peripheral sympathetic neurons through modulation of N-type Ca2+ channels in rat CG neurons.
DISCUSSION
The present study demonstrates that PAR-2 activation in rat CG neurons inhibits N-type Ca2+ channels, which may play a role in suppression of peripheral sympathetic activity.
Al-Ani et al. (1995) first reported that PAR-2 agonists caused an endothelium-dependent relaxation in rat aortic rings, and that this vasorelaxing effect was partially prevented by the nitric oxide (NO) synthase inhibitor L-NAME. Since then, PAR-2 activation has been reported to be involved in vasorelaxation under NO-dependent and -independent manners in various vascular tissues, such as porcine coronary arteries (Hamilton et al., 1998; Hwa et al., 1996), rat femoral arteries (Emilsson et al., 1997; Roy et al., 1998), rat renal mesenteric pulmonary arteries (Roy et al., 1998), and basilar arteries (Sobey and Cocks, 1998; Sobey et al., 1999). In addition, it has been shown in vivo that PAR-2 agonist peptides (SL-NH2 or SLIGKV-NH2) applied by intravenous injection cause hypotension in anesthetized rats (Cicala et al., 1999; Emilsson et al., 1997) and mice (Cheung et al., 1998; Damiano et al., 1999), and that this PAR-2-induced hypotension is also partially inhibited by L-NAME, consistent with in vitro results (Cicala et al., 2001; Emilsson et al., 1997). As for NO-independent vasorelaxation by PAR-2 activation, though several mechanisms have been suggested to explain NO-independent relaxation by PAR-2 (Hughes et al., 2013; McGuire, 2004; McGuire et al., 2002), a detailed mechanism remains to be elucidated.
The peripheral sympathetic nervous system plays a significant role in the regulation of blood flow by modulating peripheral vascular resistance. The postganglionic fibers from peripheral sympathetic ganglionic neurons, which innervate resistance arterial beds, are involved in the regulation of peripheral vascular tone through NA release from sympathetic nerve terminals. In addition, voltage-dependent N-type Ca2+ channels, which are located at peripheral sympathetic nerve terminals, play a pivotal role in modulating peripheral sympathetic activity in postganglionic sympathetic neurons (Molderings et al., 2000; Shimosawa et al., 2004). Increases in intracellular Ca2+ concentration, caused by Ca2+ influx through N-type voltage-gated Ca2+ channels, trigger NA release in peripheral sympathetic nerve terminals. Thus, if PAR-2 activation inhibits NA release by regulating N-type Ca2+ channel activity in peripheral sympathetic nerve terminals, this can cause suppression of peripheral sympathetic tone, which in turn reduces blood pressure. In fact, our group recently observed that PAR-2 agonists suppress neurogenic contraction and overflow of NA release evoked by electrical field stimulation, which mimics vasoconstriction, and that this suppression was almost completely prevented by w-CgTx, a potent N-type Ca2+ channel blocker (unpublished data). These results strongly suggest that activation of PAR-2 may suppress peripheral sympathetic outflow by modulating N-type Ca2+ channel activity in peripheral sympathetic nerve terminals, which appear to be involved in PAR-2-induced hypotension. Therefore, we sought direct evidence for a relationship between N-type Ca2+ channels and PAR-2 in peripheral sympathetic neurons. In the present study, PAR-2 agonists irreversibly diminished ICa in rat CG neurons, while thrombin had little effect on ICa. In addition, blockage of N-type Ca2+ channels with ω-CgTx almost completely prevented ICa inhibition by PAR-2 agonists. Furthermore, PAR-2 agonists reduced the membrane excitability of CG neurons in an irreversible manner, and the blockage of N-type Ca2+ channels with ω-CgTx almost completely prevented inhibition of AP firing by PAR-2 agonists. These results, therefore, suggest that the suppression of peripheral sympathetic activity by PAR-2 agonists is mediated mainly by the inhibition of N-type Ca2+ channels located in peripheral sympathetic nerve terminals.
In sympathetic ganglia neurons, Ca2+ influx through N-type Ca2+ channels is needed to maintain repetitive AP firing. In fact, ω-CgTx, GVIA, or Ca2+-free buffers significantly suppress the repetitive APs evoked by current injection in bronchial ganglion neurons (Myers, 1998). Thus, inhibition of ICa-N by neurotransmitters may attenuate the excitability of the peripheral sympathetic nervous system. In the present study, PAR-2 agonists reduced the membrane excitability of CG neurons in an irre versible manner (Figs. 7A and 7B). Consistent with previous reports, ω-CgTx significantly reduced firing frequency of APs evoked by current injection in rat CG neurons. Blocking N-type Ca2+ channels with ω-CgTx almost completely abolished the inhibition of AP firing by PAR-2 agonists (Figs. 7C and 7D). This result suggests that ICa-N inhibition by PAR-2 activation may reduce the excitability of peripheral sympathetic ganglion neurons and the relay of excitatory stimuli to sympathetic nerve terminals. In addition, if N-type Ca2+ channels are functionally coupled to PAR-2 at sympathetic nerve terminals in rat CG neurons, as they are in the soma, the activation of PAR-2 may suppress the release of NA by decreasing the intracellular Ca2+ concentration through inhibition N-type Ca2+ channels directly, as in Dunlap & Fischbach’s general concept of presynaptic inhibition (Shimosawa et al., 2004). As for the irreversible nature of the suppression of ICa-N by PAR-2 activation, we cannot suggest a feasible explanation based on the results obtained from this study. PAR-2 activated by irreversible proteolysis that results in continuous stimulation; however, cleaved PAR-2 is known to be rapidly ubiquitinated at the C-terminus, which mediates down-regulation of PAR-2 to terminate signaling. A more detailed study is needed to clearly explain this phenomenon.
In conclusion, the present findings demonstrate that the activation of PAR-2 suppresses peripheral sympathetic outflow by modulating N-type Ca2+ channel activity in peripheral sympathetic nerve terminals, which appear to be involved in PAR-2-induced hypotension. In addition, PAR-2 activation inhibits N-type Ca2+ channel activity in a voltage-independent manner, and this inhibition attenuates repetitive AP firing in CG neurons. These results provide a detailed neuronal mechanism for PAR-2-induced hypotension and a basic and theoretical framework that could lead to the development of new agents for the treatment of hypertension.
Acknowledgments
This research was supported by the Pioneer Research Center Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning (2012-0009525) and a faculty research grant from Yonsei University of Medicine for 2011 (6-2011-0164).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- al-Ani B., Saifeddine M., Hollenberg M.D. Detection of functional receptors for the proteinase-activated-receptor-2-activating polypeptide, SLIGRL-NH2, in rat vascular and gastric smooth muscle. Can. J. Physiol. Pharmacol. 1995;73:1203–1207. doi: 10.1139/y95-172. [DOI] [PubMed] [Google Scholar]
- Carrier G.O., Ikeda S.R. TTX-sensitive Na+ channels and Ca2+ channels of the L- and N-type underlie the inward current in acutely dispersed coeliac-mesenteric ganglia neurons of adult rats. Pflugers Arch. 1992;421:7–16. doi: 10.1007/BF00374726. [DOI] [PubMed] [Google Scholar]
- Cheung W.M., Andrade-Gordon P., Derian C.K., Damiano B.P. Receptor-activating peptides distinguish thrombin receptor (PAR-1) and protease activated receptor 2 (PAR-2) mediated hemodynamic responses in vivo. Can. J. Physiol. Pharmacol. 1998;76:16–25. doi: 10.1139/cjpp-76-1-16. [DOI] [PubMed] [Google Scholar]
- Chien E.K., Sweet L., Phillippe M., Marietti S., Kim T.T., Wolff D.A., Thomas L., Bieber E. Protease-activated receptor isoform expression in pregnant and nonpregnant rat myometrial tissue. J. Soc. Gynecol. Investig. 2003;10:460–468. doi: 10.1016/s1071-5576(03)00148-5. [DOI] [PubMed] [Google Scholar]
- Chung S., Ahn D.S., Kim Y.H., Kim Y.S., Joeng J.H., Nam T.S. Modulation of N-type calcium currents by presynaptic imidazoline receptor activation in rat superior cervical ganglion neurons. Exp. Physiol. 2010;95:982–993. doi: 10.1113/expphysiol.2010.053355. [DOI] [PubMed] [Google Scholar]
- Cicala C., Pinto A., Bucci M., Sorrentino R., Walker B., Harriot P., Cruchley A., Kapas S., Howells G.L., Cirino G. Protease-actibated receptor-2 involvement in hypotension in normal and endotoxemic rats in vivo. Circulation. 1999;99:2590–2597. doi: 10.1161/01.cir.99.19.2590. [DOI] [PubMed] [Google Scholar]
- Cicala C., Morello S., Santagada V., Caliendo G., Sorrentino L., Cirino G. Pharmacological dissection of vascular effects caused by activation of protease-activated receptors 1 and 2 in anesthetized rats. FASEB J. 2001;15:1433–1435. doi: 10.1096/fj.00-0633fje. [DOI] [PubMed] [Google Scholar]
- Damiano B.P., Cheung W.M., Santulli R.J., Fung-Leung W.P., Ngo K., Ye R.D., Darrow A.L., Derian C.K., de Garavilla L., Andrade-Gordon P. Cardiovascular responses mediated by protease-activated receptor-2 (PAR-2) and thrombin receptor (PAR-1) are distinguished in mice deficient in PAR-2 or PAR-1. J. Pharmacol. Exp. Ther. 1999;288:671–678. [PubMed] [Google Scholar]
- Elmslie K.S., Zhou W., Jones S.W. LHRH and GTP-gamma-S modify calcium current activation in bullfrog sympathetic neurons. Neuron. 1990;5:75–80. doi: 10.1016/0896-6273(90)90035-e. [DOI] [PubMed] [Google Scholar]
- Emilsson K., Wahlestedt C., Sun M.K., Nystedt S., Owman C., Sundelin J. Vascular effects of proteinase-activated receptor 2 agonist peptide. J. Vasc. Res. 1997;34:267–272. doi: 10.1159/000159233. [DOI] [PubMed] [Google Scholar]
- Emilsson V., Arch J.R., de Groot R.P., Lister C.A., Cawthorne M.A. Leptin treatment increases suppressors of cytokine signaling in central and peripheral tissues. FEBS Lett. 1999;455:170–174. doi: 10.1016/s0014-5793(99)00874-1. [DOI] [PubMed] [Google Scholar]
- Hamilton J.R., Nguyen P.B., Cocks T.M. Atypical protease-activated receptor mediates endothelium-dependent relaxation of human coronary arteries. Circ. Res. 1998;82:1306–1311. doi: 10.1161/01.res.82.12.1306. [DOI] [PubMed] [Google Scholar]
- Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Hollenberg M.D., Saifeddine M., al-Ani B. Proteinase-activated receptor-2 in rat aorta: structural requirements for agonist activity of receptor-activating peptides. Mol. Pharmacol. 1996;49:229–233. [PubMed] [Google Scholar]
- Holz G.G.t., Rane S.G., 4th, Dunlap K. GTP-binding proteins mediate transmitter inhibition of voltage-dependent calcium channels. Nature. 1986;319:670–672. doi: 10.1038/319670a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes K.H., Wijekoon E.P., Valcour J.E., Chia E.W., McGuire J.J. Effects of chronic in-vivo treatments with protease-activated receptor 2 agonist on endothelium function and blood pressures in mice. Can. J. Physiol. Pharmacol. 2013;91:295–305. doi: 10.1139/cjpp-2012-0266. [DOI] [PubMed] [Google Scholar]
- Hwa J.J., Ghibaudi L., Williams P., Chintala M., Zhang R., Chatterjee M., Sybertz E. Evidence for the presence of a proteinase-activated receptor distinct from the thrombin receptor in vascular endothelial cells. Circ. Res. 1996;78:581–588. doi: 10.1161/01.res.78.4.581. [DOI] [PubMed] [Google Scholar]
- Kawabata A., Kuroda R., Hollenberg M.D. Physiology of protease-activated receptors (PARs): involvement of PARs in digestive functions. Nihon Yakurigaku Zasshi. 1999;114:173P–179P. doi: 10.1254/fpj.114.supplement_173. [DOI] [PubMed] [Google Scholar]
- Kawabata A., Nakaya Y., Kuroda R., Wakisaka M., Masuko T., Nishikawa H., Kawai K. Involvement of EDHF in the hypotension and increased gastric mucosal blood flow caused by PAR-2 activation in rats. Br. J. Pharmacol. 2003;140:247–254. doi: 10.1038/sj.bjp.0705433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magazine H.I., King J.M., Srivastava K.D. Protease activated receptors modulate aortic vascular tone. Int. J. Cardiol. 1996;53 Suppl:S75–80. doi: 10.1016/0167-5273(96)02569-7. [DOI] [PubMed] [Google Scholar]
- McGuire J.J. Proteinase-activated Receptor 2 (PAR2): a challenging new target for treatment of vascular diseases. Curr. Pharm. Des. 2004;10:2769–2778. doi: 10.2174/1381612043383656. [DOI] [PubMed] [Google Scholar]
- McGuire J.J., Dai J., Andrade-Gordon P., Triggle C.R., Hollenberg M.D. Proteinase-activated receptor-2 (PAR2): vascular effects of a PAR2-derived activating peptide via a receptor different than PAR2. J. Pharmacol. Exp. Ther. 2002;303:985–992. doi: 10.1124/jpet.102.040352. [DOI] [PubMed] [Google Scholar]
- Molderings G.J., Likungu J., Gothert M. N-Type calcium channels control sympathetic neurotransmission in human heart atrium. Circulation. 2000;101:403–407. doi: 10.1161/01.cir.101.4.403. [DOI] [PubMed] [Google Scholar]
- Myers A.C. Ca2+ and K+currents regulate accommodation and firing frequency in guinea pig bronchial ganglion neurons. Am. J. Physiol. 1998;275:L357–364. doi: 10.1152/ajplung.1998.275.2.L357. [DOI] [PubMed] [Google Scholar]
- Nystedt S., Emilsson K., Wahlestedt C., Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. USA. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S.S., Saifeddine M., Loutzenhiser R., Triggle C.R., Hollenberg M.D. Dual endothelium-dependent vascular activities of proteinase-activated receptor-2-activating peptides: evidence for receptor heterogeneity. Br. J. Pharmacol. 1998;123:1434–1440. doi: 10.1038/sj.bjp.0701726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifeddine M., al-Ani B., Cheng C.H., Wang L., Hollenberg M.D. Rat proteinase-activated receptor-2 (PAR-2): cDNA sequence and activity of receptor-derived peptides in gastric and vascular tissue. Br. J. Pharmacol. 1996;118:521–530. doi: 10.1111/j.1476-5381.1996.tb15433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield G.G. Norepinephrine inhibits a Ca2+ current in rat sympathetic neurons via a G-protein. Eur. J. Pharmacol. 1991;207:195–207. doi: 10.1016/0922-4106(91)90031-c. [DOI] [PubMed] [Google Scholar]
- Schofield G.G., Ikeda S.R. Sodium and calcium currents of acutely isolated adult rat superior cervical ganglion neurons. Pflugers Arch. 1988;411:481–490. doi: 10.1007/BF00582368. [DOI] [PubMed] [Google Scholar]
- Shapiro M.S., Wollmuth L.P., Hille B. Modulation of Ca2+ channels by PTX-sensitive G-proteins is blocked by N-ethylmaleimide in rat sympathetic neurons. J. Neurosci. 1994;14:7109–7116. doi: 10.1523/JNEUROSCI.14-11-07109.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimosawa T., Takano K., Ando K., Fujita T. Magnesium inhibits norepinephrine release by blocking N-type calcium channels at peripheral sympathetic nerve endings. Hypertension. 2004;44:897–902. doi: 10.1161/01.HYP.0000146536.68208.84. [DOI] [PubMed] [Google Scholar]
- Sobey C.G., Cocks T.M. Activation of protease-activated receptor-2 (PAR-2) elicits nitric oxide-dependent dilatation of the basilar artery in vivo. Stroke. 1998;29:1439–1444. doi: 10.1161/01.str.29.7.1439. [DOI] [PubMed] [Google Scholar]
- Sobey C.G., Moffatt J.D., Cocks T.M. Evidence for selective effects of chronic hypertension on cerebral artery vasodilatation to protease-activated receptor-2 activation. Stroke. 1999;30:1933–1940. doi: 10.1161/01.str.30.9.1933. discussion 1941. [DOI] [PubMed] [Google Scholar]
- Sosa Z.Y., Casais M., Rastrilla A.M., Aguado L. Adrenergic influences on coeliac ganglion affect the release of progesterone from cycling ovaries: characterisation of an in vitro system. J. Endocrinol. 2000;166:307–318. doi: 10.1677/joe.0.1660307. [DOI] [PubMed] [Google Scholar]
- Whorlow S.L., Angus J.A., Wright C.E. SELECTIVITY OF ω-CONOTOXIN GVIA FOR N-TYPE CALCIUM CHANNELS IN RAT ISOLATED SMALL MESENTERIC ARTERIES. Clin. Exp. Pharmacol. Physiol. 1996;23:16–21. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.