

Abstract
Context:
Idiopathic hypogonadotropic hypogonadism (IHH) results from defective synthesis, secretion, or action of GnRH. Kisspeptin is a potent stimulus for GnRH secretion.
Objective:
We probed the functional capacity of the GnRH neuronal network in patients with IHH.
Participants:
Eleven subjects with congenital IHH (9 men and 2 women) and one male subject who underwent reversal of IHH were studied. Six of the twelve subjects had an identified genetic cause of their IHH: KAL1 (n = 1), FGFR1 (n = 3), PROKR2 (n = 1), GNRHR (n = 1).
Intervention:
Subjects underwent q10 min blood sampling to measure GnRH-induced LH secretion at baseline and in response to intravenous boluses of kisspeptin (0.24 nmol/kg) and GnRH (75 ng/kg) both pre- and post-six days of treatment with exogenous GnRH (25 ng/kg sc every 2 h).
Results:
All subjects with abiding IHH failed to demonstrate a GnRH-induced LH response to exogenous kisspeptin. In contrast, the subject who achieved reversal of his hypogonadotropism demonstrated a robust response to kisspeptin.
Conclusions:
The functional capacity of the GnRH neuronal network in IHH patients is impaired, as evidenced by their inability to respond to the same dose of kisspeptin that effects a robust GnRH-induced LH response in healthy men and luteal-phase women. This impairment is observed across a range of genotypes, suggesting that it reflects a fundamental property of GnRH neuronal networks that have not been properly engaged during pubertal development. In contrast, a patient who had experienced reversal of his hypogonadotropism responded to exogenous kisspeptin.
Idiopathic hypogonadotropic hypogonadism (IHH) is a clinical syndrome characterized by abnormal pubertal progression and low sex steroids in the setting of low/normal gonadotropins. In the vast majority of cases, IHH is due to abnormal GnRH synthesis, secretion or action (1, 2). Thus, the administration of exogenous pulsatile GnRH can stimulate pituitary gonadotropin and gonadal sex steroid secretion, as well as folliculogenesis and spermatogenesis (1, 2). In addition to its use as a fertility treatment, pulsatile GnRH can also be used as an investigative tool to probe the physiology of pituitary gonadotropin release. For example, administration of pulsatile GnRH to IHH patients has revealed novel insights into the effect of GnRH frequency and amplitude on differential gonadotropin release, as well as the relative roles of sex steroids and nonsteroidal hormones on negative feedback at the level of the hypothalamus and pituitary (3–9). While exogenous GnRH administration has served as a powerful probe of pituitary physiology, the absence of a comparable probe for hypothalamic GnRH neurons has limited investigation into the biology of the GnRH neuron and the underlying pathophysiology of IHH in humans.
Nonetheless, genetic studies of hypogonadotropic patients, either with anosmia (Kallmann syndrome) or with a normal sense of smell (normosmic idiopathic hypogonadotropic hypogonadism), have revealed much about the underlying pathophysiology that can affect the GnRH neurons as they differentiate, migrate, and secrete GnRH. Mutations in several genes responsible for these processes have been uncovered over the last 20 years (10), including but not limited to KAL1 (11, 12), which influences GnRH neuronal migration (13), GNRH1, which affects GnRH synthesis (14, 15), and KISS1R, which impacts GnRH secretion (16, 17). Incomplete expressivity and variable penetrance are characteristic of mutations in several of the genes implicated in congenital hypogonadotropism (18, 19); in a subset of cases, mutations in more than one gene may be required for phenotypic manifestations (20, 21). While most IHH patients have lifelong hypogonadotropism, ie, “abiding” IHH, up to 22% of patients with IHH undergo “reversal,” characterized by spontaneous activation of the hypothalamic-pituitary-gonadal axis as evidenced by normalization of sex-steroid levels, increased testicular volume, menstrual cycles, and/or fertility in the absence of any therapy (22, 23). A direct physiologic probe of GnRH function may inform genotype-phenotype correlations, assess propensity for reversal, and uncover new insights into the pathophysiology of IHH.
Discovered as a key gatekeeper of the timing of sexual maturation across mammalian species (16, 17), the hypothalamic neuropeptide kisspeptin is potentially one such probe. Kisspeptin is a potent stimulus for GnRH secretion (24), and evidence suggests it may be secreted in a pulsatile manner (25, 26). Considerable data regarding the neuroendocrine response to exogenous kisspeptin administration in healthy men and women has been assembled. In men, regardless of the formulation or method of administration, kisspeptin has been shown by our group and others to induce a robust GnRH-induced LH response (27–30, 59). In contrast, in reproductive age women, kisspeptin's effects are more variable and depend on the phase of the menstrual cycle (59). Kisspeptin administration in the luteal phase results in an LH response comparable to that observed in healthy men; however, administration in the follicular phase results in an almost negligible rise in LH (59). While this variability in response to kisspeptin administration has not been fully explained, enough data has been assembled to serve as a normative backdrop against which data from pathophysiologic populations can be compared. Thus, just as the discovery of GnRH allowed investigators to chart the biology of the pituitary gonadotrope, exogenous kisspeptin administration was utilized in this study as an in vivo probe of the GnRH neuronal network in patients with IHH. We hypothesized that patients with genetic mutations leading to an absence of GnRH neurons (ie, KAL1), would not demonstrate any GnRH-induced LH response to exogenous kisspeptin. In contrast, we hypothesized that patients with mutations leading to a reduced complement of GnRH neurons or reduced secretory capacity would exhibit an attenuated response to this hypothalamic hormone.
Materials and Methods
Subjects and eligibility criteria
This study was approved by the Institutional Review Board of Massachusetts General Hospital (MGH)/Partners Healthcare, and all subjects gave written informed consent. All subjects had been diagnosed with congenital IHH as evidenced by absent or incomplete pubertal development by age 18 years and low sex steroids in the setting of low or inappropriately normal gonadotropins. DNA extracted from subjects' blood had previously been screened for rare sequence variants in CHD7 (MIM 608892), FGF8 (MIM 600483), FGFR1 (MIM 136350), GNRH1 (MIM 152760), GNRHR (MIM 138850), HS6ST1 (MIM 604846), KAL1 (MIM 300836), KISS1 (MIM 603286), KISS1R (MIM 604161), NSMF, previously called NELF (MIM 60813), PROK2 (MIM 607002), PROKR2 (MIM 607123), TAC3 (MIM 162330), and TACR3 (MIM 162332) by PCR amplification of exons followed by Sanger sequencing, as described previously (21). Rare sequence variants (RSVs) were defined as having a minor allele frequency of less than 1% in the 1000 Genomes Project and the National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project (32). RSVs were reported if they were predicted to be damaging by at least 3 out of 4 in silico prediction programs: PolyPhen-2 (33), SIFT (34), Mutation Taster (35), Panther (36). To determine whether multiple mutations were located on one allele or separate alleles, PCR products were cloned using the TOPO TA Cloning Kit for Sequencing (Life Technologies), followed by amplification and sequencing of individual clones.
Study design
The main study protocol, schematized in Figure 1, consisted of two admissions to the Harvard Catalyst Clinical Research Center (CRC) at Massachusetts General Hospital. Prior to the first CRC admission, the subjects' sex-steroid therapy was withheld to allow them to return to their hypogonadal state; this “washout” period lasted 2 weeks for transdermal testosterone, 6 weeks for injected testosterone enanthate or cypionate, and 8 weeks for oral contraceptive (OC) pills.
Figure 1.
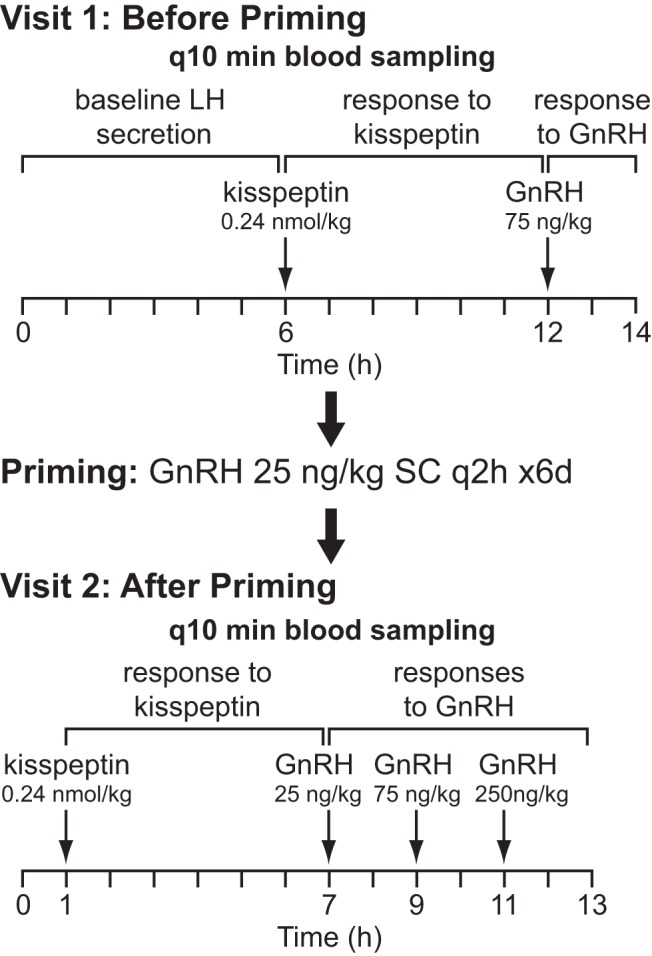
Schematic of the study protocol.
During the first CRC admission, blood sampling was performed every 10 min for 12–14 h. The first 6 h assessed the subjects' baseline LH secretion in the absence of any intervention. Subjects then received an intravenous (IV) bolus of kisspeptin-10 0.24 nmol/kg (0.313 μg/kg), followed by 4–6 h of blood sampling. This dose of kisspeptin was chosen because prior work by our group demonstrated that this dose consistently elicits GnRH-induced LH pulses of physiologic amplitude in healthy men and healthy luteal phase women (29, 31). A bolus of GnRH 75 ng/kg was then given, followed by an additional 2 h of blood sampling. Kisspeptin and GnRH were synthesized by Polypeptides, Inc., and the 10-amino-acid isoform of kisspeptin (corresponding to amino acids 112–121 of the preprohormone) was used in these studies.
Although the vast majority of patients with hypogonadotropic hypogonadism have hypothalamic and not pituitary defects in hormone secretion, many patients will fail to respond to an initial bolus of exogenous GnRH. Therefore, even if kisspeptin induced a GnRH secretory event, the subsequent GnRH stimulation of a “sleepy” pituitary gland might result in absence of an LH response. To avoid the possibility of such false negative interpretations, we capitalized on the long-standing knowledge that exposure to intermittent, exogenous GnRH administration “primes” pituitary gonadotrophs (37). Thus, either immediately or within a few days after discharge from their first CRC admission, 8 subjects underwent pituitary “priming” with GnRH 25 ng/kg administered subcutaneously every 2 h by a Crono F portable infusion pump (Canè S.p.A) for 6 days. The last dose of GnRH was given 48 h prior to a second, “postpriming” admission to allow circulating sex steroids to return to baseline concentrations. During this second admission, subjects underwent q10 min blood sampling × 1 h, kisspeptin-10 0.24 nmol/kg IV administration followed by 6 h of blood sampling, and finally three boluses of GnRH (25, 75, 250 ng/kg in ascending order) IV followed by 2 h of blood sampling after each bolus.
Four subjects returned for additional studies in which they received multiple boluses of kisspeptin at doses ranging from 0.24 to 2.4 nmol/kg. Subject 10 received five boluses and subject 7 received two boluses of the same dose of kisspeptin used in the single-bolus protocol above (0.24 nmol/kg). Subjects 9 and 12 received escalating doses of kisspeptin at 0.24, 0.72, and 2.4 nmol/kg.
Laboratory assays and pulse analysis
LH was measured at each time point, on 2-h study pools, and on an all-study quality control (QC) pool, and FSH and either testosterone or estradiol were measured on 2-h study pools. Serum LH, FSH, estradiol and testosterone concentrations were determined by direct immunoassay using the automated Abbott ARCHITECT system (Abbott Laboratories, Inc.).
LH levels are expressed in units of the second International Pituitary Standard WHO [80/552]. The minimal detectable dose (lowest dose distinguishable from zero; 95% confidence interval (CI) of a blank detection) for the LH assay is 0.07 mIU/mL. The limit of quantitation (functional sensitivity defined as lowest concentration with a CV <20%), which was used for reporting all values, for the LH assay is 0.1 mIU/mL. Assay reproducibility was monitored using commercial controls that ranged in LH concentrations from 4 to 50 mIU/mL; the CV for all these controls was <5%. Study specific variance was determined by repeated LH testing of aliquots of a pool made from the timed draws for each study. The normal reference range for adult men is 2.0–12.0 mIU/mL and the normal reference range for normally cycling women in the luteal phase is 0.6–19.0 mIU/mL.
FSH levels are expressed in units of the first International Pituitary Standard WHO [92/510]. The minimal detectable dose (lowest dose distinguishable from zero; 95% CI of a blank detection) for the FSH assay was 0.05 mIU/mL. The limit of quantitation for the FSH assay was 0.1 mIU/mL. In addition, QC sera containing 5–75 mIU/mL were tested daily and CVs were <5%. The normal reference range for adult men is 1.0–8.7 mIU/mL and the normal reference range for normally cycling women in the luteal phase is 2–13 mIU/mL.
Estradiol was measured by second generation immunoassay traceable to mass spectrometry-based assays (38). Functional sensitivity of the estradiol assay was 15 pg/mL; minimal detectable difference was 5 pg/mL. CV was <8% at low control of 45 pg/mL. Testosterone was measured by a second generation immunoassay traceable to mass spectrometry-based assays (39). Functional sensitivity of the testosterone assay was 4.33 ng/dL; the minimal detectable difference was 2.67 ng/dL. CV was <10% for testosterone levels >15 ng/dL.
A validated modification of the method of Santen and Bardin, incorporating the study specific variance, was used to identify LH pulses (40, 41). LH amplitude was calculated as the maximal LH achieved minus the LH at baseline. For the subject who underwent reversal of his IHH and who therefore produced endogenous LH pulses, the binomial probability was used to calculate the likelihood that kisspeptin administration coincided with endogenous pulses by chance. P values less than 0.05 were considered significant. Unless otherwise noted, values are reported as mean ± standard deviation.
Results
Subject characteristics
Ten men and two women with IHH participated in this study, and their clinical characteristics are described in Table 1. Seven men and one woman had Kallmann syndrome; the remaining three men and one woman had normosmic IHH. One man with normosmic IHH had undergone reversal of his condition, first noted at age 21 as evidenced by normalization of serum testosterone (275 ng/dL) and increased testicular volume (42). Six subjects carried rare sequence variants in IHH genes that were predicted to be deleterious: FGFR1 (N = 3, p.[F747L; D768H] [normosmic IHH]; p.G687R [KS]; p.L630P [KS]), GNRHR (N = 1 p.[R262Q];[L286P] [normosmic IHH]), KAL1 (N = 1, p.L601YfsX18 [KS]), KISS1 (N = 1, p.C53R [KS]), and PROKR2 (N = 1, p.L173R [KS]) (Table 1). The remaining six subjects had no identified rare sequence variants predicted to be deleterious.
Table 1.
Subject Characteristics
| # | Sex | Age (y) | BMI (kg/m2) | Diagnosis | Age at Presentation (y)a | Rare Sequence Variantb | TV (mL) | Change in LH Amplitude in Response to GnRH (mIU/mL)c | Prior Hormone Treatment | Other |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 43 | 38.6 | KS | 16 | KAL1 p.L601YfsX18 | 9, 8 | 1.3 | GnRH | |
| T | ||||||||||
| 2 | M | 59 | 29.0 | KS | 18 | None identified | 2,2 | 0.1 | GnRH | Rheumatoid arthritis |
| hCG | ||||||||||
| FSH | ||||||||||
| T | ||||||||||
| 3 | M | 20 | 35.1 | KS | 14 | None identified | 2, 3 | 2.6 | hCG | |
| T | ||||||||||
| 4 | M | 38 | 45.7 | KS | 17 | PROKR2 p.L173R | 4, 5 | 0.5 | GnRH | |
| FSH | ||||||||||
| T | ||||||||||
| 5 | M | 53 | 28.6 | KS | 25 | None identified | 2, 2 | 0.5 | GnRH | |
| T | ||||||||||
| 6 | M | 32 | 31.9 | nIHH | 12 | FGFR1 p.[F747L; D768H] | 6, 6 | 1.4 | T | |
| 7 | M | 42 | 33.5 | nIHH | 16 | None identified | 10, 12 | 6.8 | hCG | |
| FSH | ||||||||||
| T | ||||||||||
| 8 | M | 47 | 28.5 | nIHH | 21 | None identified | 15, 10 | 5.9 | GnRH | |
| hCG | ||||||||||
| T | ||||||||||
| 9 | M | 22 | 22.7 | KS | 17 | FGFR1 p.G687R | 4, 5 | 4.6 | T | |
| KISS1 p.C53R | ||||||||||
| 10 | F | 54 | 31.0 | KS | 18 | FGFR1 p.L630P | NA | 6.3 | None | Hypothyroidism |
| 11 | F | 22 | 30.2 | nIHH | 14 | GNRHR p.[R262Q]; [L286P] | NA | 0.3 | Estrogen/progesterone | |
| OCP | ||||||||||
| 12d | M | 23 | 40.5 | nIHH | 19 | None identified | 15, 20 | 7.3 | T | Reversal |
Age at presentation for delayed puberty. Subjects under the age of 18 were re-evaluated after the age of 18 to confirm the diagnosis of IHH, which was further re-confirmed for subjects 1–11 through participation in this study. Subjects 3 and 6 had been followed from birth due to cryporchidism and therefore presented at an earlier age.
Variants are listed if they were predicted to be deleterious by at least three out of four prediction programs.
Change in LH amplitude in response to GnRH is peak LH - baseline LH in response to 75 ng/kg bolus of intravenous GnRH prior to pituitary priming.
This subject attained serum testosterone in normal adult range in the absence of exogenous treatment. BMI, body mass index; KS, Kallmann syndrome; nIHH, normosmic idiopathic hypogonadotropic hypogonadism; NA, not applicable; OCP, oral contraceptive pills; T, testosterone; TV, testicular volume.
“Pre-priming”: initial responses to kisspeptin and GnRH
Subjects were admitted to the MGH Clinical Research Center for frequent blood sampling at 10 min intervals to chart endogenous LH secretion and responses to kisspeptin and GnRH (Supplemental Figure 1). Of the 11 subjects with abiding hypogonadotropism, whether normosmic or with KS, none exhibited any LH pulses at baseline. Seven of 11 subjects had LH levels near or at the limit of detection (mean ± SD LH 0.1 ± 0.1 mIU/mL). In contrast, four of 11 subjects had detectable but nonpulsatile LH secretion (LH 1.1 ± 0.1 mIU/mL). Of note, these same 4 subjects demonstrated robust responses to exogenous GnRH 75 ng/kg (LH pulse amplitude 5.9 ± 0.9 mIU/mL) suggesting that their pituitaries were in a greater state of “readiness,” likely due to endogenous stimulation from an intact but enfeebled GnRH secretory program (Table 1, subjects 7–10).
After receiving a kisspeptin bolus of 0.24 nmol/kg, no subject with abiding hypogonadotropism responded with an LH pulse. The average change in LH after kisspeptin was 0.1 ± 0.1 mIU/mL. This is in marked contrast to our experience in healthy men, in which administration of the same dose of kisspeptin-10 resulted in an LH pulse with an average amplitude of 5.0 ± 3.0 mIU/mL (29). The lack of response to kisspeptin administration in subjects with abiding hypogonadotropism was seen regardless of their baseline LH levels and their responses to exogenous GnRH (Figure 2, A and C and Supplemental Figure 1).
Figure 2.

Baseline LH secretion and responses to kisspeptin and GnRH in two representative male (♂) subjects. The phenotype, Kallmann syndrome (KS) or normosmic IHH (nIHH), and genotype are noted for each subject. Results for subject 1 before and after priming with exogenous GnRH are shown in panels A and B, respectively. Similarly, results for subject 6 before and after priming are shown in panels C and D, respectively. Arrows indicate times of boluses. T, testosterone levels during the study. FSH values measured from first 2-h pool.
“Post-priming”: responses to kisspeptin and GnRH
Eight subjects underwent pituitary priming with exogenous pulsatile GnRH × 6 days. This resulted in successful priming in all subjects (except the subject with GNRHR mutations described below), with robust responses to boluses of GnRH administered during the second “post-priming” CRC admission. Because LH levels from a single GnRH bolus did not return to baseline before the next bolus was administered, it was only possible to quantify the LH response to the first GnRH dose of 25 ng/kg. The amplitude of LH pulses in response to this first bolus of GnRH was 7.7 ± 8.0 mIU/mL (mean ± SD).
Despite this evidence for successful pituitary priming, subjects with abiding hypogonadotropism still failed to respond to kisspeptin 0.24 ng/kg iv (change in LH 0.1 ± 0.1 mIU/mL) (Figure 2, B and D and Supplemental Figure 1).
“Post priming”: a subject bearing GNRHR mutations serves as a negative control
One normosmic subject had compound heterozygous mutations in GNRHR: p.R262Q, which has been shown to be deleterious in functional studies (43), and p.L286P which was predicted to be deleterious in 4 of 4 prediction programs. Consistent with her genotype, despite priming with pulsatile GnRH, she failed to respond to exogenous GnRH and exogenous kisspeptin (Figure 3). Moreover, this patient's lack of response to kisspeptin suggests that kisspeptin does not trigger the release of the LH at the level of the pituitary.
Figure 3.
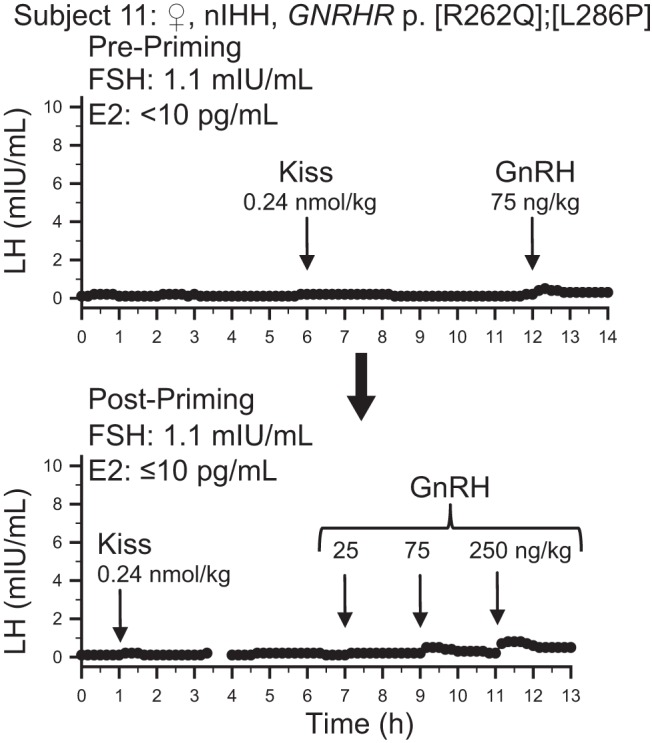
Baseline LH secretion and responses to kisspeptin and GnRH in a female (♀) subject with normosmic IHH (nIHH) and GnRH resistance due to the specified GNRHR mutations. Upper panel: results before priming with exogenous GnRH (pre-priming). Lower panel: results after priming with a failure to prime due to GnRH resistance (post-priming). Arrows indicate times of boluses. E2, estradiol levels during the study. FSH values measured from the first 2-h pool.
From pituitary gonadotrope priming to GnRH neuronal priming
To test the possibility that the GnRH neuronal network itself requires priming with repetitive exposure to kisspeptin, three subjects returned for a third visit to the CRC and received multiple boluses of kisspeptin. These patients were selected to participate in this additional arm of the protocol because they had evidence for an intact albeit enfeebled GnRH secretory program at baseline (measurable baseline LH levels and responsiveness to exogenous GnRH before formal priming). Thus, they did not require priming with the GnRH pulsatile pump. Despite these repeated and/or escalating doses of kisspeptin, no LH response to kisspeptin was seen (Figure 4).
Figure 4.
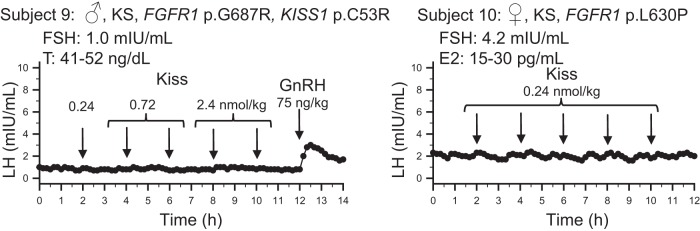
Responses to multiple boluses kisspeptin in a male (♂) and a female (♀) subject with Kallmann syndrome (KS) and evidence of endogenous GnRH neuronal activity. Arrows indicate times of boluses. Note escalating doses of kisspeptin provided to subject 9. T, testosterone; E2, estradiol levels during the study. FSH values measured from the first 2-h pool.
Exploring the sub-phenotype of hypogonadotropic reversal: response to kisspeptin
Subject #12 had previously been diagnosed with normosmic IHH based on low serum testosterone (16 ng/dL), low gonadotropin levels, and small testicular volumes at age 19 (42). No protein-altering genetic mutations were found in 14 IHH genes. Over the next 4 years he demonstrated evidence for spontaneous activation of his hypothalamic-pituitary-gonadal cascade. By age 23, he had a testosterone level of 918 ng/dL and testicular volumes of 15 and 20 mL. During his initial admission to the CRC, he exhibited pulsatile LH secretion at baseline (2 pulses in 6 h, with amplitudes of 3.0 and 0.9 mIU/mL) (Figure 5).
Figure 5.
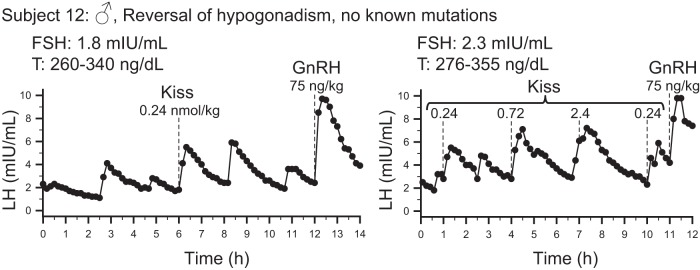
Intact responses to kisspeptin in a male (♂) subject with reversal of IHH. Both studies were performed without pituitary priming. The left panel shows the single bolus protocol and demonstrates the presence of endogenous pulsatile LH secretion and a response to kisspeptin. The right panel shows the study performed with multiple kisspeptin boluses and demonstrates consistent pulses of LH secretion in response to kisspeptin. Dashed lines mark the times kisspeptin or GnRH were given. T, testosterone levels during the study. FSH values measured from the first 2-h pool.
After receiving a bolus of exogenous kisspeptin, this subject demonstrated a robust response with an LH pulse amplitude of 3.7 mIU/mL (Figure 5).
Given the robust nature of this subject's endogenous GnRH pulse generator, it was possible that the administration of kisspeptin in this initial study coincided with the nadir of an endogenous GnRH pulse. This subject therefore returned for a second study during which he received multiple boluses of kisspeptin. Each of these boluses induced an LH pulse (Figure 5). Based on the subject's endogenous pulse frequency, it is improbable that all five kisspeptin boluses coincided with endogenous LH pulses (P = .001).
Discussion
Contrary to our hypothesis that exogenous kisspeptin administration could be used to distinguish patients with an absent GnRH neuronal complement from those with a reduced or impaired neuronal network, all subjects with abiding IHH failed to respond to kisspeptin, regardless of their genotype. In contrast, one subject who had undergone reversal of his hypogonadotropism demonstrated robust responses to kisspeptin.
The range of genotypes represented in this study provides an important dimension to the interpretation of the findings. Subject 1 carried a hemizygous frameshift mutation in KAL1; nearly all patients with KAL1 mutations exhibit lifelong hypogonadotropism attributed to abnormal GnRH neuronal migration (44). Consistent with a lack of GnRH neurons, subject 1 had no response to kisspeptin. Another subject, subject 11, carried compound heterozygous mutations in GNRHR. Consistent with a lack of GnRH signaling in the pituitary, she also exhibited no response to kisspeptin. In contrast to the these two individuals, several other subjects (subjects 4, 6, 9, 10) carried mutations in genes characterized by variable expressivity and incomplete penetrance (PROKR2 or FGFR1) but importantly, none of these subjects responded to kisspeptin. Hypomorphic and knockout mouse models for these genes have revealed that mice with mutations in these genes still have an intact or only partially attenuated GnRH neuronal complement in the hypothalamus (45–47). By extrapolation, it is possible that patients bearing heterozygous mutations in PROKR2 or FGFR1 also retain some GnRH neurons. Indeed, several of the subjects in this study (subjects 7–10, two of whom have heterozygous FGFR1 mutations) had evidence of some endogenous GnRH secretion, though not enough to sustain normal reproductive function. Thus, our results demonstrate that this residual GnRH neuronal complement is not sufficient to confer responsiveness to exogenous kisspeptin stimulation at a dose that evinces a robust response in healthy men and luteal phase women. It is important to note that patients bearing mutations in the neurokinin B pathway have been previously reported to be responsive to exogenous kisspeptin (48). However, those patients received a continuous infusion of kisspeptin as opposed to the single boluses administered in this study. Moreover, the magnitude of the LH response was quite modest, with LH and sex steroids remaining below the normal range and far below the magnitude of the response observed in healthy men undergoing a similar protocol, who achieved LH approximately two times higher than the upper limit of the normal range (30). Thus, in patients with abiding IHH, there is no clinically meaningful response to kisspeptin.
There are several possible explanations for why patients with abiding hypogonadotropism fail to respond to boluses of kisspeptin. First, it is possible that mutations exist in KISS1R, which encodes the kisspeptin receptor; while we screened for coding mutations in KISS1R, regulatory or intronic mutations would have escaped detection. Second, GnRH neurons may require “priming” with repeated exposure to kisspeptin to exhibit robust responses. However, our attempt to perform priming with the administration of multiple kisspeptin boluses failed to effect a GnRH-induced LH response. In addition, Kiss1−/− mice respond to their first exposure to kisspeptin (49), as do prepubertal rhesus monkeys (50), further arguing against the need for GnRH neuronal priming. Third, in mice, 5–40% of GnRH neurons do not respond to kisspeptin (51) and some GnRH neurons do not express Kiss1r (52), raising the possibility that different subsets of GnRH neurons may be differentially affected in IHH. In other words, congenital hypogonadotropism might preferentially affect those GnRH neurons that respond to kisspeptin. Finally, GnRH neurons form a complex network; kisspeptin may be able to signal at kisspeptin receptors on specific GnRH neurons, but other factors may prevent a coordinated response such that there is no significant LH secretion from the pituitary. Whatever the factors are that contribute to the lack of a response to exogenous administration of kisspeptin, they seem to supersede the specific genetic signature of each subject (with the exception of possible undetected regulatory mutations in KISS1R).
The fact that follicular phase women, who have physiologically low estradiol levels, demonstrate minimal responses to kisspeptin (28, 31, 53) suggests that sex steroids may play a role in modulating responsiveness to this neuropeptide. However, robust responses to exogenous kisspeptin have been observed in women with hypothalamic amenorrhea (low estradiol levels) (54, 55), in contrast to the lack of response to kisspeptin in IHH subjects observed in this study. Similarly, robust responses to kisspeptin are seen in animal models of GnRH deficiency akin to hypothalamic amenorrhea, such as rats undergoing food deprivation and mice with leptin pathway mutations (56, 57). While sex steroids can modify kisspeptin responsiveness (50, 53), both IHH and HA subjects were studied in the hypogonadal state, raising the need to consider other factors that might account for the differences in their ability to respond to kisspeptin. Most patients with HA undergo a normal “minipuberty” of infancy and normal timing of sexual maturation. Hypothetically, the physiology underlying these developmental milestones may itself change the hypothalamic architecture regulating GnRH release in a permanent way, so that the ability to respond to kisspeptin is retained even in the face of stressors such as excessive exercise or caloric deprivation.
In contrast to the subjects with abiding IHH, one subject who underwent reversal of IHH responded robustly to kisspeptin. Although it is impossible to know from these studies when this patient acquired kisspeptin responsiveness, it appears reasonable to conclude that the activation of the hypothalamic-pituitary-gonadal cascade that characterizes the reversal state in this patient appears to be mediated through kisspeptin signaling. Prospective research examining kisspeptin responsiveness in IHH patients before and after reversal will determine whether kisspeptin responsiveness exists in these patients prior to reversal or is acquired during the process of reversal.
The strengths of this study include the use of patients with a rare but prismatic disease model that speaks directly to GnRH secretory function, incorporation of contemporary genetic testing, q10 min blood sampling to capture any pulsatile activity if present, and pituitary priming with exogenous pulsatile GnRH. Limitations of this study include the inability to study a patient with homozygous mutations in KISS1, as such patients are extremely rare (58). Kisspeptin was given as IV boluses to mimic the reported physiologic pulsatile secretion of kisspeptin. It remains possible that even higher doses or infusions of kisspeptin may elicit responses in IHH patients, though the physiologic significance of such findings would be unclear.
In summary, patients with congenital hypogonadotropism do not respond to a dose of kisspeptin that, when given to men and women in the luteal phase, stimulates a physiologic GnRH-induced LH response. This study raises the possibility that even if GnRH neurons reach the hypothalamus in patients with IHH, their functional integrity is compromised. In contrast, in a patient who underwent reversal of his hypogonadotropism, the capacity of GnRH neurons to respond to kisspeptin appears to be intact. Additional research is needed to determine whether reversal is caused by the acquisition of the ability to respond to kisspeptin.
Acknowledgments
We thank the research subjects, members of the Massachusetts General Hospital Reproductive Endocrine Unit for discussions and reading of the manuscript, staff of the Harvard Catalyst Clinical Research Center for assistance with the frequent sampling studies, the Massachusetts General Hospital Clinical Laboratory Research Core and Patrick Sluss for assistance with assays, and Dr Sadaf Farooqi for early discussions about the design of this study. We also thank the physicians who referred the research subjects: Lynn Bennion, William F. Crowley, Siri Atma W. Greeley, Frances Hayes, Susan Kirsch, Rose Schneier, and Margaret Wierman. We also thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-295 102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-296 103010).
This work was supported by Grants No. R01 HD043341 and U54 HD028138 from the Eunice K. Shriver National Institute for Child Health and Human Development (NICHD), a Clinical Scholars Award from the Pediatric Endocrine Society, and the Harvard Catalyst/Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health Awards No. UL1 RR 025758 and UL1 TR000170 and financial contributions from Harvard University and its affiliated academic health care centers). S.B.S. was also supported by K24 HD067388. Y-M.C. was supported by K12 HD052896, a Charles A. King Trust postdoctoral fellowship, a Career Development Award from Boston Children's Hospital, a Charles H. Hood Foundation Child Health Research Award, and a Doris Duke Clinical Scientist Development Award. M.F.L. was supported by T32 DK007028 and a Postdoctoral Fellowship Award for Clinical Research from the Massachusetts General Hospital Executive Committee on Research Fund for Medical Discovery. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centers, or the National Institutes of Health.
Clinicaltrials.gov registration number: NCT00914823.
Disclosure Summary: The authors have nothing to disclose.
References
- 1. Crowley WF, Jr., McArthur JW. Simulation of the normal menstrual cycle in Kallman's syndrome by pulsatile administration of luteinizing hormone-releasing hormone (LHRH). J Clin Endocrinol Metab. 1980;51:173–175. [DOI] [PubMed] [Google Scholar]
- 2. Hoffman AR, Crowley WF., Jr Induction of puberty in men by long-term pulsatile administration of low-dose gonadotropin-releasing hormone. N Engl J Med. 1982;307:1237–1241. [DOI] [PubMed] [Google Scholar]
- 3. Santoro N, Filicori M, Crowley WF., Jr Hypogonadotropic disorders in men and women: diagnosis and therapy with pulsatile gonadotropin-releasing hormone. Endocr Rev. 1986;7:11–23. [DOI] [PubMed] [Google Scholar]
- 4. Spratt DI, Finkelstein JS, O'Dea LS, et al. Long-term administration of gonadotropin-releasing hormone in men with idiopathic hypogonadotropic hypogonadism. A model for studies of the hormone's physiologic effects. Ann Intern Med. 1986;105:848–855. [DOI] [PubMed] [Google Scholar]
- 5. Spratt DI, Finkelstein JS, Butler JP, Badger TM, Crowley WF., Jr Effects of increasing the frequency of low doses of gonadotropin-releasing hormone (GnRH) on gonadotropin secretion in GnRH-deficient men. J Clin Endocrinol Metab. 1987;64:1179–1186. [DOI] [PubMed] [Google Scholar]
- 6. Finkelstein JS, Badger TM, O'Dea LS, Spratt DI, Crowley WF. Effects of decreasing the frequency of gonadotropin-releasing hormone stimulation on gonadotropin secretion in gonadotropin-releasing hormone-deficient men and perifused rat pituitary cells. J Clin Invest. 1988;81:1725–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Finkelstein JS, O'Dea LS, Whitcomb RW, Crowley WF., Jr Sex steroid control of gonadotropin secretion in the human male. II. Effects of estradiol administration in normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab. 1991;73:621–628. [DOI] [PubMed] [Google Scholar]
- 8. Finkelstein JS, Whitcomb RW, O'Dea LS, Longcope C, Schoenfeld DA, Crowley WF., Jr Sex steroid control of gonadotropin secretion in the human male. I. Effects of testosterone administration in normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab. 1991;73:609–620. [DOI] [PubMed] [Google Scholar]
- 9. Sheckter CB, McLachlan RI, Tenover JS, et al. Stimulation of serum inhibin concentrations by gonadotropin-releasing hormone in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 1988;67:1221–1224. [DOI] [PubMed] [Google Scholar]
- 10. Sykiotis GP, Pitteloud N, Seminara SB, Kaiser UB, Crowley WF., Jr. Deciphering genetic disease in the genomic era: the model of GnRH deficiency. Sci Transl Med. 2010;2:32rv32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Franco B, Guioli S, Pragliola A, et al. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:529–536. [DOI] [PubMed] [Google Scholar]
- 12. Legouis R, Hardelin JP, Levilliers J, et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell. 1991;67:423–435. [DOI] [PubMed] [Google Scholar]
- 13. Schwanzel-Fukuda M, Bick D, Pfaff DW. Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Brain Res Mol Brain Res. 1989;6:311–326. [DOI] [PubMed] [Google Scholar]
- 14. Bouligand J, Ghervan C, Tello JA, et al. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med. 2009;360:2742–2748. [DOI] [PubMed] [Google Scholar]
- 15. Chan YM, de Guillebon A, Lang-Muritano M, et al. GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA. 2009;106:11703–11708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA. 2003;100:10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627. [DOI] [PubMed] [Google Scholar]
- 18. Pitteloud N, Meysing A, Quinton R, et al. Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol. 2006;254–255:60–69. [DOI] [PubMed] [Google Scholar]
- 19. Sarfati J, Dodé C, Young J. Kallmann syndrome caused by mutations in the PROK2 and PROKR2 genes: pathophysiology and genotype-phenotype correlations. Front Horm Res. 2010;39:121–132. [DOI] [PubMed] [Google Scholar]
- 20. Pitteloud N, Quinton R, Pearce S, et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest. 2007;117:457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sykiotis GP, Plummer L, Hughes VA, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci USA. 2010;107:15140–15144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Raivio T, Falardeau J, Dwyer A, et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med. 2007;357:863–873. [DOI] [PubMed] [Google Scholar]
- 23. Sidhoum VF, Chan YM, Lippincott MF, et al. Reversal and relapse of hypogonadotropic hypogonadism: resilience and fragility of the reproductive neuroendocrine system. J Clin Endocrinol Metab. 2014;99:861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. George JT, Seminara SB. Kisspeptin and the hypothalamic control of reproduction: lessons from the human. Endocrinology. 2012;153:5130–5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keen KL, Wegner FH, Bloom SR, Ghatei MA, Terasawa E. An increase in kisspeptin-54 release occurs with the pubertal increase in luteinizing hormone-releasing hormone-1 release in the stalk-median eminence of female rhesus monkeys in vivo. Endocrinology. 2008;149:4151–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith JT, Rao A, Pereira A, Caraty A, Millar RP, Clarke IJ. Kisspeptin is present in ovine hypophysial portal blood but does not increase during the preovulatory luteinizing hormone surge: evidence that gonadotropes are not direct targets of kisspeptin in vivo. Endocrinology. 2008;149:1951–1959. [DOI] [PubMed] [Google Scholar]
- 27. Dhillo WS, Chaudhri OB, Patterson M, et al. Kisspeptin-54 stimulates the hypothalamic-pituitary gonadal axis in human males. J Clin Endocrinol Metab. 2005;90:6609–6615. [DOI] [PubMed] [Google Scholar]
- 28. Dhillo WS, Chaudhri OB, Thompson EL, et al. Kisspeptin-54 stimulates gonadotropin release most potently during the preovulatory phase of the menstrual cycle in women. J Clin Endocrinol Metab. 2007;92:3958–3966. [DOI] [PubMed] [Google Scholar]
- 29. Chan YM, Butler JP, Pinnell NE, et al. Kisspeptin resets the hypothalamic GnRH clock in men. J Clin Endocrinol Metab. 2011;96:E908–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. George JT, Veldhuis JD, Roseweir AK, et al. Kisspeptin-10 is a potent stimulator of LH and increases pulse frequency in men. J Clin Endocrinol Metab. 2011;96:E1228–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chan YM, Butler JP, Sidhoum VF, Pinnell NE, Seminara SB. Kisspeptin administration to women: a window into endogenous kisspeptin secretion and GnRH responsiveness across the menstrual cycle. J Clin Endocrinol Metab. 2012;97:E1458–E1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Exome Variant Server. NHLBI Grand Opportunity Exome Sequencing Project (ESP), Seattle, WA: http://evs.gs.washington.edu/EVS/6500 samples, April, 2014. [Google Scholar]
- 33. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. [DOI] [PubMed] [Google Scholar]
- 36. Thomas PD, Kejariwal A, Guo N, et al. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 2006;34:W645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Crowley WF., Jr An overview of LHRH and its analogues: clinical uses. Ups J Med Sci. 1984;89:3–12. [DOI] [PubMed] [Google Scholar]
- 38. Sluss PM, Hayes FJ, Adams JM, et al. Mass spectrometric and physiological validation of a sensitive, automated, direct immunoassay for serum estradiol using the Architect. Clin Chim Acta. 2008;388:99–105. [DOI] [PubMed] [Google Scholar]
- 39. Bui HN, Sluss PM, Blincko S, Knol DL, Blankenstein MA, Heijboer AC. Dynamics of serum testosterone during the menstrual cycle evaluated by daily measurements with an ID-LC-MS/MS method and a 2nd generation automated immunoassay. Steroids. 2013;78:96–101. [DOI] [PubMed] [Google Scholar]
- 40. Santen RJ, Bardin CW. Episodic luteinizing hormone secretion in man. Pulse analysis, clinical interpretation, physiologic mechanisms. J Clin Invest. 1973;52:2617–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hayes FJ, McNicholl DJ, Schoenfeld D, Marsh EE, Hall JE. Free alpha-subunit is superior to luteinizing hormone as a marker of gonadotropin-releasing hormone despite desensitization at fast pulse frequencies. J Clin Endocrinol Metab. 1999;84:1028–1036. [DOI] [PubMed] [Google Scholar]
- 42. Gianetti E, Tusset C, Noel SD, et al. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab. 2010;95:2857–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Roux N, Young J, Misrahi M, et al. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997;337:1597–1602. [DOI] [PubMed] [Google Scholar]
- 44. Hu Y, Bouloux PM. X-linked GnRH deficiency: role of KAL-1 mutations in GnRH deficiency. Mol Cell Endocrinol. 2011;346:13–20. [DOI] [PubMed] [Google Scholar]
- 45. Pitteloud N, Zhang C, Pignatelli D, et al. Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA. 2007;104:17447–17452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chung WC, Moyle SS, Tsai PS. Fibroblast growth factor 8 signaling through fibroblast growth factor receptor 1 is required for the emergence of gonadotropin-releasing hormone neurons. Endocrinology. 2008;149:4997–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matsumoto S, Yamazaki C, Masumoto KH, et al. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc Natl Acad Sci USA. 2006;103:4140–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Young J, George JT, Tello JA, et al. Kisspeptin restores pulsatile LH secretion in patients with neurokinin B signaling deficiencies: physiological, pathophysiological and therapeutic implications. Neuroendocrinology. 2013;97:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lapatto R, Pallais JC, Zhang D, et al. Kiss1−/− mice exhibit more variable hypogonadism than Gpr54−/− mice. Endocrinology. 2007;148:4927–4936. [DOI] [PubMed] [Google Scholar]
- 50. Guerriero KA, Keen KL, Millar RP, Terasawa E. Developmental changes in GnRH release in response to kisspeptin agonist and antagonist in female rhesus monkeys (Macaca mulatta): implication for the mechanism of puberty. Endocrinology. 2012;153:825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dumalska I, Wu M, Morozova E, Liu R, van den Pol A, Alreja M. Excitatory effects of the puberty-initiating peptide kisspeptin and group I metabotropic glutamate receptor agonists differentiate two distinct subpopulations of gonadotropin-releasing hormone neurons. J Neurosci. 2008;28:8003–8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Herbison AE, de Tassigny Xd, Doran J, Colledge WH. Distribution and postnatal development of Gpr54 gene expression in mouse brain and gonadotropin-releasing hormone neurons. Endocrinology. 2010;151:312–321. [DOI] [PubMed] [Google Scholar]
- 53. George JT, Anderson RA, Millar RP. Kisspeptin-10 stimulation of gonadotrophin secretion in women is modulated by sex steroid feedback. Hum Reprod. 2012;27:3552–3559. [DOI] [PubMed] [Google Scholar]
- 54. Jayasena CN, Nijher GM, Chaudhri OB, et al. Subcutaneous injection of kisspeptin-54 acutely stimulates gonadotropin secretion in women with hypothalamic amenorrhea, but chronic administration causes tachyphylaxis. J Clin Endocrinol Metab. 2009;94:4315–4323. [DOI] [PubMed] [Google Scholar]
- 55. Jayasena CN, Nijher GM, Abbara A, et al. Twice-weekly administration of kisspeptin-54 for 8 weeks stimulates release of reproductive hormones in women with hypothalamic amenorrhea. Clin Pharmacol Ther. 2010;88:840–847. [DOI] [PubMed] [Google Scholar]
- 56. Quennell JH, Howell CS, Roa J, Augustine RA, Grattan DR, Anderson GM. Leptin deficiency and diet-induced obesity reduce hypothalamic kisspeptin expression in mice. Endocrinology. 2011;152:1541–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Castellano JM, Navarro VM, Fernández-Fernández R, et al. Changes in hypothalamic KiSS-1 system and restoration of pubertal activation of the reproductive axis by kisspeptin in undernutrition. Endocrinology. 2005;146:3917–3925. [DOI] [PubMed] [Google Scholar]
- 58. Topaloglu AK, Tello JA, Kotan LD, et al. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. N Engl J Med. 2012;366:629–635. [DOI] [PubMed] [Google Scholar]
- 59. Jayasena CN, Nijher GM, Comninos AN, Abbara A, Januszewki A, Vaal ML, Sriskandarajah L, Murphy KG, Farzad Z, Ghatei MA, Bloom SR, Dhillo WS. The effects of kisspeptin-10 on reproductive hormone release show sexual dimorphism in humans. J Clin Endocrinol Metab. 2011;96:E1963–E1972. [DOI] [PMC free article] [PubMed] [Google Scholar]