

Abstract
The most common mutation causing cystic fibrosis (CF) is deletion of phenylalanine residue 508 in the cystic fibrosis transmembrane regulator conductance (CFTR) protein. Small molecules that are able to correct the misfolding of defective ΔF508-CFTR have considerable promise for therapy. Reported here are the design, preparation, and evaluation of five more hydrophilic bisazole analogs of previously identified bithiazole CF corrector 1. Interestingly, bisazole ΔF508-CFTR corrector activity was not increased by incorporation of more H-bond acceptors (O or N), but correlated best with the overall bisazole molecular geometry. The structure activity data, together with molecule modeling, suggested that active bisazole correctors adopt a U-shaped conformation, and that corrector activity depends on the molecule’s ability to access this molecular geometry.
Keywords: cystic fibrosis; correctors; transmembrane regulator; C,C-linked bisazoles
Cystic fibrosis (CF) is a genetic disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator protein (CFTR). The most common CFTR mutation is deletion of a phenylalanine residue at position 508, ΔF508, which results in a misfolded CFTR that is retained at the endoplasmic reticulum and rapidly degraded.1 Failure to provide functioning CFTR, an ATP-gated chloride channel, in epithelial cells in the lungs, pancreas, and other tissues leads to impaired chloride transport. Defective chloride transport results in bacterial growth in the lung due to the accumulation of viscous mucus, and pancreatic malfunction.2 Intensive efforts have been taken to discover small molecules that are able to correct the folding of the ΔF508-CFTR and restore its normal transportation and ion channel function3 with two investigational correctors currently in clinical trial.4 The recent work of Yoo et al. indicates that bithiazole 1, a 4-(thiazol-5-yl)thiazole derivative, has significant corrector action on the mutant ΔF508-CFTR.5 A bithiazole-focused structure-activity-relationship study by Yu et al. showed that an s-cis coplanar conformation of the bithiazole, a peripheral pivolyl group, and a substituted aniline moiety are crucial structural features in eliciting ΔF508-CFTR corrector activity with the bithiazole chemo type.6 Indeed, the s-cis coplanar conformation, as exemplified by bithiazole 1a wherein the two thiazole rings are constrained by an alkyl chain, is required for corrector activity.6,7
However, bithiazoles are relatively hydrophobic (cLogP = 5.60 and 6.35 for 1 and 1a, respectively; Figure 1) and so may not be suitable for development of an orally administered drug (cf., Lipinski’s “rule of five”).8 Considering that cLogP values for oxazole, oxadiazole, and thiodiazole are much lower (−0.18, −1.41, and −0.60, respectively) than that of a thiazole ring (0.49), we set out to replace one of the two thiazole rings in 1 with these more hydrophilic five-membered aromatic heterocycles to explore different chemo types in the hope of identifying new CFTR correctors (e.g., 2) with less hydrophobicity. Given the importance of the peripheral pivalamide and 5-chloro-2-meth-oxyphenylamine moieties in conveying corrector activity, these two structural motifs were retained in the design of these new bisazole correctors. This ring replacement strategy, in conjunction with considering the relative accessibility of each target molecule, led to a series of bisazole analogs – the cLogP for which is lowered to a range of from 3.95 to 5.20 (Table 1). In addition to increased hydrophilicity, another possible benefit associated with oxygen and nitrogen-rich bisazole rings might be increased and stronger H-bonding interaction within the ΔF508-CFTR binding site. Herein, we report the synthesis of thiazole-tethered imidazolone, oxazole, oxadiazole and thiodiazole bisazole analogs, their ΔF508-CFTR corrector activity, and structure-activity-relationships in this series of bisazoles.
Figure 1.

cLogP and CFTR corrector lead development.
Table 1.
Corrector activity, Vmax, and cLogP data for bisazoles.
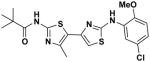 1 1
|
EC50(μM)a = 0.87 Vmax(μM/s)b = 97.97 CLogP = 5.60 |
 2a′ 2a′
|
EC50(μM)a = not active Vmax(μM/s)b = not active CLogP = 3.95 |
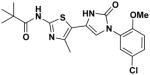
 2b 2b
|
EC50(μM)a = 0.74 Vmax(μM/s)b = 84.46 CLogP = 5.20 |
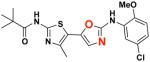
 2c 2c
|
EC50(μM)a = 0.66 Vmax(μM/s)b = 65.00 CLogP = 4.39 |
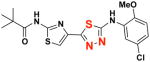 2d 2d
|
EC50(μM)a = not active Vmax(μM/s)b = not active CLogP = 5.04 |
 2e 2e
|
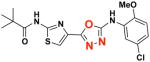
EC50(μM)a = 0.59 Vmax(μM/s)b = 35.46 CLogP = 4.39 |
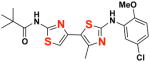 23 23
|
EC50(μM)a = not active Vmax(μM/s)b = not active CLogP = 5.60 |
Concentration where the increased I− influx is 50% of Vmax.
Vmax is the maximum increase in I− influx due to compound dosing.
While some directly-linked bisazoles have been reported,9,10,11 the thiazole-thethered bisazoles (2) targeted here have not been reported in the literature. Indeed, the preparation of directly-linked bisazoles with amide substituents are more difficult than expected as azole formation is problematic in the presence of interfering amino functionalities. With this backdrop, our first target, oxazolyl analog 2a, was initially expected to be available as depicted in Scheme 1 where isocyanate 3 is converted to urea 4 which would then be condensed with 1-(2-amino-4-methylthiazol-5-yl)-2-bromoethanone to deliver thiazole-oxazole 5. Chemoselective N-acylation with pivaloyl chloride would then deliver give 2a. An advantage of this planned route would be the opportunity to incorporate different acid chlorides in this final step. Unfortunately, all attempts at formation of oxazole 5 from urea 4 and thiazole 9 by reference methods were unsuccessful.12
Scheme 1.
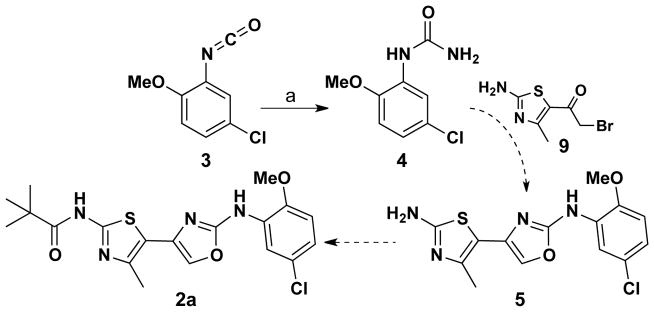
Reagents & conditions: a) NH3, THF, room temperature; 81%.
These failed oxazole-forming reactions (4 + 9 → 5) prompted us to investigate the alternative route to 4-(thiazol-5-yl)oxazole 2a detailed in Scheme 2. In this approach, the amino group of thiazole 6 was N-acylated first to give pivalamide 7 in a CDI-mediated coupling reaction (83% yield). We reasoned that making the amide first would decrease electron-donation into the thiazole ring (e.g., 6 vs. 7), thereby increasing the reactivity of the corresponding α-bromoketone 8 with urea 4. Unfortunately, while bromination of 7 with pyridinium tribromide gave 8 in 90% yield, subsequent reaction of this α-bromo ketone with urea 4 delivered 4-(thiazol-5-yl)-1H-imidazol-2(3H)-one 2a′ in 18% yield, instead of the targeted 4-(thiazol-5-yl)oxazole 2a.13 The Crank/Khan protocol for oxazole formation (replacing the -Br of 8 with an -OH and subsequent reaction with cyanoamide)14 also did not yield the desired oxazole.
Scheme 2.
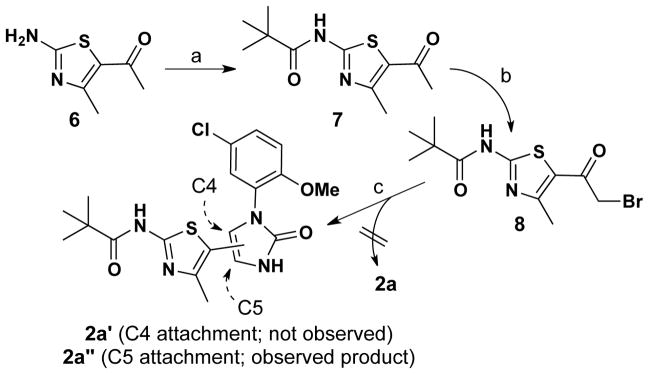
Reagents & conditions: a) pivalic acid, CDI, DMF, 80 °C; 83%. b) Pyridinium tribromide, 33% HBr in HOAc; 90%. c) 4, CaCO3, 120 °C; 18%.
We next set out to prepare oxazole 2b – the C5 analog of 2a. This thiazole-tethered oxazole analog was prepared via the three transformations depicted in Scheme 3. Treatment of bromide 9 with sodium azide gave 10 in 78% yield. Subsequent Straudinger reduction of this azide delivered the corresponding α-amino ketone as an unisolated intermediate; addition of thioisocyanate gave a urea intermediate which underwent in situ heterocyclization to give 5-(thiazol-5-yl)oxazole 11 in 44% overall yield. Acylation of 11 with pivaloyl chloride gave 2b (40% yield).
Scheme 3.
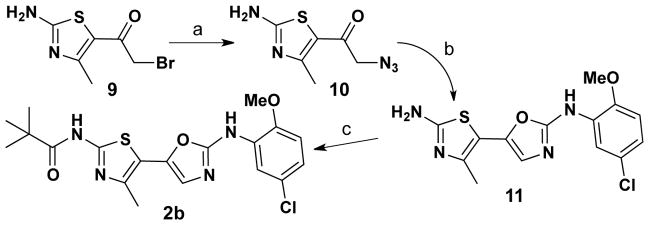
Reagents and conditions: a) NaN3, MeOH, 78%. b) PPh3, DCM; 4-chloro-2-isothiocyanato-1-methoxybenzene, DCM, 44%. c) Pivaloyl chloride, Et3N, DCM, 40%.
Our initial approach to oxadiazole and thiadiazole analogs is shown in Scheme 4, but two problems were encountered with this route. Firstly, thiazole-4-carboxylic 12 reacted effectively with hydrazinecarbothioamide (→ 13, X = S), but not hydrazinecarboxamide (→ 13, X = O). Secondly, carbothiamide 134 (X = S) could not be cyclized to thiadiazole 14 (X=S) in a practical yield under any of the protocols reported in literature.15
Scheme 4.
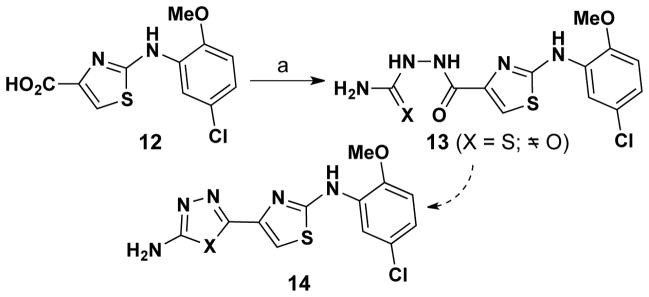
Reagents & conditions: a) NH2NHC(X)NH2 (X = O or S), EtN(iPr)2, DCC, CH2Cl2.
In light of these issues, we decided to focus on replacing the right-hand thiazole ring of lead compound 1 (see Figure 1) with oxadiazole and thiadiazole heterocycles. Both targeted analogs, 2c and 2d, were delivered via the unified route depicted in Scheme 5. In this approach, 2-aminothiazole 15 was converted to amide 16, which was then reacted with hydrazine hydrate to give thiazole-4-carbohydrazide 17 in 60% overall yield from 15. Hydrazide 17 was next reacted with 4-chloro-2-isothiocyanato-1-methoxybenzene to give 2-carbonylhydrazinecarbothioamide 18. From this common intermediate, 2-(thiazol-4-yl)-1,3,4-oxadiazole 2c was obtained in 66% yield by the reaction of 18 with tosyl chloride in pyridine, while 2-(thiazol-4-yl)-1,3,4-thiadiazole 2d was accessed via a phosphoryl trichloride-mediated cyclization process in 75% yield.
Scheme 5.

Reagents & conditions: a) Pivaloyl chloride, DCM, TEA, 50 °C, 1 h; 85%. b) NH2NH2•H2O, EtOH, 90 °C, 5 h; 71%. c) 4-Chloro-2-isothiocyanato-1-methoxybenzene, THF, rt, 18 h; 88%. d) (→ 2c) TsCl, THF, pyridine, 75 °C, 3 h; 66%. e) (→ 2d) POCl3, 110 °C, 4 h; 75%.
Finally, 2-(thiazol-4-yl)-1,3,4-oxadiazole analog 2e was prepared via the four-step process shown in Scheme 6. Treatment of N-substituted thiourea 19 with ethyl 3-bromo-2-oxopropanoate produced ethyl thiazolo-4-carboxylate 20 in 92% yield. Reacting ester 20 with hydrazine hydrate produced 21 and treatment of this hydrazide with CNBr delivered 1,3,4-oxadiazole 22 in 60% yield. Microwave irradiation of this 1,3,4-oxadiazol-2-amine with pivaloyl chloride in 1,4-dioxane (+ TEA) gave 2e in 45% yield.
Scheme 6.

Reagents & conditions: (a) ethyl 3-bromo-2-oxopropanoate, ethanol, 90 °C, 3 h; 92%; (b) NH2NH2•H2O, EtOH, 90 °C, 12 h; 77%; (c) CNBr, MeOH, rt, 18 h; 60%; (d) pivaloyl chloride, 1,4-dioxane, TEA, microwave, 40 min; 45%.
Bisazole analogs 2a′/2b–e were assayed for ΔF508-CFTR correct- or activity using our well-established cell-based corrector assay. Briefly, the influx of I− as surrogate for Cl− was measured in FRT cells co-expressing human ΔF508-CFTR employing the I−-sensitive fluorescent sensor YFP-H148Q/I152L.16,17 Following 24 h incubation with each test compound, I− influx was determined from the kinetics of YFP-H148Q/I152L quenching in response to I− addition in cells treated with a cAMP agonist and the potentiator genistein. The corrector activity of each analog was calculated from influx vs. concentration data.18 The corrector activity of bisazole analogs 2a′/2b–e, as well as lead compound 1 and reference bithiazole compound 23,2 are listed in Table 1.
Three of the five new bisazole analogs, thiazole-tethered oxazole 2b and oxdiazoles 2c and 2e, are effective at recovering the ion efflux function of ΔF508-CFTR. Among these three active bisazoles, 5-(oxazol-5′-yl)oxazole analog 2b is more potent than the oxadiazolyl analogs (2c and 2e) and its corrector activity is comparable with lead compound 1. Interestingly, thiodiazole analog 2d and heteroatom invertomer 23 are not active. Thiazole-tethered oxazole 2b is active in the corrector assay, while, not surprisingly, thiazole-tethered imidazol-2-one 2a′ is not. It is intriguing to note that the active oxazole analog 2b is an isostere of inactive bithiazole 23. Collectively, the results within this series suggest that corrector activity is tied to overall molecular geometry.
Conformational searches on 1, 2a–e and 23 were carried out using the Merck molecular force field in Spartan.19,20 The lowest energy conformers were then optimized using GAUSSIAN0921 with the M06-2X/6-31+G(d,p)22 density functional method. Relative energies were calculated both in the gas phase (as an extreme model of nonpolar environments) and water (using the SMD continuum solvation method).23 Structural drawings in Table 2 were produced using CYLView.24
Table 2.
Results of conformational analyses.
| cmpd | active or inactive? | lowest energy conformer in water | lowest energy U-shaped conformer in water |
|---|---|---|---|
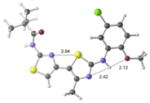
| 1 | active |
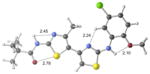
|
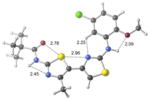 +7.1 kcal/mol −0.2 kcal/mol in gas phase |
| 2a | unknown (see 2a′) |
|
|
| 2b | active |

|
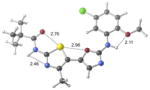 +0.2 kcal/mol −1.3 kcal/mol in gas phase |
| 2c | active |
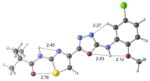
|
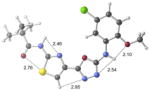 +1.7 kcal/mol +0.6 kcal/mol in gas phase |
| 2d | inactive |
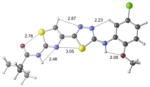
|
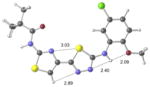 +7.3 kcal/mol +12.2 kcal/mol in gas phase |
| 2e | active |
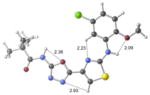
|
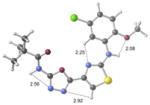 +1.0 kcal/mol −0.9 kcal/mol in gas phase |
| 23 | inactive |
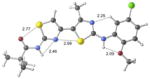
|
 +8.4 kcal/mol +5.9 kcal/mol in gas phase |
Free energies are relative to the lowest energy conformer in water.
Free energies are relative to the lowest energy conformer in the gas phase.
Our previous work,5–7 especially that with tethered bithiazoles such as 1a, provides circumstantial evidence in support of a “U-shaped” bioactive conformation (Figure 2), rigidified in part by S•••lone pair interactions.6,25 The overall lowest energy conformer found for each compound, along with the lowest energy U-shaped conformer (both in water) are shown in Table 2 (see Supporting Information for additional details, including interatomic distances). These results indicate that: (1) the preferred conformer varies from bisazole to bisazole and (2) not having a U-shaped conformer as the preferred conformation does not preclude activity (see 1 and 2b,c,e) when a U-shaped conformer can be readily accessed (i.e., gas phase U-shaped conformer within ~1 kcal/mol of the lowest energy conformer; that activity appears to track with gas phase energies suggests that the target binding site is not particularly polar). Of course, having an accessible U-shaped conformer does not guarantee of activity, since a compound could be inactive due to subtle differences in non-covalent interactions with its binding site, off-target effects or other issues with this whole cell assay. Point (2) above suggests that the putative bioactive conformation shown in Figure 2 likely needs to be accessible for activity; note that gas phase U-shaped conformers for inactive 2d and 23 are 12.2 and 5.9 kcal/mol, respectively, higher in energy than their preferred conformations. Our previous work indicates that the conformation about the inter-ring bond (see ~coplanar bisazole rings in Figure 2) correlates with activity as constrained 1a is more active than unconstrained 1.5–7 It therefore appears that access to the conformational control elements highlighted in Figure 2 are required for activity.
Figure 2.

Putative bioactive U-shaped conformation (illustrated for bithiazole 1).
Thiazole-tethered bisazoles [thiazole-oxazole (2b), thiazole-oxadiazole (2c and 2e), and thiazole-thiodiazole (2d)], which are structurally related to the bithiazole ΔF508-CFTR corrrector 1, were prepared from carbonyl-substituted thiazoles and a series of new bisazole CF correctors have been identified. While the newly identified bisazole correctors contain more oxygen and/or nitrogen atoms and have lower cLogP values than bithazole 1, their ΔF508-CFTR corrector activity does not correlate with the number of the H-bonding acceptors. SAR analysis suggested that incorporation of more H-bond acceptors can fundamentally change the molecular geometry in these bisazole systems (see 1 vs. 2b–e vs. 23). Finally, bithazole-to-bisazole atom substitution can modulate the accessibility of the U-shaped conformation required for corrector activity.
Supplementary Material
Acknowledgments
The authors thank the Tara K. Telford Fund for Cystic Fibrosis Research at the University of California, Davis, the National Institutes of Health (Grants DK072517, GM089153, and HL073856), the Petroleum Research Fund of the American Chemical Society (grant 52801-ND4), and the National Science Foundation (Grants CHE-0910870, CHE-0443516, CHE-0449845, and CHE-9808183 supporting NMR spectrometers). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation [ACI-1053575].
Footnotes
Supplementary data associated with this article (experimental details) can be found, in the online version, at XXXXX.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Hryciw DH, Guggino WB. Clin Exp Pharmacol Physiol. 2000;27:892. doi: 10.1046/j.1440-1681.2000.03356.x. [DOI] [PubMed] [Google Scholar]
- 2.Widdicombe JH. In: Cystic Fibrosis Transmembrane Conductance Regulator. Kirk KL, Dawson DC, editors. 2003. pp. 137–159. [Google Scholar]
- 3.Becq F. Curr Pharm Des. 2006;12:471. doi: 10.2174/138161206775474459. [DOI] [PubMed] [Google Scholar]
- 4.(a) Deeks ED. Drugs. 2013;73:1595. doi: 10.1007/s40265-013-0115-2. [DOI] [PubMed] [Google Scholar]; (b) Kopeikin Z, Yuksek Z, Yang H-Y, Bompadre SG. J Cyst Fibros. 2014 doi: 10.1016/j.jcf.2014.04.003. Ahead of Print. [DOI] [PubMed] [Google Scholar]
- 5.Yoo CL, Yu GJ, Yang B, Robins LI, Verkman AS, Kurth MJ. Bioorg Med Chem Lett. 2008;18:2610. doi: 10.1016/j.bmcl.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu GJ, Yoo CL, Yang B, Lodewyk MW, Meng L, El-Idreesy TT, Fettinger JC, Tantillo DJ, Verkman AS, Kurth MJ. J Med Chem. 2008;51:6044. doi: 10.1021/jm800533c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coffman KD, Nguyen HH, Phuan P-W, Hudson B, Yu GJ, Bagdasarian AL, MontgomerY D, Lodewyk MW, Yang B, Yoo CL, Verkman AS, Tantillo DJ, Kurth MJ. J Med Chem. 2014;57:6729. doi: 10.1021/jm5007885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Del Rev. 1997;23:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 9.Kim JY, Seo HJ, Lee SH, Jung ME, Ahn K, Kim J, Lee J. Bioorg Med Chem Lett. 2009;19:142. doi: 10.1016/j.bmcl.2008.10.130. [DOI] [PubMed] [Google Scholar]
- 10.Quan C, Kurth M. J Org Chem. 2004;69:1470. doi: 10.1021/jo0352124. [DOI] [PubMed] [Google Scholar]
- 11.Wang S, Meades C, Wood G, Osnowski A, Anderson S, Yuill R, Thomas M, Mezna M, Jackson W, Midgley C, Griffiths G, Fleming I, Green S, McNae I, Wu SY, McInnes C, Zheleva D, Walkinshaw MD, Fischer P. J Med Chem. 2004;47:1662. doi: 10.1021/jm0309957. [DOI] [PubMed] [Google Scholar]
- 12.For protocols, see: Pathak VN, Goyal MK, Jain M, Joshi KC. J Ind Chem Soc. 1993;70:539.Kidwai M, Dave B, Bhushan KR. Cchem Pap. 2000;54:231.Gonzalez MI, Stock HT, Pinnock RD, Pritchard MC, Wayman CP, Van der Graaf PH, Naylor AM, Higginbottom M. WO2002040008A2 2002Dabholkar VV, Mishra SKJ. Ind J Chem, Section B: Org Chem Incl Med Chem. 2006;45B:2112.Jorgensen WL, Ruiz-Caro J, Hamilton AD. WO2007038387A2 2007Moriconi A, Aramini A. WO2009050258A1 2009Allegretti M, Aramini A, Bianchini G, Cesta MC. WO2010031835A2 2010
- 13.(a) The authors thank BMCL Reviewer #1 for thoughtful comments regarding Scheme 1 and 2a versus 2a′/2a′. Using NMR chemical shift calculations {done using the GIAO13b–f method at the SCRF-mPW1PW91/6-311+G(2d,p)//M06-2X/6-31+G(d,p) level13g and empirically scaled [SCRF refers to the inclusion of solvent effects (chloroform) using the SMD continuum model]13h–i}, structures 2a and 2a′ are ruled out as the product of the reaction of 8 + 4 and 2a′ is assigned as the product (see SI for details). Mean absolute deviations were 0.4, 0.4, and 0.1 for 1H NMR and 4.6, 3.0, and 3.1 for 13C NMR (2a, 2a′, and 2a′, respectively). Furthermore, DP4 analysis13j showed 0.4% and 98.7% match for 1H NMR and 33.6% and 66.4% match for 13C NMR to experimental chemical shifts (2a′ and 2a′, respectively); 2a′ is therefore assigned as the 8 + 4 product. London F. J Phys Radium. 1937;8:397.McWeeny R. Phys Rev. 1962;126:1028.Ditchfield R. Mol Phys. 1974;27:789.Wolinski K, Hilton JF, Pulay P. J Am Chem Soc. 1990;112:8251.Cheeseman JR, Trucks GW, Keith TA, Frisch MJ. J Chem Phys. 1996;104:5497.Matsuda SPT, Wilson WK, Xiong Q. Org Biomol Chem. 2006;4:530. doi: 10.1039/b513599k.Lodewyk M, Siebert MR, Tantillo D. Chem Rev. 2012;112:1839. doi: 10.1021/cr200106v.Frisch MJ. Gaussian 09 (revision B.01) ; see SI for full author list Smith SG, Goodman JM. J Am Chem Soc. 2010;132:12946. doi: 10.1021/ja105035r.
- 14.Crank G, Khan HR. Aust J Chem. 1985;38:447. [Google Scholar]
- 15.For protocols, see: Short FW, Long LM. J Heterocycl Chem. 1969;6:707.Theocharis AB, Alexandrou NE. J Heterocycl Chem. 1990;27:1685.Supura CT, Barboiu M, Luca C, Pop E, Brewster ME, Dinculescu A. Eur J Med Chem. 1996;31:7.Sathishaa KR, Khanumb SA, Narendra Sharath Chandrac JN, Ayishab F, Balajid S, Marathee GK, Gopala S, Rangappac KS. Bioorg Med Chem Lett. 2011;19:211.
- 16.Galietta LJ, Haggie PM, Verkman AS. FEBS Lett. 2001;499:220. doi: 10.1016/s0014-5793(01)02561-3. [DOI] [PubMed] [Google Scholar]
- 17.Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, Lukacs GL, Taddei A, Folli C, Pedemonte N, Galietta LJV, Verkman AS. J Bio Chem. 2003;278:35079. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- 18.Ye L, Knapp JM, Sangwung P, Fettinger JC, Verkman AS, Kurth MJ. J Med Chem. 2010;53:3772. doi: 10.1021/jm100235h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao Y, et al. Phys Chem Chem Phys. 2006;8:3172. doi: 10.1039/b517914a. [DOI] [PubMed] [Google Scholar]
- 20.Halgren TA. J Comp Chem. 1998;17:490. [Google Scholar]
- 21.Frisch MJ, et al. Gaussian 09, Revision B.01. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 22.(a) Zhao Y, Truhlar DG. Theor Chem Acc. 2008;120:215. [Google Scholar]; (b) Zhao Y, Truhlar DG. Acc Chem Res. 2008;41:157. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 23.Marenich AV, Cramer CJ, Truhlar DG. J Phys Chem B. 2009;113:6378. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 24.Legault CY. CYLview, 1.0b. Université de Sherbrooke; 2009. ( http://www.cylview.org) [Google Scholar]
- 25.Sadchikova EV, Bakulev VA, Subbotina JO, Privalova DL, Dehaen W, Van Hecke K, Robeyns K, Van Meervelt L, Mokrushin VS. Tetrahedron. 2013;69:6987. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.