

Abstract
Mechanotransduction pathways convert forces that stress and strain structures within cells into gene expression levels that impact development, homeostasis, and disease. The levels of some key structural proteins in the nucleus, cytoskeleton, or extracellular matrix have been recently reported to scale with tissue- and cell-level forces or mechanical properties such as stiffness, and so the mathematics of mechanotransduction becomes important to understand. Here, we show that if a given structural protein positively regulates its own gene expression, then stresses need only inhibit degradation of that protein to achieve stable, mechanosensitive gene expression. This basic use-it-or-lose-it module is illustrated by application to meshworks of nuclear lamin A, minifilaments of myosin II, and extracellular matrix collagen fibers—all of which possess filamentous coiled-coil/supercoiled structures. Past experiments not only suggest that tension suppresses protein degradation mediated and/or initiated by various enzymes but also that transcript levels vary with protein levels because key transcription factors are regulated by these structural proteins. Coupling between modules occurs within single cells and between cells in tissue, as illustrated during embryonic heart development where cardiac fibroblasts make collagen that cardiomyocytes contract. With few additional assumptions, the basic module has sufficient physics to control key structural genes in both development and disease.
Introduction
Polymer physics provides fundamental explanations for how elasticity and viscosity of diverse polymer systems often scale as power laws with polymer concentration (1), even when the polymers interact or assemble (2). Living organisms are of course built from biopolymers (Fig. 1 A), and assembling proteins such as extracellular matrix (ECM) collagens, which are the most abundant proteins in metazoans, exhibit gel elasticities that indeed scale with concentration when purified and reconstituted (3). Perhaps not surprisingly, tissue stiffness not only scales with collagen levels but is also dictated by the amount of collagen, with soft tissues such as brain having much less collagen than stiffer tissues such as muscle (4). However, biopolymers that include many other key structural proteins within cells and tissues are subject to a variety of enzymatic processes of degradation and synthesis with turnover timescales that, in cell culture, can be only hours or days (5,6). How the mechanics of a tissue or cell reaches or maintains a steady state is therefore a fundamental question of biopolymer physics that impacts the form, function, and dysfunction of cells and tissues in general.
Figure 1.
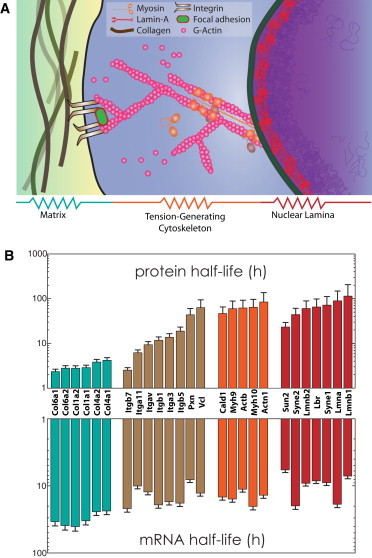
Systems-level view of structural molecules involved in mechanotransduction. (A) A living cell and its microenvironment are built from biopolymers that provide structure; these molecules assemble and are interconnected, to resist stress arising from development or disease. (B) Protein (top) and mRNA (bottom) half-lives of collagens (teal), membrane-bound integrins (brown), and cytoskeletal (orange) and nuclear envelope components (red) measured in NIH3T3 mouse fibroblasts grown on conventional rigid plastic (5). Half-lives are relatively constant within structural groups, suggesting similar dynamics.
Some of the earliest examples of tension-mediated protein stability were demonstrated in cyclically beating neonatal rat heart cells, where components of the contractile myofibril assembly such as cardiac actin (7) and myosin heavy chain (8) were found degraded when contractile activity was reduced. For collagen fibers, recent evidence suggests mechanical strain stabilizes against enzymatic degradation (9). High matrix stiffness is further associated with an increased stress or tension in the cell (10), and the key contractile protein, myosin II, responds to matrix stiffness by assembling into stress fibers and increasing in amount (11,12). More recently, we have shown that nucleoskeletal lamin-A level scales with tissue microelasticity E, with higher lamin A levels giving physically stiffer nuclei in stiffer tissues (4). Lamin A and myosin II thus seem to mechanically couple to the collagenous matrix (Fig. 1 A) in ways that are prescribed by polymer physics. These coiled-coil proteins that assemble into structural networks are prime candidates as biological tension sensors, transducing similar mechanical signals from the ECM to the nucleus (13).
Genome-wide measurements of the production and degradation dynamics of mRNA as well as protein in mouse fibroblasts (NIH 3T3; National Institutes of Health, Bethesda, MD) in standard cultures (5) have shown that mRNA and protein half-lives are fairly uniform within structural groupings of collagens, cytoskeletal and nucleoskeletal components (Fig. 1 B). Collagen and actomyosin modules differ significantly in half-lives, and the integrins exhibit intermediate half-lives consistent perhaps with these membrane proteins serving as intermediary linkages between ECM and the cytoskeleton. Even structural proteins on the nuclear envelope such as lamins exhibit largely coordinated expression as a single module, with half-lives similar to those for the actomyosin module. This seems consistent with mechanically coordinated responses of the nuclear lamina to cytoskeletal stresses (14). Much has already been learned from modeling the dependence of structure and dynamics of adhesions and the contractile cytoskeleton on matrix mechanics or forces (15–21). Here we focus on higher-level, long-time models that address mechanics-dependent trends in expression for which experimental data is just beginning to emerge.
General Methods
For a given structural gene (S) and its corresponding protein (s) in a module, typical rate equations consist of synthesis and turnover rates such that
| (1) |
| (2) |
where S and s represent the concentrations of mRNA and protein (dimensionless; normalized to total mRNA and protein levels), respectively, and whose synthesis (α, γ) and degradation (β, δ) rates dictate expression levels. The synthesis rate α is considered to be a function of protein levels (s)—a positive feedback loop that may produce bistability depending on the cooperativity of protein-induced feedback (22), but the new and perhaps important goal of modeling mechanobiology here is to somehow incorporate cell and matrix mechanics into these otherwise classic expressions for α, β, δ, and γ. Any of these rate terms could in principle be functions of S, s and other factors such as matrix elasticity or cell tension. Variations of the rate orders in Eqs. 1 and 2 were explored further in the Supporting Material. Phase plots of gene and protein levels were generated numerically (MATHEMATICA, Ver. 9; Wolfram Research, Champaign, IL) for each variation, and how each rate form could properly recapitulate matrix elasticity-dependence of structural proteins was explored. The goal here is to identify a possible minimal model that fits contemporary understanding of tension coupling to gene expression.
We consider tension-mediated stabilization of polymeric, structural proteins as the basis for systems mechanobiology, and here we explore such models mathematically for single, coupled, and population-coupled modules. Structural proteins of the extracellular matrix, cytoskeleton, or nucleoskeleton are understandably polymeric. In an effort to parsimoniously define a mechanobiological gene circuit, we assume that polymerization and depolymerization rates are relatively rapid compared to the rates defined here. Thus, tension-mediated turnover rate (δ) is assumed to depend simply on effective tension, giving protein concentration, s. Tension on various coiled-coil assemblies has been shown to suppress the affinity of a phosphorylating kinase/protease that initiates enzymatic solubilization/degradation (4,9,23,24). Like pulling on a wet rope to wring out water, tension squeezes out free volume or sterically shields binding sites via coiled-coil assembly to prevent enzyme access. Single-molecule studies of collagen have suggested tension-enhanced degradation (25), but such short polymers tend to unwind under tension, whereas ropelike polymer fibers would tend to tighten their coils and knots. Regardless of mechanism, the rate of degradation can be generally represented by Michaelis-Menten kinetics (26) as
| (3) |
where δ0 is the maximum degradation rate at saturating concentrations of substrate s, such as a coiled-coil protein assembly; Ks is an enzyme affinity for the substrate and is a function of tension; and n is a cooperativity coefficient ≥2 that is typical of multimeric interactions. By incorporating Eq. 3 into Eq. 2 and by including an experimentally supported mechanism for transcriptional control, we solve the system of differential equations at steady state and arrive at a relationship between S, s and tension-surrogate Ks. In contrast to a recent modeling study for the kinetics of RNA-interference of focal adhesions that incorporates the effect of force via a varying concentration of Rho (27), the systems-level modeling here generalizes our recent model of tension-stabilized lamin-A (4). This is a first parsimonious approach to incorporating cell tension and matrix stiffness into traditional rate equations for expression changes (Eqs. 1 and 2).
Starting with the model above applied to lamin A, we then extend the model to two structural modules that couple the nucleoskeleton to the cytoskeleton. Dynamics on the tissue-level is then illustrated with a two-cell type model of embryonic heart development. Our models thus elaborate systems-level behavior of structural modules that respond to mechanical stress. These results have implications for both the physiology and pathophysiology of diseases involving structural proteins, from early cardiac development to stress-dependent aging.
The sets of ordinary differential equations that describe the various mechanobiological systems modeled here are explicitly written in the Supporting Material. The analytical results are also derived where applicable. The values of the various rate constants, initial conditions, etc., used in each model are also included in the Supporting Material. Steady-state measurements were obtained by allowing the system to run until the levels of each species stabilize.
Results
Tension-inhibited degradation of coiled-coil proteins
With lamin-A as a representative mechanosensitive protein, we predicted systems-level trends by constructing a parsimonious model of its gene circuit that takes into account protein-mediated feedback on transcription and tension-dependent protein turnover (Fig. 2 A). Lamin-A protein is known to feed-back on its own gene through positively regulating its own transcription factor, retinoic acid receptor-γ (RARG) (4). Kinetic measurements of lamin-A changes with mechanical perturbations are clearly needed to further define the model. We found that only first-order rates can sufficiently recapitulate experimental observations. For simplicity, synthesis of messenger RNA (mRNA) and protein as well as degradation of mRNA were all assumed to be linear with rate constants of order unity (to eliminate bias on one biological process), such that the rate equations for lamin-A mRNA (L) and protein (l) are
| (4) |
| (5) |
Figure 2.
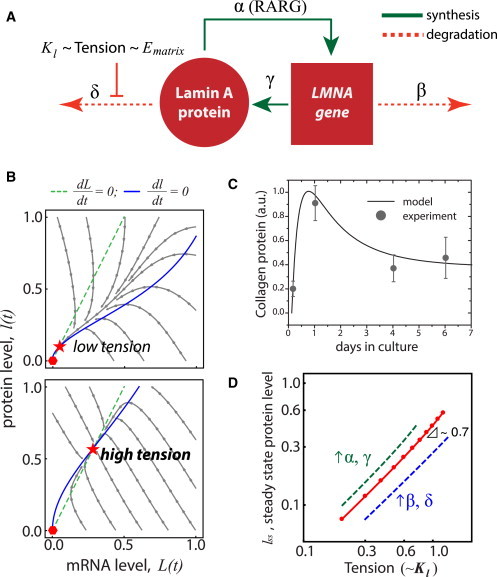
Feedback-based mechanobiological gene circuit model for lamin A exhibits polymer-physics scaling if cell tension suppresses protein turnover. (A) Nucleoskeletal Lamin-A protein regulates its own message (LMNA) and assembles in response to tension from matrix elasticity (Ematrix), which inhibits protein degradation. (B) Trajectories of lamin-A message and protein as the model converges from a range of initial conditions (arrowed lines) to where mRNA (dashed lines) and protein (solid lines) nullclines intersect to give a stable steady-state solution (star) appropriate to the tension (top, Kl = 0.5; bottom, Kl = 1.1). Null solution is unstable (hexagon). (C) Procollagen-1 expression in mesenchymal stem cells over seven days was tracked by immunolabeling and quantified. Stem cells adhere and spread on a substrate and start to synthesize collagen matrix, which stabilizes over time. The biphasic kinetic response can be recapitulated by the model that assumes tension-based inhibition of collagen protein degradation rate. (D) Setting the kinase/protease binding coefficient, Kl, to be proportional to (Tension)0.3 allows the model to generate steady-state scaling with tension that is consistent with tissue-level scaling of lamin A (4). Increasing the synthesis (α, γ) or degradation (β, δ) rates shifts steady-state lamin-A levels higher or lower, respectively. To see this figure in color, go online.
Time evolution of protein (l) and mRNA (L) can be analytically solved with example trajectories in Fig. 2 B as a phase plot of l(t) versus L(t) converging to steady-state values: {lss, Lss}. Because of how tension-mediated degradation and positive feedback are modeled, the shape of the nullclines allows for the two physiologically relevant steady states. The phase plot shows a stable, nonzero, steady-state node and a saddle point at zero where the mRNA and protein nullclines intersect, lending both mathematical consistency and biophysical relevance to the parsimonious model (further details in the Supporting Material). A set-point tension (Kl) that a cell encounters dictates the magnitude of steady-state levels.
The phase plot also suggests that the initial protein and mRNA levels dictate expression dynamics, and it should be noted that initial states and trajectories include cases where—for example—protein is high and decreasing, whereas transcript is low and increasing in seemingly uncorrelated processes. For example, for lamin A and the nucleoskeletal module, which have stable protein levels but unstable mRNA levels (5), protein dynamics for a given tension set-point (Kl) are predictably well controlled and stabilize linearly toward steady-state (Fig. 2 B, upper-left quadrant). Lamin A is of course known for tethering heterochromatin near the nuclear envelope, and thus, may need a relatively stable protein expression. Other gene groups with unstable mRNAs include RNA-binding proteins (5). On the other hand, the ECM module has the opposite trend (Fig. 1 B); the kinetics of the model predicts that, for highly stable mRNA but short-lived protein levels, the dynamics are nonlinear (Fig. 2 B, lower-right quadrant). That is, protein levels would overshoot before stabilizing at a steady-state level. The kinetics of this regime was verified by tracking collagen protein expression in mesenchymal stem cells over time from suspension to attachment, starting at low collagen protein levels but with COL1 mRNA levels remaining relatively stable among structural genes (Fig. 1 B). Cell suspension has been known to cease translational rates, and sequester mRNA for subsequent protein synthesis during anchorage recovery (28). With an initially high mRNA level (Fig. 2 B, lower-right quadrant), collagen protein tended to overshoot before stabilizing at a lower steady-state concentration, as was observed experimentally over seven days (Fig. 2 C). Other gene groups in this regime include those involved in defense response and homeostasis (5).
Ultimately, the model suggests that tension dictates the expression of structural genes such as nuclear lamin-A regardless of initial conditions, such that at steady state,
| (6) |
Equations 4 and 5 can be solved analytically for n = 2 to yield nonzero steady-state values for l and L, (details in the Supporting Material)
| (7) |
Based on the steady-state analysis above, a solution only exists if
| (8) |
Although steady-state values depend on the various rate constants, we assumed all to be important and of ∼1 (see the Supporting Material) as we focus on Kl: at high stresses where lamin-A assembly is favored, Kl increases so that phosphorylation-mediated lamin-A degradation (24) decreases. Plotting steady-state lamin-A levels lss against different values for Kl fit a power-law lss ∼ Kl2 (for n = 2). As a test of whether such a model could capture key experimental trends, computational results showed that if Kl = (Tension)0.3, then lss ∼ (Tension)0.7 (Fig. 2 D), which parallels the scaling of lamin A with tissue microelasticity E (noting that Tension ∼ E) (4). This assumed relationship of Kl and Tension is analogous to the scaling physics of solvent to polymer stability. The set of equations and trends delineated above also applies obviously to collagen because it defines tissue E itself, and it was found to scale more strongly experimentally (E1.5) (4).
Mechanical coupling of coiled-coil modules in series
We further developed our model by coupling two coiled-coil proteins in the cytoskeleton and nucleoskeleton modules, with nonmuscle myosin (e.g., MYH9) and lamin A as representatives in their respective modules. Lamin-A {L,l} and nonmuscle-myosin {M,m} message and protein circuitry is schematically presented in Fig. 3 A.
Figure 3.
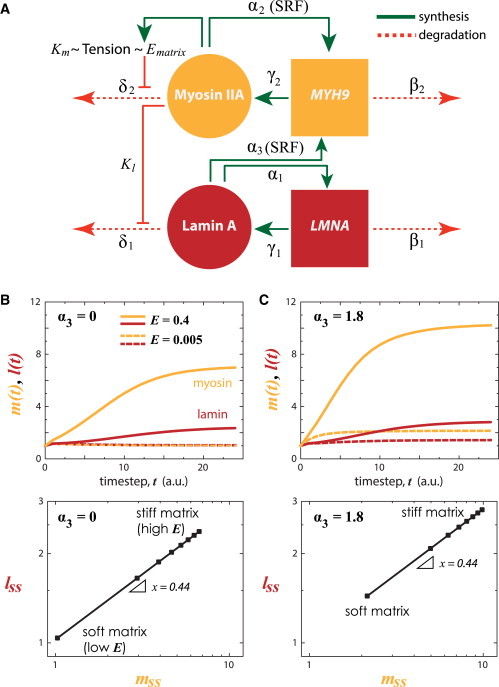
Matrix stiffness-coupled lamin A and MYH9 turnover. (A) Cytoskeletal myosin and nucleoskeletal lamin A is coupled where each regulates their own message and follows a tension-inhibited degradation mechanism. Lamin A has also been shown to regulate SRF-target genes (α3), such as MYH9 (4). (B) Kinetics (top) and steady-state (bottom) myosin (m) and lamin A (l) protein levels, assuming α3 = 0, at low (E = 0.005) or high (E = 0.4) tension. (C) With feedback of lamin A on myosin transcription (α3 > 0), both myosin and lamin levels are increased. To see this figure in color, go online.
In particular, the expression kinetics were described as coupled rate equations for respective transcripts {L,M} and proteins {l,m}:
| (9) |
| (10) |
| (11) |
| (12) |
Again, for simplicity, mRNA degradation and translation rates were assumed to be of first-order. Lamin-A protein positively regulates one of its transcription factors (RARG; Swift et al. (4)) as does MYH9 with one of its transcription factors (SRF; Miralles et al. (29)) so that each enhances its own transcription (with rate constants, and , respectively). Mechanical regulation of protein phosphorylation and turnover has been demonstrated in recent studies (4,23,24), and so we described lamin-A and myosin turnover with suitable Hill functions (rate constants δ1, δ2). Specifically, lamin-A turnover is dictated by for some x that dictates sensitivity of lamin-A degradation to myosin-generated stress. Myosin protein turnover is, in turn, dictated by matrix elasticity simply as for some y that represents the affinity for myosin degradation. Both Km and Kl effectively couple matrix mechanics to cytoskeletal stress, which in turn tenses the nucleoskeleton (Fig. 1 A; Wang et al. (13)).
Equations 9–12 were solved numerically at steady state (all derivatives = 0; see the Supporting Material). Rate constants and free parameters were adjusted collectively within an order of magnitude of each other (see the Supporting Material) to reflect the observed similar half-lives of the modules (Fig. 1 B). In the simplest case, we first assumed synthesis and degradation rate constants to be equal,
and observed that, for any given E, the range of expression for myosin is larger than that for lamin A (Fig. 3 B). This likely reflects the matrix-cytoskeleton-nucleoskeleton assembly in series (Fig. 1 A). Nonetheless, because matrix and cell tension suppresses protein phosphorylation and turnover, steady-state levels monotonically increase with matrix E, consistent with coupled mechanoregulation of lamin A and myosin (24).
If we consider a first-order effect of lamin-A protein on MYH9 transcription via SRF pathway (; Swift et al. (4)), numerical analysis showed that the dynamic range of myosin increased further due to matrix E and lamin-A contributions, but not the slope of the myosin–lamin-A response to matrix E (Fig. 3 C). The relative sensitivity of each module is instead dependent on x and y. If sensitivity of lamin A to degradation (x) is increased (or decreased), then more (lesser) myosin protein is required to generate the same stress that maintains the original lamin-A protein level (Fig. 4 A). Increasing x is similar to decoupling nucleoskeletal and cytoskeletal modules, as was done experimentally by ectopic expression of SUN2, a nuclear membrane protein that connects the nucleoskeleton to the cytoskeleton, which led to reduced lamin-A levels (4). On the other hand, if sensitivity of myosin to degradation (y) is decreased, then at low E the steady-state levels of myosin (and indirectly, lamin A) is much higher than at sufficiently high matrix E, where myosin (and lamin) levels remain relatively unperturbed (Fig. 4 B). Experiments that observed this prediction include a nonphosphorylatable myosin mutant, which localizes to assembled stress fibers and overrides soft-matrix (low E) effects on stem-cell phenotype; conversely, a phosphomimetic myosin mutant that enriches in the soluble pool fails to completely override stiff-matrix (high E) effects (23).
Figure 4.
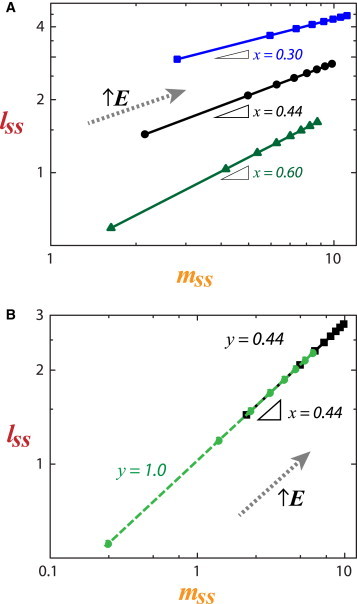
Sensitivity analysis of coupled structural modules. (A) By varying x, we can perturb the (slope) sensitivity of lamin A to degradation in response to myosin-generated stress. (B) Varying y perturbs the degradation sensitivity of myosin, but not its coupling with lamin A. To see this figure in color, go online.
Logistic coupling between collagen and myosin in cardiac development
Systems biology can potentially build an integrated understanding of the electrophysiological and physical processes involved in cardiac physiology and pathophysiology. It might also help identify therapeutic targets. Understanding how the balance between mechanical stiffness and contractile ability of the myocardium is achieved with age and pathological changes, ultimately requires a systems-level model to guide hypotheses. The expression of actomyosin contractility proteins and collagen, among hundreds of abundant proteins, parallel myocardial stiffening in development (30). Both static/cyclic and uniaxial/biaxial strains encourage collagen matrix deposition by cardiac fibroblasts (31), inasmuch as passive and active contraction increase throughout cardiac development. However, as contractility (or myosin levels) increasingly strains the developing heart tissue, we postulate that fibroblast proliferation is ultimately limited by the stiffness of their environment, which correlates strongly with collagen-1 levels (4,30). The various components of the developing heart matrix and cytoskeleton, and any other functionally relevant signaling proteins, must be integrated into a realistic physical model of the observed mechanics. Thus, we considered using our simplified model that focuses on the mechanical interaction between the collagenous matrix deposited by cardiac fibroblasts and the contractile activity of cardiomyocytes.
To explore possible general mechanisms within the developing myocardium, a coupled network of myosin {M,m} and collagen {C,c} mRNAs and proteins can be modeled within the developing myocardium (Fig. 5 A). With collagen produced primarily by cardiac fibroblasts, the rate of collagen mRNA production is assumed to be proportional to the fibroblast population, which is in turn limited by tissue stiffness imparted by collagen matrix density, such that
| (13) |
The collagen mRNA production rate of fibroblasts increases at low collagen matrix densities up to a critical collagen concentration (and hence matrix stiffness), then it decreases thereafter; the critical points and amplitudes of this biphasic behavior are modulated by nf and kf, respectively (Fig. 5 B).
Figure 5.
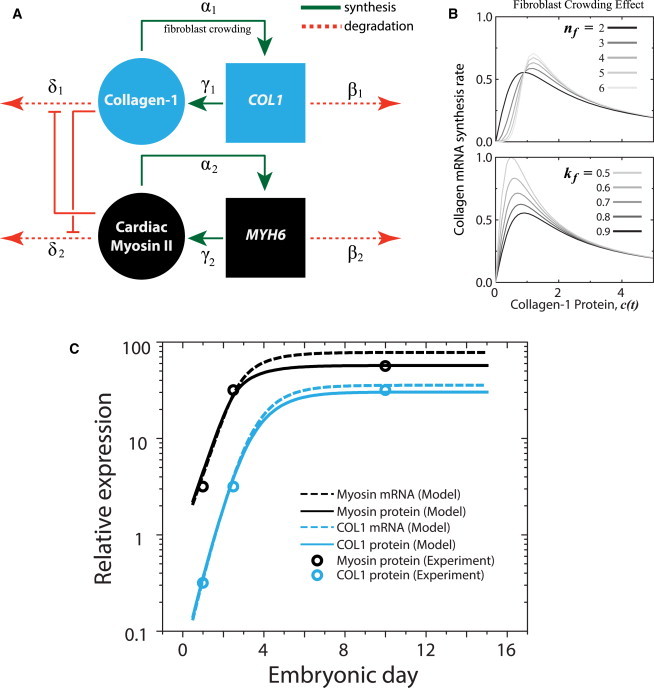
Cardiomyocytes and fibroblasts balance contraction and matrix production during heart development. (A) Assumptions in matrix stiffness limiting fibroblast population α1 (and hence, collagen expression), while encouraging myofibril organization in cardiomyocytes (and hence, myosin expression) are incorporated in a model where tension derived from each module inhibits degradation of the other. (B) Fibroblast crowding is modeled by a Hill function of collagen protein (and hence tension) (13) with nf and kf as the critical point and amplitude, respectively, of the collagen mRNA synthesis rate. (C) Experimentally measured changes in both collagen-1 (COL1) and cardiac myosin expression (circles) in a developing embryonic chick heart (30) can be recapitulated by the model (protein, lines; mRNA, dashed lines). To see this figure in color, go online.
The concept of tension-mediated degradation is again applied here for collagen protein. Collagen matrices have been shown to be stabilized (against degradation) by applied tension (9), such that
| (14) |
Cardiomyocytes are of course the primary contributors of myosin-mediated tension in cardiac tissue, such that for some x that dictates the extent of myosin-mediated collagen degradation.
Additionally, myosin-II molecules under tension remain assembled and abundant (4,30), with some evidence of tension-suppressed phosphorylation of nonmuscle myosin-II suggesting an intermediate step (23). Striated muscle myosin-II is certainly turned-over in vivo (32), and its disuse probably favors degradation and muscle atrophy. Thus, the transcript (M) and protein (m) rate equations for cardiac myosin are coupled to collagen such that
| (15) |
| (16) |
where , for some y.
With rate constants and free parameters adjusted within an order of magnitude of each other, Eqs. 13–16 were solved numerically (see the Supporting Material) and obtained best-agreement with the reported experimental results. By representing the basic assumptions on fibroblast population crowding (Eq. 13) and tension-stabilized proteins (Eqs. 14 and 16), the model was able to recapitulate the logistic growth kinetics (Fig. 5 C) observed experimentally for cardiac myosin and collagen protein levels in a developing heart (30). The model also predicted, perhaps not surprisingly, that mRNA levels should exhibit the same trends as the protein counterparts (Fig. 5 C).
Discussion
Cells experience or exert stresses inside and out. Studies that look into cell mechanics responses on timescales of hours or less (e.g., Webster et al. (33)) need to be somehow linked in future work to half-lives of relevant components, which seem to be on much longer timescales, particularly actin-myosin cytoskeleton components with half-lives of 10–100 h (Fig. 1 B). Although here we focused on intracellular tension that increases with matrix stiffness E, externally imposed stresses such as shear flow or stretching of cells in tissue can in principle be incorporated into some functional form of K = f (E, shear stress, cell stretch,…). Experimental measurements of turnover (per Fig. 1 B) are clearly needed for these various stress conditions.
The question remains: Should tension facilitate or suppress protein degradation? From an evolutionary standpoint, multicellularity in metazoans is preceded by the appearance of genes necessary for structural organization and intercellular communication. All biomolecules degrade (“dust to dust”), but rates of decomposition of any material also depend in general on both the state of the material and the thermodynamic properties of the environment (e.g., temperature, stress, oxygen, water, etc.). From a biochemical standpoint, it is possible for cleavage sites to be exposed under tension. With purified protein, collagen-I trimers under tension seem to be degraded faster by collagenase (25), whereas collagen fibers under tension are degraded more slowly (9), which is what we assumed here for collagen (Fig. 5 A). Within cultured cells, filamin is reportedly regulated by tension-induced degradation, but an autophagy factor (BAG3) that mediates degradation also potently feeds-back on transcription, so that filamin increases with matrix stiffness and cell tension (34). The transcriptional coactivator YAP/TAZ seems to be involved (34), but TAZ is also known to be degraded more so in cells on soft matrices than stiff matrices and also by treatment with inhibitors of Rho, F-actin, and actomyosin tension (35). Detailed molecular mechanisms remain elusive, but for ropelike coiled-coil polymers, tension could sterically or conformationally prevent protease binding to collagen fibers (9) or kinase binding to myosin minifilaments (23) and the lamin-A meshwork (4). Future experimental work on expression dynamics of mechanosensitive proteins would seem likely to benefit from measurements of turnover rates (e.g., Fig. 2 B) while systematically varying extracellular stiffness or stress and intracellular tension.
The concept of tension-mediated transcriptional regulation, especially when applied to lamin-A control, also implicates epigenetic silencing of heterochromatin that is typically sequestered by the nuclear lamina. Additionally, the proposed feedforward mechanism to control LMNA necessitates another level of regulation of mechanosensing via transcription-factor control (in this case, by RARG that is modulated by soluble retinoid agonists and antagonists (4)). Regardless of mechanism, the turnover and expression of key structural proteins appears to be mechanoregulated.
In the models presented here, membrane-bound integrins were implicitly lumped together (along with matrix and cytoskeleton) in the K-parameter. Overexpressing integrin receptors does not change the ability of a cell to spread, as observed experimentally (36). However, the relative distribution of integrin types with different bond lifetimes (e.g., slip- and catch-bonds) may affect the coupling of intracellular tension to extracellular stiffness, as suggested recently by theory (27) and experiment (37). On the other hand, perturbations to the linker of nucleoskeleton and cytoskeleton (LINC) complex are understandably more complicated, inasmuch as they physically couple coiled-coil modules of myosin and lamin and influence chromatin architecture (38). Nonetheless, the LINC complex is implicitly included in the x coefficient, inasmuch as it couples cytoskeletal and nucleoskeletal responses. By applying force to nesprin-1, a component of the LINC complex, an isolated nucleus with an intact nuclear lamina stiffens (39).
To date, most experimental techniques in developmental biology fall short of characterizing the systems-level landscape. Our modeling analysis of collagen and myosin levels in cardiac tissue development (Fig. 5) demonstrates that coupled structural modules are sufficient to recapitulate the dynamics of tissue-level architecture. Given the highly interconnected signaling pathways in mammalian biology, our work distills the essential mechanobiological circuits governing not just intracellular but also tissue-level observations into a testable, theoretical framework. The inherent difficulty of detailing these circuits with accurate rate constants and functional forms will be overcome by developing more sophisticated “-omic” approaches.
Acknowledgments
Support from the AHA Award #14GRNT20490285, NIH (R01-HL124106, R01-HL062352, P01-DK032094, NCATS-8UL1TR000003, P30-DK090969) and NSF (Materials Research Science and Engineering Center) are gratefully acknowledged.
Supporting Material
References
- 1.Gennes P.G.d. Cornell University Press; Ithaca, N.Y.: 1979. Scaling concepts in polymer physics. [Google Scholar]
- 2.Doi M., Edwards S.F. Oxford University Press; Oxford, England: 1986. The theory of polymer dynamics. [Google Scholar]
- 3.Yang Y.L., Leone L.M., Kaufman L.J. Elastic moduli of collagen gels can be predicted from two-dimensional confocal microscopy. Biophys. J. 2009;97:2051–2060. doi: 10.1016/j.bpj.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swift J., Ivanovska I.L., Discher D.E. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwanhäusser B., Busse D., Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 6.Eden E., Geva-Zatorsky N., Alon U. Proteome half-life dynamics in living human cells. Science. 2011;331:764–768. doi: 10.1126/science.1199784. [DOI] [PubMed] [Google Scholar]
- 7.Sharp W.W., Terracio L., Samarel A.M. Contractile activity modulates actin synthesis and turnover in cultured neonatal rat heart cells. Circ. Res. 1993;73:172–183. doi: 10.1161/01.res.73.1.172. [DOI] [PubMed] [Google Scholar]
- 8.Byron K.L., Puglisi J.L., Samarel A.M. Myosin heavy chain turnover in cultured neonatal rat heart cells: effects of [Ca2+]i and contractile activity. Am. J. Physiol. 1996;271:C01447–C01456. doi: 10.1152/ajpcell.1996.271.5.C01447. [DOI] [PubMed] [Google Scholar]
- 9.Flynn B.P., Bhole A.P., Ruberti J.W. Mechanical strain stabilizes reconstituted collagen fibrils against enzymatic degradation by mammalian collagenase matrix metalloproteinase 8 (MMP-8) PLoS ONE. 2010;5:e12337. doi: 10.1371/journal.pone.0012337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Discher D.E., Janmey P., Wang Y.L. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 11.Rehfeldt F., Brown A.E.X., Discher D.E. Hyaluronic acid matrices show matrix stiffness in 2D and 3D dictates cytoskeletal order and myosin-II phosphorylation within stem cells. Integr. Biol. (Camb) 2012;4:422–430. doi: 10.1039/c2ib00150k. [DOI] [PubMed] [Google Scholar]
- 12.Engler A.J., Sen S., Discher D.E. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 13.Wang N., Tytell J.D., Ingber D.E. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 2009;10:75–82. doi: 10.1038/nrm2594. [DOI] [PubMed] [Google Scholar]
- 14.Houben F., Ramaekers F.C.S., Broers J.L.V. Role of nuclear lamina-cytoskeleton interactions in the maintenance of cellular strength. Biochim. Biophys. Acta. 2007;1773:675–686. doi: 10.1016/j.bbamcr.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 15.Wang N., Butler J.P., Ingber D.E. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 16.Kumar S., Maxwell I.Z., Ingber D.E. Viscoelastic retraction of single living stress fibers and its impact on cell shape, cytoskeletal organization, and extracellular matrix mechanics. Biophys. J. 2006;90:3762–3773. doi: 10.1529/biophysj.105.071506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paul R., Heil P., Schwarz U.S. Propagation of mechanical stress through the actin cytoskeleton toward focal adhesions: model and experiment. Biophys. J. 2008;94:1470–1482. doi: 10.1529/biophysj.107.108688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan C.E., Odde D.J. Traction dynamics of filopodia on compliant substrates. Science. 2008;322:1687–1691. doi: 10.1126/science.1163595. [DOI] [PubMed] [Google Scholar]
- 19.Walcott S., Sun S.X. A mechanical model of actin stress fiber formation and substrate elasticity sensing in adherent cells. Proc. Natl. Acad. Sci. USA. 2010;107:7757–7762. doi: 10.1073/pnas.0912739107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maître J.L., Berthoumieux H., Heisenberg C.P. Adhesion functions in cell sorting by mechanically coupling the cortices of adhering cells. Science. 2012;338:253–256. doi: 10.1126/science.1225399. [DOI] [PubMed] [Google Scholar]
- 21.Novikova E.A., Storm C. Contractile fibers and catch-bond clusters: a biological force sensor? Biophys. J. 2013;105:1336–1345. doi: 10.1016/j.bpj.2013.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griffith J.S. Mathematics of cellular control processes. II. Positive feedback to one gene. J. Theor. Biol. 1968;20:209–216. doi: 10.1016/0022-5193(68)90190-2. [DOI] [PubMed] [Google Scholar]
- 23.Raab M., Swift J., Discher D.E. Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J. Cell Biol. 2012;199:669–683. doi: 10.1083/jcb.201205056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buxboim A., Swift J., Discher D.E. Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr. Biol. 2014;24:1909–1917. doi: 10.1016/j.cub.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adhikari A.S., Chai J., Dunn A.R. Mechanical load induces a 100-fold increase in the rate of collagen proteolysis by MMP-1. J. Am. Chem. Soc. 2011;133:1686–1689. doi: 10.1021/ja109972p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michaelis L., Menten M.L. The kinetics of effective invertin [Die kinetik der invertinwirkung] Biochem. Z. 1913;49:333–369. [Google Scholar]
- 27.Hoffmann M., Schwarz U.S. A kinetic model for RNA-interference of focal adhesions. BMC Syst. Biol. 2013;7:2. doi: 10.1186/1752-0509-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benecke B.J., Ben-Ze’ev A., Penman S. The control of mRNA production, translation and turnover in suspended and reattached anchorage-dependent fibroblasts. Cell. 1978;14:931–939. doi: 10.1016/0092-8674(78)90347-1. [DOI] [PubMed] [Google Scholar]
- 29.Miralles F., Posern G., Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–342. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 30.Majkut S., Idema T., Discher D.E. Heart-specific stiffening in early embryos parallels matrix and myosin expression to optimize beating. Curr. Biol. 2013;23:2434–2439. doi: 10.1016/j.cub.2013.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacKenna D., Summerour S.R., Villarreal F.J. Role of mechanical factors in modulating cardiac fibroblast function and extracellular matrix synthesis. Cardiovasc. Res. 2000;46:257–263. doi: 10.1016/s0008-6363(00)00030-4. [DOI] [PubMed] [Google Scholar]
- 32.Ball R.D., Krus D.L., Alizadeh B. Myosin degradation fragments in skeletal muscle. J. Mol. Biol. 1987;193:47–56. doi: 10.1016/0022-2836(87)90625-5. [DOI] [PubMed] [Google Scholar]
- 33.Webster K.D., Ng W.P., Fletcher D.A. Tensional homeostasis in single fibroblasts. Biophys. J. 2014;107:146–155. doi: 10.1016/j.bpj.2014.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ulbricht A., Eppler F.J., Höhfeld J. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr. Biol. 2013;23:430–435. doi: 10.1016/j.cub.2013.01.064. [DOI] [PubMed] [Google Scholar]
- 35.Dupont S., Morsut L., Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 36.Engler A., Bacakova L., Discher D. Substrate compliance versus ligand density in cell on gel responses. Biophys. J. 2004;86:617–628. doi: 10.1016/S0006-3495(04)74140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schiller H.B., Hermann M.R., Fässler R. β1- and αv-class integrins cooperate to regulate myosin II during rigidity sensing of fibronectin-based microenvironments. Nat. Cell Biol. 2013;15:625–636. doi: 10.1038/ncb2747. [DOI] [PubMed] [Google Scholar]
- 38.Dahl K.N., Ribeiro A.J.S., Lammerding J. Nuclear shape, mechanics, and mechanotransduction. Circ. Res. 2008;102:1307–1318. doi: 10.1161/CIRCRESAHA.108.173989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guilluy C., Osborne L.D., Burridge K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 2014;16:376–381. doi: 10.1038/ncb2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.