

Abstract
Cystic fibrosis pulmonary disease is characterized by excessive and prolonged inflammation. CF Pulmonary disease severity exhibits considerable variation that, to some extent, appears to be due to the presence of modifier genes. Several components of the inflammatory response are known to have altered regulation in the CF lung. Genetic variants in 52 inflammatory genes were tested for associations with lung disease indices in a CF patient population (n=737) homozygous for the ΔF508 cystic fibrosis transmembrane conductance regulator mutation. Variants in 3 inflammatory genes showed significant genotypic associations with CF lung disease severity, including IL8 and previously reported TGFβ11 (p≤0.05). When analyzed by gender, it was apparent that IL8 variant associations were predominantly due to males. The IL8 variants were tested in an additional CF population (n=385) and the association in males verified (p≤0.01). The IL8 variants were in strong linkage disequilibrium with each other (R2≥0.82), while variants in neighboring genes CXCL6, RASSF6 and PF4V1 did not associate (p≥0.26) and were in weaker LD with each other and with the IL8 variants (0.01≤R2≤0.49). Studies revealed differential expression between the IL8 promoter variant alleles (p<0.001). These results suggest that IL8 variants modify CF lung disease severity and have functional consequences.
Keywords: cystic fibrosis, genetic modifier, interleukin-8, pulmonary
INTRODUCTION
Cystic fibrosis (CF) is an autosomal recessive disease that is caused by mutations in cystic fibrosis transmembrane conductance regulator (CFTR)2. Although CF is considered to be a classic monogenic disease, there is considerable phenotypic variation in CF among patients that share the same CFTR mutations; this is especially true with regards to pulmonary disease progression.3 It has been suggested that the majority of the variation observed in pulmonary disease is due to genetic heterogeneity, independent of CFTR genotype4. Our previous work has revealed that transforming growth factor β1 (TGF-β1), which is involved in immune responses and contributes to the progression of lung disease in animals, is a genetic modifier of CF lung disease1; however, TGFβ1 is predicted to only account for a small amount of the disease variation.
CF pulmonary disease is characterized by persistent inflammation and recurrent infection.5 Several components of the inflammatory response (chemokines, cytokines, transcription factors, etc.) are known to have altered expression in the CF lung.6–8 In general, dysregulation of the inflammatory responses is considered to be the primary source of much of the pathophysiology in CF.9–13 The gender of a patient may also contribute to this phenomenon, as females are reported to display worse lung disease than males, which is demonstrated by earlier colonization with P. aeruginosa14, reduced lung function14, and shorter lifespan14–16.
Here we report a survey of genetic variation in inflammatory pathway components, along with the effect of gender, and their effect on the severity of CF pulmonary disease. We selected 181 variants from a panel of 52 inflammatory genes and tested them for association with CF disease severity (see Supplementary Data). Common single nucleotide polymorphisms (SNPs) in these inflammatory genes were genotyped first in a large CF patient population (n=737) in which each subject is homozygous for the ΔF508 CFTR mutation, and associating variants then tested in a second cohort of 385 patients. Variants in the IL8 gene showed significant association with disease severity in males in both cohorts of CF patients. In vitro functional studies revealed differences in transcription due to the rs4073 IL8 promoter variant.
RESULTS
Inflammatory Gene Screen
One hundred eighty-one single nucleotide polymorphisms (SNPs) in 52 genes involved in inflammation were examined for skewing of genotype frequency between severely and mildly affected ΔF508-homozygous CF patients ascertained by lung disease status17. These 52 genes were a subset of a larger project that included 810 variants in 300 gene regions, all selected as candidate modifiers of CF disease severity based on specific pathways thought to contribute to CF pathophysiology. Using a χ2 test of independence, genotype frequencies of 49 of the 810 polymorphisms (6%) were significantly different between mild and severe patients (P < 0.05) while only 4 of the 181 inflammatory gene variants (2.2%) were significant. These 4 SNPs represented 4 different genes, including previously reported TGFB11, and therefore only the remaining 3 are included here, namely interleukin 8 (IL8), interleukin 10 receptor B (IL10RB), and transforming growth factor beta-induced protein (TGFBI).
In addition to the test of independence, the cohort was assessed for possible population stratification using a genome control method (see Methods). The values of λ ranged from 1.0 to 1.29, suggesting stratification is minimal in this population and the adjusted p values are given for genotypes in Table 1.
Table 1.
Significantly associating SNPs in the initial inflammatory gene screen
| Gene | SNP | n | Genotypic p-value |
|---|---|---|---|
| IL8 | rs2227307 | 732 | 0.04 |
| IL10RB | rs1467845 | 733 | 0.04 |
| TGFBI | rs248158 | 657 | 0.04 |
Defining the associating interval
Alleles of 4 of the 52 inflammation-related genes tested showed significant associations with lung disease severity (p<0.05). Of the newly reported associations, IL8 was selected for further characterization because its variants have been reported to associate with susceptibility to other airway disorders18, 19. Additionally, IL-8 is an integral component of CF lung disease; it is elevated, with substantial variation, in the lungs of CF patients6, 20. Because of the reported genetic association with CF lung function and the biological plausibility of IL8 as a genetic modifier of CF airway pathophysiology, the IL8 genomic region was investigated in more detail.
The strategy employed was to first define the genetic interval with the most significant association and evaluate the candidate interval in a second group of patients. To this end, we first examined markers in genes neighboring IL8 to determine which interval contained the most significant association. As IL8 is in proximity to several other chemokine genes in the chromosome 4q12–q13 region, and work by others suggests that some of these genes may be in linkage disequilibrium (LD) with IL821, one could not rule out flanking genes as the source of association in our initial screen.
There are 13 known CXC-family chemokine genes in the human genome, apparently the result of gene duplication22. Eight of these genes are in the IL8 genomic region (4q13–21), therefore functional redundancy is of concern. Guided by the findings of Hull et. al. showing three distinct LD blocks (r2>0.3) encompassing IL8 and the genes immediately flanking it.23, we selected the following variants for further evaluation: rs13131954 from RASSF6 and rs941758 from PF4V1 to represent the LD blocks adjacent to IL823 and rs4694639 from CXCL6, which is immediately 3′ of IL8, but for which the LD relationship has not been reported. The arrangement of these genes and the location of markers is shown in Figure 1. The pairwise linkage disequilibrium pattern was determined for all eight markers. The four IL8 markers were all in strong LD (R2>0.82, Table 2). The CXCL6 marker is in moderate LD with the IL8 markers (0.39≤R2≤0.47), but the RASSF6 and PF4V1 markers are essentially in equilibrium with IL8 and CXCL6 (R2≤0.16). Our data are concordant with those of Hull23 and show that not only are the IL8 markers in very strong LD with each other, but also that the CXCL6 marker is in modest LD with the IL8 alleles (Table 2).
Figure 1.
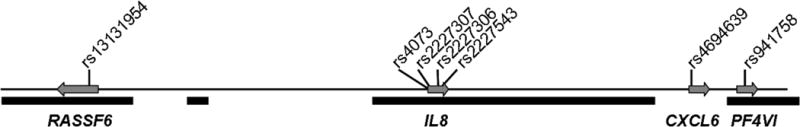
The surveyed IL8 genomic region (240 kb). Markers in the three genes most proximal to IL8 were tested for an association with pulmonary disease severity. Arrows indicate direction of gene transcription. Solid black bars denote haplotype blocks as defined by the Haplotype Map Project34.
Table 2.
Correlation coefficients (R2) from pairwise disequilibrium analysis
|
RASSF6 (rs13131954) |
IL8 (rs4073) |
IL8 (rs2227307) |
IL8 (rs2227306) |
IL8 (rs2227543) |
CXCL6 (rs4694639) |
PF4VI (rs941758) |
|
|---|---|---|---|---|---|---|---|
| RASSF6 (rs13131954) | 1.00 | ||||||
| IL8 (rs4073) | 0.00 | 1.00 | |||||
| IL8 (rs2227307) | 0.00 | 0.95 | 1.00 | ||||
| IL8 (rs2227306) | 0.00 | 0.82 | 0.85 | 1.00 | |||
| IL8 (rs2227543) | 0.00 | 0.86 | 0.88 | 0.95 | 1.00 | ||
| CXCL6 (rs4694639) | 0.01 | 0.50 | 0.47 | 0.39 | 0.40 | 1.00 | |
| PF4VI(rs941758) | 0.01 | 0.12 | 0.10 | 0.10 | 0.09 | 0.16 | 1.00 |
The markers across this region (240 kb) were tested for association with the lung disease phenotypes, severe and mild (see Methods), and the most significant associations were restricted to those markers in the IL8 gene (Table 3). There was no significant association between the RASSF6 (p=0.25), CXCL6 (p=0.26), or PF4V1 (p=0.29) markers and lung function. These analyses indicate that the most significant markers are those in high linkage disequilibrium with each other, and corresponding to the IL8 gene, strongly implicating only the IL8 interval.
Table 3.
Analysis of association testing for SNPs in IL8-flanking genes
| Gene | SNP | n | Genotypic p-value |
|---|---|---|---|
| RASSF6 | rs13131954 | 717 | 0.54 |
| rs4073 | 733 | 0.07 | |
| rs2227307 | 732 | 0.04 | |
| IL8 | |||
| rs2227306 | 727 | 0.19 | |
| rs2227543 | 737 | 0.06 | |
| CXCL6 | rs4694639 | 709 | 0.27 |
| PF4V1 | rs941758 | 716 | 0.40 |
Because of the differences in pulmonary disease observed between males and females, we assessed males and females separately. Table 4 shows that the most significant findings are found in the male sub-population and do not extend outside of the IL8 markers (Table 3).
Table 4.
Comparison of rs4073 genotypic p-values between cohort 1 and 2 reveals gender effect
| rs4073 | rs2227307 | rs2227306 | rs2227543 | |
|---|---|---|---|---|
| Patient Cohort 1: | ||||
| Total (n=737) | 0.07 | 0.04 | 0.19 | 0.06 |
| Males (n=392) | 0.07 | 0.06 | 0.15 | 0.12 |
| Females (n=345) | 0.25 | 0.12 | 0.36 | 0.19 |
| Patient Cohort 2: | ||||
| Total (n=385) | 0.25 | 0.15 | 0.18 | 0.52 |
| Males (n=219) | 0.01 | 0.08 | 0.01 | 0.06 |
| Females (n=166) | 0.82 | 0.59 | 0.84 | 0.70 |
Testing the association in additional populations
As Table 4 indicates, the evidence for association is more significant in the males than females, but is modest even in males. Therefore, an additional population was examined to determine if the association, particularly in males, could be replicated. Three of the IL8 markers were typed in a second cohort. Patients in Cohort 2 (n=385, 219 males) were ascertained by the same phenotypic criteria as Cohort 1, but were not all homozygous for the ΔF508 mutation. They did, however, carry at least one copy of the ∆F508 CFTR mutation and another pancreatic insufficiency-associated mutation24 on the second allele. As Table 5 shows, the males in these two cohorts are not significantly different across indices of pulmonary function.
Table 5.
Clinical characteristics of Study Cohorts
| Variable | Severe Males | Mild Males | ||
|---|---|---|---|---|
|
| ||||
| Cohort 1 (n=130) | Cohort 2 (n=69) | Cohort 1 (n=303) | Cohort 2 (n=151) | |
| Age – Mean (± SD) (yrs) | 16.3 ± 4.5 | 17.1 ± 5.1 | 29.7 ± 9.9 | 31.5 ± 10.3 |
| Age – Range (yrs) | 8.1–32 | 8–25.9 | 15.1–54.1 | 15.1–58.2 |
| FEV1 (± SD) (% predicted) | 47.0 ± 17.1 | 46.5 ± 17.8 | 69.8 ± 30.0 | 64.0 ± 29.9 |
| FEV1 (± SD) (decline %/year) | −3.59 ± 2.27 | −3.63 ± 2.01 | −1.41 ± 1.52 | −1.30 ± 1.59 |
| EBint20 (± SD) | 34.5 ± 17.4 | 35.0 ± 17.3 | 84.8 ± 15.3 | 82.0 ± 17.0 |
| Body mass Index (± SD) (percentile) | 17.5 ± 2.05 | 17.8 ± 2.80 | 22.4 ± 2.76 | 22.4 ± 2.78 |
| P. aeruginosa Positive (%) | 85.4 | 85.7 | 85.8 | 90.4 |
| % ΔF508/ΔF508 | 100.0 | 63.0 | 100.0 | 66.7 |
Males and females in Cohort 2 were assessed separately using a χ2 test of association. In accordance with the first population, only male genotypes showed significant associations (0.01>p>0.08) in this second cohort (Table 4). The reproducibility of the association only in males indicates that gender is a major factor in IL8’s role as a genetic modifier of CF. We also examined the IL8 SNPs for departures from Hardy-Weinberg proportions and found no evidence of such departures in either of the cohorts, nor in males alone (P > 0.6).
The IL8 markers display high LD with each other and are therefore largely redundant in their information. An example of the genotype distribution in the studied subjects is provided for the promoter polymorphism A-251T (rs4073) in Table 6. The frequency of males homozygous for the A allele in the mild group is greater than in the severe group (25.6% vs. 18.9%), while the frequency of T homozygotes is greater for the male subjects in the severe group (38.9% vs. 25.1%). Heterozygosity is found more often in the mildly affected males (49.3% vs. 42.1%), suggesting a dominant or semi-dominant effect of the A allele on the mild phenotype.
Table 6.
Comparison of rs4073 genotypes between cohort 1 and 2 reveals gender effect
| %AA | %AT | %TT | Genotypic p-value | |
|---|---|---|---|---|
| Patient Cohort 1: | ||||
| All (n=733): | ||||
| Mild (n=490) | 25.6 | 48.8 | 25.6 | 0.07 |
| Severe (n=243) | 19.2 | 50.8 | 30.0 | |
| Males (n=389): | ||||
| Mild (n=271) | 26.5 | 50.0 | 23.5 | 0.07 |
| Severe (n=118) | 19.5 | 46.9 | 33.6 | |
| Patient Cohort 2: | ||||
| All (n=379): | 22.7 | 46.7 | 30.6 | 0.25 |
| Mild (n=242) | 20.4 | 42.3 | 37.2 | |
| Severe (n=137) | ||||
| Males (n=219): | ||||
| Mild (n=149) | 23.6 | 47.9 | 28.6 | 0.01 |
| Severe (n=70) | 17.5 | 33.3 | 49.2 | |
| Patient Cohorts 1 and 2: | ||||
| All(n=l 112): | ||||
| Mild (n=732) | 24.7 | 48.2 | 27.1 | 0.02 |
| Severe (n=380) | 19.6 | 47.9 | 32.5 | |
| Males (n=608): | ||||
| Mild (n=420) | 25.6 | 49.3 | 25.1 | 0.001 |
| Severe (n= 18 8) | 18.9 | 42.1 | 38.9 |
Effect of other loci and phenotypes on IL8 association
We previously reported an association between variants in the transforming growth factor-β1 gene, TGFB1, and pulmonary function. Logistic regression was used to determine if there were evidence of interaction between these two genes, Using the TGFB1 promoter and codon 10 polymorphisms, no significant interactions were identified with any of the 4 IL8 variants (0.96 > P > 0.46).
To determine if the association found in males might be reflective of other pulmonary conditions, the incidence of physician-reported asthma was assessed by genotype as well and was not significant (0.05 < P <0.4).
Effect of the IL8 promoter variant on expression
A significant statistical association between IL8 gene polymorphisms and CF pulmonary disease has been demonstrated in two populations; however these associations do not address the functional mechanism by which these variants modify phenotype.
A luciferase-reporter assay was used to assess the IL8 promoter variant for effects at the level of transcription. IL8 promoter-luciferase constructs with either the A or T allele at variant rs4073 (rs4073A or rs4073T) were transiently transfected into 9HTEo− cells (pCEP-2), treated with a pro-inflammatory cytokine mix, and luciferase output measured at 6, 12, and 24 hours post-treatment.
Treatment with the cytokine mix induced a significant increase in luciferase expression for both constructs (p<0.001) (Figure 2A), but under all conditions the T allele expressed 2–3 times more than the A allele (p<0.001) (Figure 2B). The fold induction of both constructs by the cytokine mix was similar and non-significant, however (p>0.56, data not shown).
Figure 2.
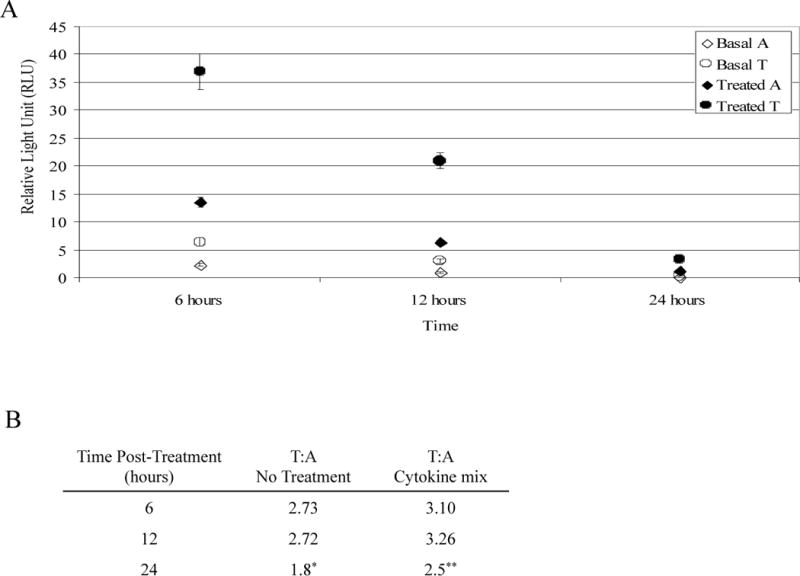
pCEP-2 cells transfected with IL8 promoter-luciferase constructs (rs4073A or rs4073T) were treated with a pro-inflammatory cytokine mix. (a) Luciferase expression at 6, 12, and 24 hours post-treatment. Treatment with the cytokine mix induced a significant increase in luciferase expression for both constructs, (b) Values represent relative luciferase activity of the rs4073T allele to the rs4073A allele (*p=0.08 p=, **p=0.01, all others p<0.0001).
DISCUSSION
Pulmonary disease in CF, which is characterized by recurrent infections and excessive inflammation5, 12, exhibits marked phenotypic variability, due in part to genetic variation, or so-called modifier genes.25–27 In general, a dysregulation of inflammatory responses (with an overall increase in pro-inflammatory and decrease in anti-inflammatory response) is thought to be a key contributor to the lung pathophysiology in CF10, 11. Due to the central role of inflammation in the lung, we attempted to determine whether variants in inflammatory genes could act as modifiers of CF lung disease.
A panel of polymorphisms in candidate inflammatory genes (n=52) thought to be relevant to cystic fibrosis (CF), as determined through a literature review, were screened for associations with level of lung function in a cohort of CF patients (n=737). Polymorphisms in three inflammatory pathway genes showed nominal associations in the initial survey: IL10 receptor (IL10R), TGFβ-induced protein (TGFβI), and interleukin-8 (IL8). IL8 was selected for further characterization because its variants have been reported to associate with susceptibility to other airway disorders18, 19. Additionally, IL-8 is an integral component of CF lung disease; it is elevated, with substantial variation, in the lungs of CF patients6, 20. The primary function of IL-8 is to mediate neutrophil chemotaxis, and CF lung inflammation is largely characterized by neutrophilic infiltrates, leading to subsequent tissue damage28–31. Thus, the role of IL8 polymorphisms in CF pathophysiology should be more readily understood than that of the other candidates.
Our studies found that three polymorphisms within IL8 associate with CF pulmonary disease severity, rs4073, rs2227306 and rs2227307, and the effect was most obvious in males. With respect to pulmonary phenotype, associations between the rs4073 variant and infant susceptibility to bronchiolitis subsequent to infection with respiratory syncytial virus (RSV)18, 32 and bronchial asthma19 have been identified. The studies by Hull and Hacking found that homozygosity for the rs4073A allele is associated with increased occurrence of bronchiolitis following acute RSV infection, while the rs4073T allele is protective against this condition18, 32. In our CF groups, we find that the rs4073A allele is protective and the rs4073T allele is associated with the more severe phenotype. A similar finding was reported in a study in which the rs4073T allele was associated with bronchial asthma, which is a chronic pulmonary condition19. That the same variant seems to have contradicting associations suggests that the functional impact of this variant is context dependent. We propose that the context of inflammation (i.e., acute as in innate defense versus chronic stimulation) may dictate the direction of the association.
The reason for the observed gender effect is not clear but a phenomenon that is not novel in CF. Females with CF have an overall worse prognosis as demonstrated by earlier colonization with P. aeruginosa14, reduced lung function14, and more rapid decline than males, suggesting that the inflammatory burden is greater in the female lung than the male14, 15, 34–36 and culminating in shorter lifespan14–16 for females. Sex hormones are able to regulate cytokine expression33, but there is no evidence that IL8 expression is directly regulated by sex hormones or that there are no known sex hormone-responsive elements in the IL8 promoter. Thus, the sex-specific association is likely more complicated than hormone-responsive transcription. Whether our results reflect an ascertainment bias due to our study design (selecting males and females using the same criteria for both may impose a bias) or a true sex-specific effect could be addressed by replication in populations without exclusion of intermediate phenotypes. In the populations studied here, the relative risk of having severe lung disease for patients homozygous for the “severe” allele of rs4073 is 1.7 (p=0.04) but increases to 2.25 when considering only males (P=0.001).
As there are no known common variants in the coding sequence one of IL8, variation is most likely due to quantitative effects. Two of the variants are located in introns (rs2227307 and rs2227543) while the remaining variant (rs4073) is found in the promoter region of IL8 and has been shown to associate with several pulmonary phenotypes18, 19, 32 and to have demonstrated functional effects18, 32. We therefore felt this variant to be the most likely candidate to impart the functional effect and rs4073 was therefore the target for functional testing. We found that the rs4073T variant induces a 2–3 fold increase in transcriptional activity relative to the rs4073A allele. Although such a modest fold change may not be usually considered biologically significant, context must be considered when investigating genetic modifiers of disease. As stated, CF patients are known to have abnormally high levels of IL-8 in their lungs6, 20. Given a scenario in which elevated IL-8 is associated with severe pulmonary disease, it is entirely plausible that a moderate, yet significant, and chronic increase in IL-8 could account for more severe lung disease. Based on our findings, we propose that regardless of colonization or exacerbation status, CF patients with the rs4073T allele have higher levels of IL8 expression and are thus more likely to have severe pulmonary disease.
Interpretation of previous evidence regarding IL8 transcript regulation is somewhat unclear. Hacking et. al.32 found that when assessed in basal and TNF-α stimulated reporter constructs, the T allele at −251 (rs4073T), which was predominant in our severe phenotypic groups, has more than twice the promoter activity of the A allele, which is similar to our findings32. However, when endogenous transcript levels are compared in primary epithelial cells, the TNF-α stimulation results in marginally higher levels from the haplotype containing the A allele32.
Our reporter construct assays are in accord with similar studies, but we have not yet assessed endogenous transcript levels. Sequence analysis suggests the rs4073 variant alters binding for the Oct-1 transcription factor, which has been shown to regulate the expression of several cytokines37, 38 including IL832, 39. The IL8 variants do appear to alter expression levels, but mechanism of the regulation may be context dependent and will be explored in the future.
Other studies have identified potential genetic modifiers of CF; Hull et. al. showed variants in tumor necrosis factor-α (TNF-α) associate with FEV1 in CF patients,40–43 although we have not been able to replicate these findings in a larger group of subjects1. We have, however, found that variants within the transforming growth factor-β1 (TGF-β1) gene correlate with CF patient disease severity1. The findings presented here show that variants in IL8 associate reproducibly with lung disease severity in males with cystic fibrosis and that there is functional variation associated with one of the polymorphism. Taken in context with the biological role of IL-8, this gene likely modifies lung function in CF through different levels of neutrophil chemotaxis and subsequent differences in destruction of lung tissue.
METHODS
Patient Recruitment
The initial study cohort was made up of 737 CF patients enrolled in the Gene Modifier Study (GMS), ascertained by methods described previously1, 17. Briefly: All study subjects in cohort 1 are CF patients homozygous for the ΔF508 CFTR mutation. Based on data obtained from the National CF Patient Registry, upper and lower quartiles of lung function for age were determined. Using these values as a guide, subjects were designated as “mild” (n=491) or “severe” (n=246), respectively, to reflect their lung function profile. A case-control study design was used to compare these groups, with patients with severe lung disease arbitrarily set as the case population and the mild group as the control population.
A second cohort ascertained for the same clinical criteria as the first, but including ΔF508 compound heterozygous patients, were used for the replication study (n=385). Cohort 2 patients not homozygous for ΔF508 carry one copy of the ΔF508 CFTR mutation and a mutation recognized as severe on the second allele, with severe defined as being associated with pancreatic exocrine insufficiency1. The same criteria for “mild” and “severe” used for cohort 1 were applied to the second cohort.
Both cohorts were assessed for indices of pulmonary function, including average forced expiratory volume in 1 second (FEV1), rate of decline in FEV1 and predicted FEV1 at age 20 (EBint2017), as well as bacterial colonization and body mass index.
Genotyping
In the initial screen, genotyping was performed by SNP BeadArray™ (Illumina, Incorporated, San Diego, CA) technology and included the following polymorphic loci within IL8 (position relative to the transcriptional start site, number of patients genotyped): rs4073 (A-251T, n=733), rs2227307 (G396T, n=732), and rs2227543 (T1632C, n=737). Patients not genotyped by Illumina, Inc. were genotyped at the following IL8 loci using restriction fragment length polymorphism (RFLP) assays. Primers and restriction enzymes used for the RFLP genotyping were (SNP, forward, reverse, product size, restriction endonuclease): rs4073, 5′-TGAAAAGTTGTAGTATGCCCC-3′, 5′-CATTTAAAATACTGAAGCTCCACAATTTGGTGAATTATCgA-3′, 310 bp, ClaI; and rs2227543, 5′-TGGTTAAAAAAAAAGGAATAGCATCAATAGTGAGTTTGTTGTcCT-3′, 5′-ACCCTACAACAGACCCACACAATA-3′, 420 bp, MnlI. Lower case letters indicate a sequence change in order to introduce a restriction endonuclease recognition site. Digestion products were analyzed on a 2% agarose gel. Reactions were performed using Invitrogen Platinum Taq polymerase enzyme, 10× buffer, deoxyribonucleotides (2.5mM), and MgCl2 (25mM). Annealing temperature, MgCl2 concentration, and amplification cycles were optimized for each primer set.
Variants in the RASSF6 (rs13131954, n=717), IL8 (rs2227306, n=727), CXCL6 (rs4694639, n=709), and PF4VI (rs941758, n=716) genes were genotyped using an Assay-on-Demand™ assay (Applied Biosystems, Foster City, CA).
Statistical Analysis
The distribution of the marker alleles and genotypes in the severe group was compared to that in the mild group using a chi-square test of independence. A goodness of fit test for Hardy-Weinberg Equilibrium (HWE) was performed by comparing the genotype frequencies with those expected on the basis of the observed allele frequencies44. SNPs with a p<0.05 for the test of HWE were noted as possibly requiring careful consideration of the possible causes of departure from HWE, but were not removed from the analysis. The IL8 and flanking polymorphisms were found to exist in HWE, 0.27<p<0.96. All analyses were conducted in SPlus version X.0 (Insightful Corporation, Seattle, WA).
Population stratification
The current study was part of a larger project in which 737 subjects were typed for 810 markers. The genomic control method45 was performed using these markers to examine the potential impact of population structure in this study. The observed values of the inflation factor λ ranged from 1 to 1.29, and were used to adjust the p-values of the IL-8 regional markers.
Pairwise Linkage Disequilibrium (LD)
The pattern of pairwise LD between SNPs was measured by the correlation coefficient (R2)46. R2 varies from 0 to 1 and measures the correlation between alleles on the same chromosome. R2 will take the value of 1 when the allele at one locus can always be predicted by the allele at the second locus (that is, if only two haplotypes are present).
IL8 Promoter Luciferase Assay
The IL8 promoter (1.5kb) was cloned into a pGL3 vector (Promega Corporation, Madison, Wisconsin) containing the firefly luciferase gene. Using site-directed mutagenesis, two versions of the construct were generated to reflect the polymorphism at the −251 position (relative to the start of transcription), corresponding to SNP rs4073. The constructs are designated −251A/rs4073A and −251T/rs4073T.
These constructs were transiently co-transfected with the Renilla luciferase reporter construct (Promega) into control (pCEP-2) and CF-like (pCEP-RF) 9HTEo- cell lines47 using FuGene 6 Transfection Reagent (Roche Applied Science, Indianapolis, Indiana). As results from the pCEP-2 and pCEP-RF cells lines were similar, only data from pCEP-2 cells are shown.
To induce IL8-promoter driven luciferase expression48, transfected cells were treated with physiologically relevant49 quantities of a pro-inflammatory cytokine mixture (interleukin-1β [0.167 ng/μl] and tumor necrosis factor-α [0.33 ng/μl]) in serum-free culture media 24 hours after transfection. Treatment with the cytokine mix was intended to mimic the pro-inflammatory state in CF airway in which there are increased levels of IL-1β and TNF-α, both of which are considered key pro-inflammatory cytokines. Cells were also treated with hydrogen peroxide (H2O2), as oxidative stress has been shown promote inflammation50, which yielded a similar expression pattern to cytokine mix treatment (data not shown). Control cells were not exposed to stimulus, but rather to the phosphate-buffered saline diluent.
Cell lysates were harvested using the Dual-Luciferase Kit (Promega) at 6, 12, 24, 36 and 48, 60, and 72 hours after treatment and luciferase activity measured in a luminometer (Turner BioSystems, Sunnyvale, California). All samples returned to baseline by 24 hours and thus data are shown for 6, 12, and 24 hour time points only. The average relative luminescence was obtained for each condition (n=30 wells). Comparisons were made between treated and untreated cells, as well as between alleles. In all cases comparisons were assessed by a two-tailed, student’s t-test.
The cytokine mix was found to significantly repress Renilla expression (p<0.04 – p<0.005), thereby falsely inflating the magnitude of the observed response to the cytokine mix. Therefore, Renilla expression was only used to determine if transfection efficiencies were comparable for each time point. While expression estimates from the control samples were normalized to the co-transfected Renilla construct, the level of expression from the treated samples was not. Rather, multiple replicates were performed to ensure accuracy (30 individually transfected wells for each condition).
Acknowledgments
This work was supported by NIH grants HL68890, P30 DK27651, T32 HL07515, DK066368 GCRC RR00046, and grants from the Cystic Fibrosis Foundation.
References
- 1.Drumm ML, Konstan MW, Schluchter MD, Handler A, Pace R, Zou F, et al. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443–53. doi: 10.1056/NEJMoa051469. [DOI] [PubMed] [Google Scholar]
- 2.Welsh MJ, R B, Accurso FJ, Cutting GR. In: Cystic fibrosis. Scriver CR, B A, Sly WS, Valle D, editors. McGraw Hill; New York: 2000. [Google Scholar]
- 3.Kerem E, Kerem B. Genotype-phenotype correlations in cystic fibrosis. Pediatr Pulmonol. 1996;22:387–95. doi: 10.1002/(SICI)1099-0496(199612)22:6<387::AID-PPUL7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 4.Vanscoy LL, Blackman SM, Collaco JM, Bowers A, Lai T, Naughton K, et al. Heritability of lung disease severity in cystic fibrosis. Am J Respir Crit Care Med. 2007;175:1036–43. doi: 10.1164/rccm.200608-1164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chmiel JF, Berger M, Konstan MW. The role of inflammation in the pathophysiology of CF lung disease. Clin Rev Allergy Immunol. 2002;23:5–27. doi: 10.1385/CRIAI:23:1:005. [DOI] [PubMed] [Google Scholar]
- 6.Bonfield TL, Konstan MW, Berger M. Altered respiratory epithelial cell cytokine production in cystic fibrosis. J Allergy Clin Immunol. 1999;104:72–8. doi: 10.1016/s0091-6749(99)70116-8. [DOI] [PubMed] [Google Scholar]
- 7.Colombo C, Crosignani A, Battezzati PM. Liver involvement in cystic fibrosis. J Hepatol. 1999;31:946–54. doi: 10.1016/s0168-8278(99)80299-2. [DOI] [PubMed] [Google Scholar]
- 8.Hubeau C, Puchelle E, Gaillard D. Distinct pattern of immune cell population in the lung of human fetuses with cystic fibrosis. J Allergy Clin Immunol. 2001;108:524–9. doi: 10.1067/mai.2001.118516. [DOI] [PubMed] [Google Scholar]
- 9.Chmiel JF, Konstan MW, Knesebeck JE, Hilliard JB, Bonfield TL, Dawson DV, et al. IL-10 attenuates excessive inflammation in chronic Pseudomonas infection in mice. Am J Respir Crit Care Med. 1999;160:2040–7. doi: 10.1164/ajrccm.160.6.9901043. [DOI] [PubMed] [Google Scholar]
- 10.Stecenko AA, King G, Torii K, Breyer RM, Dworski R, Blackwell TS, et al. Dysregulated cytokine production in human cystic fibrosis bronchial epithelial cells. Inflammation. 2001;25:145–55. doi: 10.1023/a:1011080229374. [DOI] [PubMed] [Google Scholar]
- 11.Corvol H, Fitting C, Chadelat K, Jacquot J, Tabary O, Boule M, et al. Distinct cytokine production by lung and blood neutrophils from children with cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2003;284:L997–1003. doi: 10.1152/ajplung.00156.2002. [DOI] [PubMed] [Google Scholar]
- 12.Konstan MW. Treatment of airway inflammation in cystic fibrosis. Curr Opin Pulm Med. 1996;2:452–6. [PubMed] [Google Scholar]
- 13.Konstan MW, Davis PB. Pharmacological approaches for the discovery and development of new anti-inflammatory agents for the treatment of cystic fibrosis. Adv Drug Deliv Rev. 2002;54:1409–23. doi: 10.1016/s0169-409x(02)00146-1. [DOI] [PubMed] [Google Scholar]
- 14.Demko CA, Byard PJ, Davis PB. Gender differences in cystic fibrosis: Pseudomonas aeruginosa infection. J Clin Epidemiol. 1995;48:1041–9. doi: 10.1016/0895-4356(94)00230-n. [DOI] [PubMed] [Google Scholar]
- 15.Davis PB. The gender gap in cystic fibrosis survival. J Gend Specif Med. 1999;2:47–51. [PubMed] [Google Scholar]
- 16.Rosenfeld M, Davis R, FitzSimmons S, Pepe M, Ramsey B. Gender gap in cystic fibrosis mortality. Am J Epidemiol. 1997;145:794–803. doi: 10.1093/oxfordjournals.aje.a009172. [DOI] [PubMed] [Google Scholar]
- 17.Schluchter MD, Konstan MW, Drumm ML, Yankaskas JR, Knowles MR. Classifying severity of cystic fibrosis lung disease using longitudinal pulmonary function data. Am J Respir Crit Care Med. 2006;174:780–6. doi: 10.1164/rccm.200512-1919OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hull J, Thomson A, Kwiatkowski D. Association of respiratory syncytial virus bronchiolitis with the interleukin 8 gene region in UK families. Thorax. 2000;55:1023–7. doi: 10.1136/thorax.55.12.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinzmann A, Ahlert I, Kurz T, Berner R, Deichmann KA. Association study suggests opposite effects of polymorphisms within IL8 on bronchial asthma and respiratory syncytial virus bronchiolitis. J Allergy Clin Immunol. 2004;114:671–6. doi: 10.1016/j.jaci.2004.06.038. [DOI] [PubMed] [Google Scholar]
- 20.Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, et al. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med. 1995;152:2111–8. doi: 10.1164/ajrccm.152.6.8520783. [DOI] [PubMed] [Google Scholar]
- 21.Hull J, Ackerman H, Isles K, Usen S, Pinder M, Thomson A, et al. Unusual haplotypic structure of IL8, a susceptibility locus for a common respiratory virus. Am J Hum Genet. 2001;69:413–9. doi: 10.1086/321291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Donovan N, Galvin M, Morgan JG. Physical mapping of the CXC chemokine locus on human chromosome 4. Cytogenet Cell Genet. 1999;84:39–42. doi: 10.1159/000015209. [DOI] [PubMed] [Google Scholar]
- 23.Hull J, Rowlands K, Lockhart E, Sharland M, Moore C, Hanchard N, et al. Haplotype mapping of the bronchiolitis susceptibility locus near IL8. Hum Genet. 2004;114:272–9. doi: 10.1007/s00439-003-1038-x. [DOI] [PubMed] [Google Scholar]
- 24.Tsui LC. The spectrum of cystic fibrosis mutations. Trends Genet. 1992;8:392–8. doi: 10.1016/0168-9525(92)90301-j. [DOI] [PubMed] [Google Scholar]
- 25.Cutting GR. Modifier genetics: cystic fibrosis. Annu Rev Genomics Hum Genet. 2005;6:237–60. doi: 10.1146/annurev.genom.6.080604.162254. [DOI] [PubMed] [Google Scholar]
- 26.Slieker MG, Sanders EA, Rijkers GT, Ruven HJ, van der Ent CK. Disease modifying genes in cystic fibrosis. J Cyst Fibros. 2005;4(Suppl 2):7–13. doi: 10.1016/j.jcf.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Davies JC, Griesenbach U, Alton E. Modifier genes in cystic fibrosis. Pediatr Pulmonol. 2005;39:383–91. doi: 10.1002/ppul.20198. [DOI] [PubMed] [Google Scholar]
- 28.Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med. 1996;154:1229–56. doi: 10.1164/ajrccm.154.5.8912731. [DOI] [PubMed] [Google Scholar]
- 29.Berger M. Inflammation in the lung in cystic fibrosis. A vicious cycle that does more harm than good? Clin Rev Allergy. 1991;9:119–42. doi: 10.1007/978-1-4612-0475-6_8. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura H, Yoshimura K, McElvaney NG, Crystal RG. Neutrophil elastase in respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene expression in a human bronchial epithelial cell line. J Clin Invest. 1992;89:1478–84. doi: 10.1172/JCI115738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Devaney JM, Greene CM, Taggart CC, Carroll TP, O’Neill SJ, McElvaney NG. Neutrophil elastase up-regulates interleukin-8 via toll-like receptor 4. FEBS Lett. 2003;544:129–32. doi: 10.1016/s0014-5793(03)00482-4. [DOI] [PubMed] [Google Scholar]
- 32.Hacking D, Knight JC, Rockett K, Brown H, Frampton J, Kwiatkowski DP, et al. Increased in vivo transcription of an IL-8 haplotype associated with respiratory syncytial virus disease-susceptibility. Genes Immun. 2004;5:274–82. doi: 10.1038/sj.gene.6364067. [DOI] [PubMed] [Google Scholar]
- 33.Trotter A, Muck K, Grill HJ, Schirmer U, Hannekum A, Lang D. Gender-related plasma levels of progesterone, interleukin-8 and interleukin-10 during and after cardiopulmonary bypass in infants and children. Crit Care. 2001;5:343–8. doi: 10.1186/cc1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The International HapMap Project. Nature. 2003;426:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 35.Parad RB, Gerard CJ, Zurakowski D, Nichols DP, Pier GB. Pulmonary outcome in cystic fibrosis is influenced primarily by mucoid Pseudomonas aeruginosa infection and immune status and only modestly by genotype. Infect Immun. 1999;67:4744–50. doi: 10.1128/iai.67.9.4744-4750.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenfeld M, D R, FitzSimmons S, Pepe M, Ramsey B. Gender gap in cystic fibrosis mortality. Am J Epidemiol. 1997;145:794–803. doi: 10.1093/oxfordjournals.aje.a009172. [DOI] [PubMed] [Google Scholar]
- 37.Kaushansky K, Shoemaker SG, O’Rork CA, McCarty JM. Coordinate regulation of multiple human lymphokine genes by Oct-1 and potentially novel 45 and 43 kDa polypeptides. J Immunol. 1994;152:1812–20. [PubMed] [Google Scholar]
- 38.Knight JC, Udalova I, Hill AV, Greenwood BM, Peshu N, Marsh K, et al. A polymorphism that affects OCT-1 binding to the TNF promoter region is associated with severe malaria. Nat Genet. 1999;22:145–50. doi: 10.1038/9649. [DOI] [PubMed] [Google Scholar]
- 39.Wu GD, Lai EJ, Huang N, Wen X. Oct-1 and CCAAT/enhancer-binding protein (C/EBP) bind to overlapping elements within the interleukin-8 promoter. The role of Oct-1 as a transcriptional repressor. J Biol Chem. 1997;272:2396–403. [PubMed] [Google Scholar]
- 40.Hull J, Thomson AH. Contribution of genetic factors other than CFTR to disease severity in cystic fibrosis. Thorax. 1998;53:1018–21. doi: 10.1136/thx.53.12.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Courtney JM, Plant BJ, Morgan K, Rendall J, Gallagher C, Ennis M, et al. Association of improved pulmonary phenotype in Irish cystic fibrosis patients with a 3′ enhancer polymorphism in alpha-1-antitrypsin. Pediatr Pulmonol. 2006;41:584–91. doi: 10.1002/ppul.20416. [DOI] [PubMed] [Google Scholar]
- 42.Davies JC, Turner MW, Klein N. Impaired pulmonary status in cystic fibrosis adults with two mutated MBL-2 alleles. Eur Respir J. 2004;24:798–804. doi: 10.1183/09031936.04.00055404. [DOI] [PubMed] [Google Scholar]
- 43.Blaisdell CJ, Howard TD, Stern A, Bamford P, Bleecker ER, Stine OC. CLC-2 single nucleotide polymorphisms (SNPs) as potential modifiers of cystic fibrosis disease severity. BMC Med Genet. 2004;5:26. doi: 10.1186/1471-2350-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sasieni PD. From genotypes to genes: doubling the sample size. Biometrics. 1997;53:1253–61. [PubMed] [Google Scholar]
- 45.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 46.Hill WG, Robertson A. Linkage disequilibrium in finite populations. Theor Appl Genet. 1968;38:226–231. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- 47.Bryan R, Kube D, Perez A, Davis P, Prince A. Overproduction of the CFTR R domain leads to increased levels of asialoGM1 and increased Pseudomonas aeruginosa binding by epithelial cells. Am J Respir Cell Mol Biol. 1998;19:269–77. doi: 10.1165/ajrcmb.19.2.2889. [DOI] [PubMed] [Google Scholar]
- 48.Kelley TJ, Elmer HL, Corey DA. Reduced Smad3 protein expression and altered transforming growth factor-beta1-mediated signaling in cystic fibrosis epithelial cells. Am J Respir Cell Mol Biol. 2001;25:732–8. doi: 10.1165/ajrcmb.25.6.4574. [DOI] [PubMed] [Google Scholar]
- 49.Osika E, Cavaillon JM, Chadelat K, Boule M, Fitting C, Tournier G, et al. Distinct sputum cytokine profiles in cystic fibrosis and other chronic inflammatory airway disease. Eur Respir J. 1999;14:339–46. doi: 10.1034/j.1399-3003.1999.14b17.x. [DOI] [PubMed] [Google Scholar]
- 50.Adcock IM, Ito K, Barnes PJ. Histone deacetylation: an important mechanism in inflammatory lung diseases. Copd. 2005;2:445–55. doi: 10.1080/15412550500346683. [DOI] [PubMed] [Google Scholar]