

A fundamental uncertainty in cellular cholesterol homeostasis is the role of cholesterol mass transport by aqueous diffusion between hydrophobic lipid membranes such as the cell plasma membrane and lipoprotein in extra-cellular space1. The solubility of cholesterol in water is low (estimated at 200 nM)2 and the probable significance of mass transfer by aqueous diffusion to the kinetics of specific cholesterol trafficking pathways is not understood. This communication reports cholesterol oxidase modified platinum micro-cavity electrodes for detection of plasma membrane cholesterol efflux. The data are constant with the notion that cholesterol in the cell plasma membrane undergoes exchange with cholesterol in solution.
The micro-cavity electrode is positioned in contact with the cell plasma membrane dictating a fixed distance between the cell surface and the enzyme-modified electrode surface (Figure 1). The mouth of the micro-cavity electrode defines a point of contact at the cell surface where efflux occurs. Enzymatic consumption of cholesterol at the recessed electrode surface lowers the concentration of cholesterol in solution in the cavity volume. This results in a decreased rate of unidirectional influx of cholesterol from solution in the cavity to the plasma membrane. Net efflux of cholesterol from the plasma membrane to solution in the cavity is, thus, driven by enzymatic consumption of cholesterol at the electrode surface.
Figure 1.
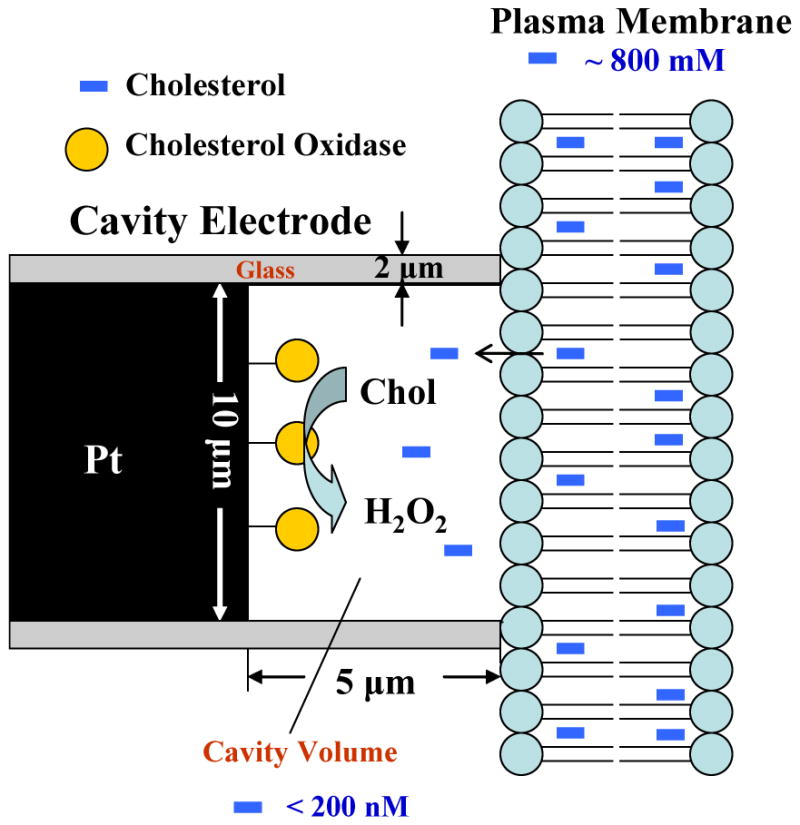
Scheme of micro-cavity electrode positioned in contact with cell plasma membrane.
The distance between the cell surface and the electrode surface (i.e., the 5 μm cavity depth) is one factor controlling the cholesterol concentration gradient at the cell surface and thus the rate of cholesterol efflux. The other major factors controlling the efflux rate are enzymatic activity at the electrode surface and the efflux rate exhibited by the cell plasma membrane at the point of contact (theoretically the activity of cholesterol in the membrane3).
With the micro-cavity electrode positioned in direct contact with the plasma membrane surface (as indicated by a characteristic drop in electrode capacitance of about 10%) of a single neuron in the buccal ganglion of Aplysia, the electrode is poised at an experimentally determined potential that does not oxidize or reduce hydrogen peroxide. This hydrogen peroxide collection potential is held for 5 minutes to allow cholesterol efflux from the plasma membrane and accumulation of a measurable concentration of hydrogen peroxide in the cavity volume. Two sequential potential step experiments are preformed for analog chrono-coulombmetric analysis of the accumulated hydrogen peroxide (Figure 2A). The first potential step (Figure 2A, step 1) is from 200 mV (the hydrogen peroxide collection potential) to 600 mV for complete oxidization of the hydrogen peroxide confined in the cavity volume (Figure 2B; trace labeled step 1). The potential is stepped back to 200 mV for 30 seconds and a replicate of the first potential step is conducted (Figure 2A, step 2) to gauge the background charge (Figure 2B, trace labeled step 2). Subtraction of the background charge measured in the second potential step from the charge past in the first potential step is an estimate of enzymatic oxidation of cholesterol and accumulation of hydrogen peroxide in the cavity volume during the 5 minute hold time (Figure 2C, trace labeled Enzyme modified). The background charge can drift over minutes and this sequential potential step method allows the background to be continuously tracked and subtracted to give charge from hydrogen peroxide oxidation. The method provides an absolute charge measurement so data acquisition does not rely on a reference current as is the case with the amperometric disk electrode experiments reported earlier4.
Figure 2.

(A) Voltage perturbation for two sequential potential step experiments; (B) Overlaid charge collected for the two potential steps; (C) Background subtracted (difference) charge collected at the enzyme modified electrode (red) and bare electrode (black) at the cell (1, 2, 3 indicate the chronological order); (D) Average difference charge for four consecutive determinations at the cell and cyclodextrin (500 μM for one hour) treated cell using the enzyme modified electrode and bare electrode (difference charge taken at 1 second).
The background charge measured in step 2 must contain a contribution from hydrogen peroxide being generated during the 30 second quite time between the first and second potential step experiments. This is a systematic error where signal is lost. Also, the rate at which hydrogen peroxide escapes from the cavity is uncertain and is also assumed to be a systematic error. Thus, the efflux rate estimated below is a lower limit for the actual efflux rate. Optimization of the coulombmetry is discussed in the Supplemental Information.
The difference charge at one second also reflects faradaic reactions of other species. This faradaic background charge is estimated using the bare platinum micro-cavity electrode (electrode prior to enzyme modification) positioned in contact with the same cell (Figure 2C, trace labeled Bare). Within one second, the difference charge from the enzyme-modified electrode reaches a maximum value relative to the difference charge from the bare cavity electrode (Figure 2C, 1–3 seconds). The larger charge for the enzyme-modified electrode is due to hydrogen peroxide oxidation. Hydrogen peroxide oxidation is driven at a mass transfer controlled rate at 600 mV and the cavity volume is quickly depleted. Comparing averaged data from four sequential and replicate experiments conducted at the bare and enzyme modified electrode surfaces suggests that about 75% of the difference charge past at the enzyme-modified electrode at 1 second (Figure 2D, bars 1 and 2, respectively) is from oxidation of hydrogen peroxide. The average efflux rate (lower limit) is calculated to be 1 pmol/cm2 s, where the efflux site (contact area of the cavity mouth) is 0.5% of the total area of the cell surface. Assuming linear diffusion of cholesterol from the cavity mouth to the electrode surface, a concentration gradient of Δ 7–8 nM is predicted across the 0.4 pL cavity volume. The total estimated amount of cholesterol extracted from the cell over 5 minutes is about 240 million molecules; 1.2 times the amount contained at the contact site (plasma membrane outer leaflet covering the cavity mouth) and 0.6% of the estimated total cholesterol in plasma membrane outer leaflet.
Cells partially depleted of cholesterol using cyclodextrin solution are studied to verify that the measurements are cholesterol dependent. After cholesterol depletion, the electrode showed a smaller difference charge indicating a decreased rate of cholesterol oxidation during the hold time (Figure 2D, bar 3) and, thus, slower efflux of cholesterol from the cell plasma membrane. Exposure of the cells to cyclodexrin solution with higher concentration results in an additional decrease in cholesterol efflux.
It is not yet possible to reproduce the surface area of the etched platinum micro-cavity electrodes. This causes variation in the amount of enzyme immobilized on different electrodes. Micro-cavity electrodes with higher surface area (as gauged by double layer charge) show higher activity for oxidation of cholesterol in solution and larger responses at cells (supporting information). The faradaic background charge also correlates with electrode surface area.
Characterization of micro-cavity electrodes for enzymatic activity is conducted in cholesterol/Triton X-100 solution. Figure 3A shows the difference charge for an enzyme modified micro-cavity electrode in buffer and in 300 μM cholesterol solution. Three sequential determinations of the difference charge are shown overlaid for the buffer and cholesterol solution. Figure 3B is a bar graph including average data for 100,300 and 600 μM solutions.
Figure 3.
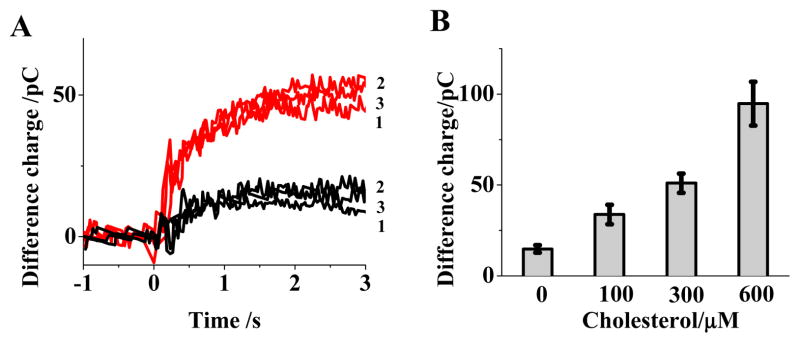
(A) Difference charge at an enzyme-modified electrode exposed to 300 μM cholesterol (red) and buffer solution (black) (B) Bar graph for average difference charge measured at the enzyme modified electrode exposed to 0, 100, 300 and 600 μM cholesterol solutions.
While the solution data provide a measure of enzymatic activity, it is not possible to directly compare the solution data with data collected at the cell surface. For the cell experiments, the plasma membrane likely acts as a barrier slowing escape of hydrogen peroxide from the cavity volume. For the solution experiments, the mouth of the cavity is open and hydrogen peroxide can readily escape. Additionally, for the solution experiments, the cholesterol concentration gradient at the electrode surface extends beyond the cavity mouth into solution. For the cell experiments, the plasma membrane gates cholesterol mass transport to the cavity volume and, therefore, it is proposed that the cavity depth largely dictates the thickness of the diffusion gradient.
That is, it is proposed that mass transfer of cholesterol in the plasma membrane is fast relative to the rate of efflux. A consequence of the cavity electrode geometry and the defined diffusion distance between the electrode surface and the plasma membrane is that cholesterol efflux occurs at a relatively slower rate compared to the disk electrode measurements (estimated to be 3–4 times slower). Slower efflux at the micro-cavity electrode, while maintaining the small size of the efflux site, leads to a measurement that is expected to be more reflective of the efflux step and to be less influenced by depletion of cholesterol at the electrode contact site.
The degree to which the micro-cavity measurement locally depletes plasma membrane cholesterol is of concern regarding the relevance of the measured efflux rate to the unidirectional efflux rate occurring at pseudo-equilibrium. The literature regarding cholesterol efflux from cells and model lipid membranes is consistent with the existence of two pools of plasma membrane cholesterol. Most cholesterol molecules in the plasma membrane efflux slow to cyclodextrin solution (t1/2: ca. 20 minutes)5 and are likely associated with phospholipids.3,6 A smaller amount of the cholesterol, however, effluxes fast to cyclodextrin solution (t1/2: ca. 15 seconds) and likely reflects excess cholesterol above the stoichiometric amount that can be accommodated through phospholipid association3,6. This amount of “excess” cholesterol probably serves as a switch in cell signaling for cholesterol trafficking.3
The aqueous efflux model for the micro-cavity electrode measurements assumes no direct contact between the plasma membrane and the electrode surface. It is also assumed that no enzyme is immobilized on the glass walls of the cavity or at parameter of the cavity mouth. This issue and the consequences on the physical contact between the electrode tip and the cell surface are uncertain. Still, the charge measurement at the micro-cavity electrode provides a qualitative estimate of the homeostatic plasma membrane cholesterol content for living cells.
Acknowledgments
This work was supported by the Cystic Fibrosis Foundation and the Department of Chemistry, Case Western Reserve University. Helpful discussions with Professors Barry Miller, Dan Scherson, and Theodore Steck are acknowledged.
Footnotes
Supporting Information available: Details for electrode fabrication and method optimization.
References
- 1.Rothblat GH, Phillips MC. J Biol Chem. 1982;257:4775–2. [PubMed] [Google Scholar]
- 2.Haberland ME, Reynolds JA. Proc Natl Acad Sci U S A. 1973;70:2313–6. doi: 10.1073/pnas.70.8.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radhakrishnan A, McConnell HM. Biochemistry. 2000;39:8119–24. doi: 10.1021/bi0005097. [DOI] [PubMed] [Google Scholar]
- 4.Jiang DC, Devadoss A, Palencsar MS, Fang DJ, White NM, Kelley TJ, Smith JD, Burgess JD. J Am Chem Soc. 2007;129:11352–3. doi: 10.1021/ja074373c. [DOI] [PubMed] [Google Scholar]
- 5.Haynes MP, Phillips MC, Rothblat GH. Biochemistry. 2000;39:4508–17. doi: 10.1021/bi992125q. [DOI] [PubMed] [Google Scholar]
- 6.Lange Y, Ye J, Steck TL. Proc Natl Acad Sci U S A. 2004;101:11664. doi: 10.1073/pnas.0404766101. [DOI] [PMC free article] [PubMed] [Google Scholar]